Introduction
Acute myeloid leukemia (AML) is a heterogeneous
disease characterized by clonal expansion of myeloid progenitors,
called blasts (1). Its median
survival time without treatment is 17 weeks (2) and its median age at diagnosis is 69
years (3). The incidence is
reported as 4 cases per 100,000 population (4).
Although there is rapidly increasing knowledge about
the pathogenesis of AML, particularly regarding the role of
genetics (5–7), the treatment of AML has not changed
fundamentally in the last four decades (1) and the ‘7+3’ chemotherapy regimen is
still the standard of care (4).
Targeted therapies, such as midostaurin, a
multikinase inhibitor (8), or
enasidenib, an IDH2-inhibitor (9),
are slow to be established in the clinical setting and are only
approved for a small cohort of patients so far (9).
Molecular therapies have also failed to prove
themselves as universal remedy until now (10), so other ways of dealing with this
life-threatening disease are the subject of intensive research.
Among these, therapeutic antibodies which attack malignant cells,
e.g., gemtuzumab ozogamicin, a CD33 immunotoxin conjugate (11), are particularly promising candidates
for the effective treatment of AML.
Phage display is an effective technique for the
development of novel antibodies (12,13).
This in vitro method uses the genotype to phenotype linkage
of bacteriophages to express different types of recombinant
antibodies on the surface of phage particles (14). Antibody fragments are selected
during several rounds of panning by their affinity to a given, not
necessarily known, but relevant antigen, such as the surface of
intact cells, cell lysates, fixed tissues or membrane fractions
(15–19). Furthermore, in vitro
selection techniques allow optimizing the biopanning conditions,
such as modifying the negative (depletion) and positive (selection)
steps to facilitate the isolation of antibodies with desired
binding properties. However, selection and depletion conditions
vary tremendously between different laboratories regarding e.g.,
applied biopanning protocols, phage antibody libraries, and target
antigens.
Particularly the depletion step (DS), i.e., the
absorption of non-specific antibody phage particles to minimize
false-positive rates and, therefore, to exclude cross-reactive
antibodies, is performed very differently in different labs. While
it has been determined that mononuclear cells (PBMCs) are widely
established as depletion antigen fraction during whole-cell panning
procedures (19–22), the efficiencies of PBMC-depleted
libraries are often limited. Therefore, we hypothesized that using
mononuclear cells, i.e., lymphocytes and monocytes only, for
depletion is not sufficient to achieve efficient elimination of
unspecific phage particles.
Moreover, we postulated that efficient elimination
of unspecific phage particles during the whole-cell-biopanning
process has major impact on the generation of target cell-specific
antibodies. Therefore, we examined the correlation between
depletion efficiency and depletion antigen diversity in different
depletion approaches by adding or overrepresenting different
specific cell fractions from healthy whole blood samples.
Our aim was to optimize and standardize the
biopanning methodology to generate target- specific antibodies
against primary AML blasts by comparing different panning
procedures. For this, we adapted phage selection procedures in
consideration of patient blast cell counts and found that quantity
and quality of blast cells as well as depletion antigen diversity
are critical for the selection outcome.
The result of this research will have significant
implications on the design and optimization of library selection
strategies using primary patient-derived tumor cells as target
antigens.
Materials and methods
Literature research
A literature research was performed at https://www.ncbi.nlm.nih.gov/pubmed. After
analyzing the query results, seven articles were selected which
show the most important differences among different labs.
Cells of AML patients and healthy
blood samples
All AML patients gave written informed consent, and
the clinical ethics committee of the Justus Liebig University
Giessen approved all experimental work (ref: 140/16). Healthy
leukocyte samples were obtained from voluntary donors with blood
groups A, B, and 0. If necessary, those samples were stored using
density gradient centrifugation (DGC) and cryoconservation
(Freezing medium; Life Technologies; #12648-010).
In addition, AB blood serum was received from
voluntary donors after written informed consent as well. Approval
of the clinical ethics committee of the Justus Liebig University
Giessen was also obtained for this procedure (ref: 05/00).
Cell culture and cell methods
Kasumi-1 AML cell line
The human AML-M2-derived cell line Kasumi-1 was used
as model cell line for e.g., testing of internalization behavior.
The cell line was purchased from the German Resource Centre for
Biological Material (DSMZ; ACC 220). Kasumi-1 cells were cultured
in 80% RPMI-1640 Gluta- MAX™ medium (RPMI; Life Technologies;
#61870-044) and supplemented with 20% fetal bovine serum (FBS; Life
Technologies; #10270-106) at 37°C, 5% CO2 (19).
293T cell line and transfection
The embryonic kidney cell line 293T, purchased from
the American Type Culture Collection (ATCC; CRL 3216), was cultured
in 90% RPMI containing 10% FBS and 1% penicillin-streptomycin
mixture (Life Technologies; #15140-122) (19).
The cell line was used for transfection by
incubating 5×104 cells with 1 mg pMS2-plasmid DNA
bearing the scFv-Fc fusion protein (scFv-Fc) plus 3 µl FuGene HD
Transfection Reagent (Promega; #E2311) at room temperature for 15
min. Subsequently, 293T cells were subjected to a selection
pressure by zeocin (Life Technologies; #R25001, final concentration
100 µg/ml) to yield higher amounts of transfected cells producing
scFv-Fc and secreting them into the supernatant. The final duration
of the incubation with zeocin selection pressure was three weeks at
37°C, 5% CO2. Transfection efficiency was confirmed by
flow cytometry through an enhanced green fluorescence protein
(eGFP) produced as reporter protein by those cells secreting the
scFv-Fc.
Antibody library and vectors
The well-known human scFv antibody library Tomlinson
J (Medical Research Council Laboratory) was used for the selection
process. This library is based on a single human framework (VH-3
and Vκ-1) and a chain diversity of NNK triplets which is
incorporated in the complementarity determining regions 2 and 3
(CDR2/3) (23). The library was
constructed by the pIT2-phagemid vector and stored in the E. coli
strain TG1 until usage.
After the selection procedure, scFvs were converted
from the pIT2 to the pMS2 vector to receive a bivalent, recombinant
and homodimerized scFv-Fc antibody that included the Fc-fragment of
a mouse-IgG2a-antibody.
Phage display (PD)
Phage production and rescue was carried out as
previously described (19): The
Tomlinson J antibody library was infected with M13K07∆pIII
hyperphage (Progen Biotechnik GmbH; #PRHYPE-XS) and M13K07 helper
phage (New England Biolabs; #N0315S). While M13K07∆pIII was used
during the first of three selection rounds (SR) only, M13K07 was
employed during the second and third SR.
Subtractive selection including optimized depletion
steps were performed with different healthy blood cell subsets
prior to positive selection on AML blasts to remove common and
non-specific antibody binders. An overview of the different
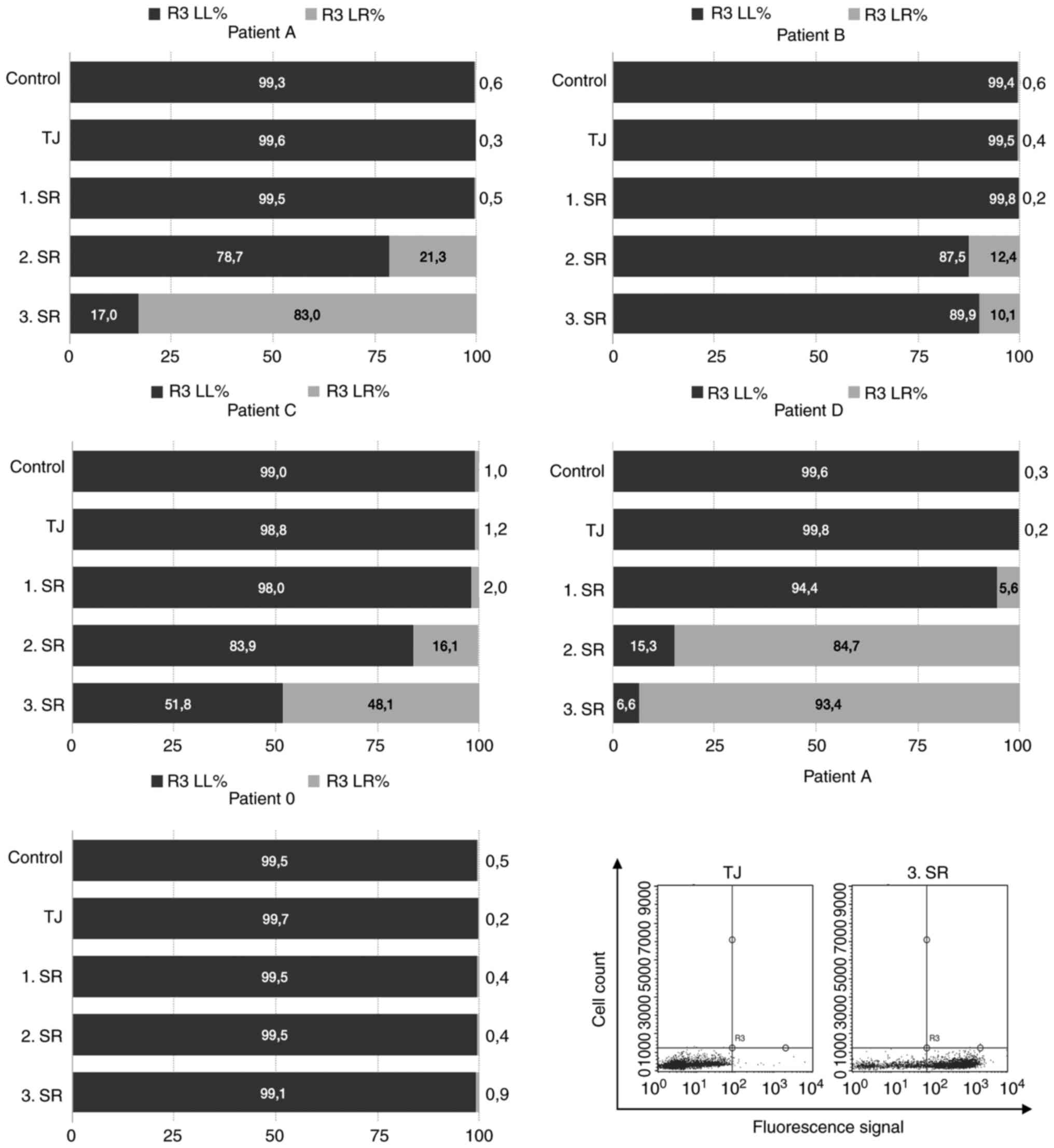
cell-based phage panning strategies applied is shown in Fig. 1.
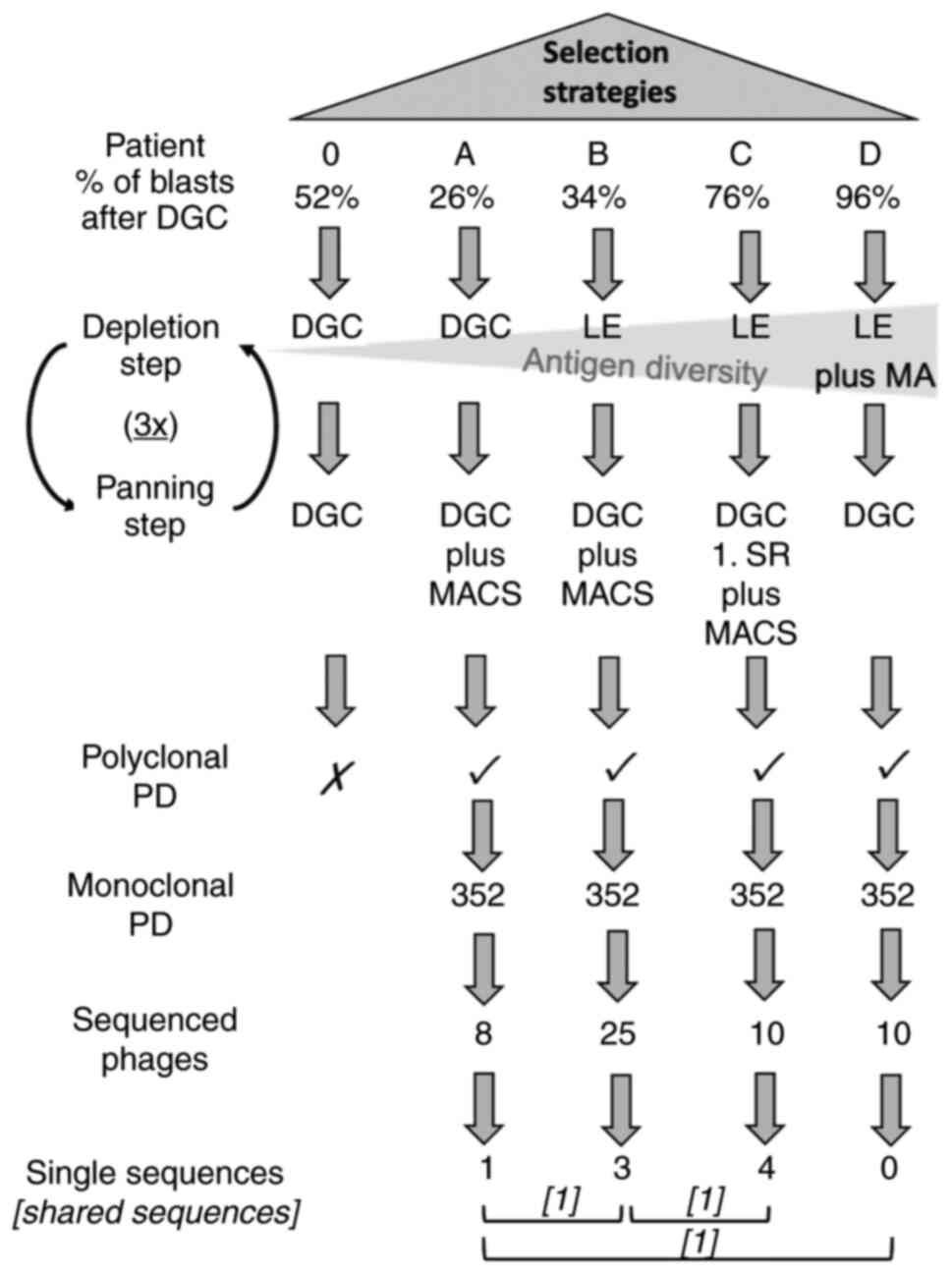 | Figure 1.Selection strategies performed. All
five patients (0, A, B, C and D) were included. The depletion steps
and planning steps are depicted. In subsequent polyclonal PD FACS
analysis patient 0 failed to show any enrichment. Hence, only the
selection strategies of patient A, B, C and D reached monoclonal
analysis where the most promising phages of each selection strategy
were chosen and sequenced. After sequencing, one unique sequence
was revealed in the sequenced phages of patient A, three unique
sequences within patient B and four single sequences in patient C.
Furthermore, three shared sequences were revealed among the
selection strategies (named consensus phages) which are symbolized
with square brackets. Cross indicates not continued. Tick indicates
continued. 3×, three times; DGC, density gradient centrifugation;
LE, lysis of erythrocytes; MA, adherence of monocytes; MACS,
magnetic-activated cell sorting; PD, phage display; SR, selection
round. |
Negative selection: Depletion step
(DS)
For efficient elimination of unspecific phage
particles during cell-panning, blood samples from healthy donors
were collected to isolate different blood cell fractions for
depletion. Samples were anti-coagulated with either heparin (used
in density gradient centrifugation and enrichment of monocytes) or
EDTA (ethylenediamine tetra-acetic acid, used in lysis of
erythrocytes). Panning started with three depletion steps for the
phage library on centrifuged or lysed blood samples. Cell counts
required for the DS (i.e., cells from a healthy donor) and the
panning step (i.e., blasts from an AML patient) were adjusted to
10×106 cells per sample. However, monocytes were
adjusted to 2.5×106 cells per sample.
Density gradient centrifugation was performed for
patients 0 and A. Lysis of erythrocytes was carried out for
patients B, C and D (Fig. 1). By
contrast, enrichment of monocytes was performed for patient D only
as second DS.
Density gradient centrifugation
(DGC)
Peripheral blood was mixed in a ratio of 1:1 with
RPMI. Subsequently, one third Lymphoprep™ (Axis-Shield; #1114544)
medium was overlaid with two thirds of the RPMI-blood-mixture and
centrifuged at 1,800 rpm for 30 min. Afterwards, separated
peripheral blood mononuclear cells (PBMC) were washed twice with
RPMI, centrifuged at 10 min and either used directly or stored at
−80°C as reviewed above.
Lysis of erythrocytes (LE)
The commercially available lysis solution (BD Pharm
Lyse™; Becton-Dickinson (BD); #555899) was mixed in a ratio of 1:10
with purified water (pH 7.3). 25 ml reagent was added to 2.5 ml
blood sample, then shaken for 12 min and subsequently centrifuged
at 1,200 rpm for 5 min. The pellet was resuspended with 25 ml PBS
(phosphate buffered saline, pH=7.4) plus 1% FBS and centrifuged
again at 1,200 rpm for 5 min.
Enrichment of monocytes via adherence
(MA)
After DGC, the obtained PBMCs were seeded in T175
cm2 culture flasks, resuspended in RPMI supplemented
with 10% FBS at a density of 2–3×106 cells/ml.
Incubation was performed at 37°C for 1 h. Subsequently, the
adherent monocytes were cleaned from remaining supernatant by
washing twice with preheated PBS (37°C). RPMI plus 10% FBS was
added and incubated at 37°C overnight.
Positive selection: Panning step
(PS)
Target cell concentration is essential in cell-based
phage display. Therefore, at least one round of CD34 magnetic
activated cell sorting (MACS) was performed to concentrate
patients' targeted blast cells if the blast percentage was below
90% after DGC. Thus, samples from patient A and B were sorted for
all three SR, patient C samples were sorted for the first SR only
(Fig. 1) and material from patient
D, which contained a blast percentage above 90%, was not sorted.
Samples from patient 0 served as control and were therefore not
CD34-sorted at all.
Enrichment of peripheral blood
CD34+ cells
Blood cells from patient samples were enriched for
CD34-positive blasts by using immunomagnetic beads according to the
manufacturer's protocol (CD34 MicroBeads Kit; Miltenyi Biotec;
#130-046-702). Purity was controlled using flow cytometry (FACS)
against the precursor antigen CD34 (CD34-antibody conjugated with
fluorescein-isothiocyanate (CD34-Fitc), Lot B213405, monoclonal,
mouse anti-human; Biolegend) (24).
CD34-positive blasts were used for cell-panning directly after
sorting. If the initial blast cell count after DGC was below 75%,
all three selection rounds were conducted on CD34-enriched
material. If cell counts were above 90%, no CD34 enrichment was
performed. Cells were only purified for the first SR if blast
counts were between 75 and 90%.
Flow cytometry-based analysis of
polyclonal phage antibody binding
Polyclonal FACS analysis was performed to determine
the AML blast-specific enrichment of antibody phages obtained after
three rounds of subtractive cell-panning (Fig. 2). Phage rescue was performed as
described above:
In brief, for each individual stain,
3×105 patient cells after DGC were used and blocked for
1 1/2 h in 2% MPBS (milk powder plus PBS) at room temperature. In
addition, each polyclonal phage solution was diluted with MPBS to
achieve a titer of 5×109 colony forming units (CFU)/ml
and was blocked for 2 h on a rotary instrument. Subsequently, 100
µl phage solution was added to the patient's cells and incubated 1
h at 4°C. After washing with washing buffer for FACS experiments
(WB-FACS, i.e. PBS containing 0.2% BSA; Sigma-Aldrich, Merck KGaA;
#A7906-100G, 0.1% NaN3) and centrifugation at 2,600 rpm
for 5 min at room temperature (RT), staining with a secondary
antibody directed against a phage protein (anti-M13, monoclonal,
mouse anti-phage, diluted 1:10.000 in 2% MPBS; GE Healthcare;
#27942001) was conducted.
After incubation for 30 min at 4°C and
centrifugation at 2,600 rpm for 5 min at RT, a third antibody
conjugated with a fluorochrome was added (i.e. a goat anti-mouse
antibody labelled with fluorescein-isothiocyanate, polyclonal, Lot
6287542; BD Biosciences; dilution 1:50). The staining procedure of
the third antibody was performed in the same manner as the
secondary antibody stain.
Finally, antibody binding was measured using the
Guava easycyte 5HT flow cytometer (Merck Millipore). All FACS data
were analyzed by InCyte 2.7 (Merck Millipore).
Isotype controls were used for setting up the
instrument (Isotype Fitc, Lot B213387, monoclonal, mouse
anti-human, Biolegend), cells without bound phage antibody were
used as negative control and Kas-1 phage antibody (against
Kasumi-1, unpublished) served as internal positive control.
Binding activity was scored positive (+), if at
least 10% of the cell population (25) were confirmed positive and if an
increase in median fluorescence intensity of at least 75% was
detectable compared to the fluorescence signal of the control
(unselected, naïve Tomlinson J library).
Flow cytometry-based analysis of
monoclonal phage antibody binding
The binding specificity of monoclonal phage
antibodies was verified by monoclonal FACS analysis. For this
purpose, randomly picked single bacterial colonies from the third
SR of each FACS-positive polyclonal phage pool were analyzed.
A 96-well plate was filled with 180 µl Yeast Extract
Tryptone Difco™ Medium (BD; #11738892) containing 100 µg/ml
ampicillin and 1% glucose. Single bacterial colonies were diluted
in this medium and incubated overnight at 37°C under agitation at
180 rpm. 2 µl bacterial solution were transferred to another
96-well plate filled with the same medium as described above and
grown for 2 h at 37°C under agitation at 180 rpm.
Subsequently, 30 µl of hyperphage solution (1:100
diluted in PBS) was added to each well, incubated at 37°C for 30
min without agitation, and 30 min with agitation. Subsequently,
samples were centrifuged for 10 min at 4,000 rpm, 4°C. The
supernatant was discarded and replaced by the same medium as
described above plus 50 mg/µl Kanamycin. Finally, samples were
incubated overnight at 30°C, 180 rpm, to complete phage rescue.
For FACS analysis, 1×105 cells were added
into each well of another 96-well plate. If blasts were tested,
they were treated in the same manner by pure density gradient
centrifugation. If healthy donors were tested, the cells were
treated the same way as they were for the correspondent selection
strategy.
All cells were blocked in 2% MPBS for at least 1 1/2
h. Rescued phages were blocked for 2 h under the same conditions.
After blocking, rescued phages were added to each cell-containing
well and incubated at 4°C for 1 h. The subsequent staining
procedure was identical to the staining procedure used during
polyclonal analysis except for the centrifugation settings, which
were adjusted to 1,500 rpm, 5 min. Promising phages were sequenced.
Unique scFv clones were investigated thoroughly as single phage
particles.
Flow cytometry-based analysis of phage
binding by titration measurements
Single phages, i.e., phages not tested in a 96-well
format which were therefore titratable, were normalized to a titer
of 5×1010 CFU/ml and phage rescue was performed as
described above. AML cells underwent a DGC. In addition, cells of
patient A and B were CD34-sorted before staining. Subsequently, the
staining was carried out as described for polyclonal analysis.
Moreover, cells of healthy donors were evaluated for
cross-reactivity by the analyzed phages (Fig. 3).
First, these healthy cells were treated by lysis of
erythrocytes. Second, cells were stained equally to the malignant
cells. Third, single phage analysis was carried out using flow
cytometry in the same way as described above and evaluated by the
same criteria as the polyclonal PD.
Production and flow cytometry analysis of
scFv-Fc fusion proteins
Sanger sequencing and scFv-Fc fusion
protein production
Specific scFv clones were sequenced by sanger
sequencing. All clones harboring an amber termination codon (TAG)
in their sequence (see 3.4) underwent site-directed mutagenesis as
previously described (26). A
glutamine (Q) was inserted instead of the amber codon to enable
eukaryotic expression (Table I).
Primers were created by LasergeneCoreSuite 14 (DNASTAR Inc.) and
ordered by Eurofins Genomics GmbH (Table II).
 | Table I.Sequence analysis of acute myeloid
leukemia-binding phage clones. |
Table I.
Sequence analysis of acute myeloid
leukemia-binding phage clones.
| A, Variable domains
of the heavy chain |
|---|
|
|---|
| Clone no. | CDR 1 | CDR 2 | CDR 3 |
|---|
| 11 | GFTFSSYA |
IQHLGSRTb | AKHATSFDY |
| 14a |
|
IQGRGHRTb | AKSSLVFDY |
| 22a |
|
|
|
| 44a |
|
|
|
| 24a |
|
ITQRGSSTb | AKGTTVFDY |
| 35a |
|
|
|
| 33a |
|
IQNRGQSTb | AKSAARFDY |
|
| B, Variable
domains of the light chain |
|
| Clone
no. | CDR 1 | CDR 2 | CDR 3 |
|
| 11 | QSISSY | RAS | QQWAIVPPT |
| 14a |
| RAS | QQAHTAPST |
| 22a |
| YAS | QQSSLTPAT |
| 44a |
| AAS | QQSYSTPNT |
| 24a |
| RAS | QQYQTGPMT |
| 35a |
|
| QQNALAPMT |
| 33a |
| NAS | QQPHTSPFT |
| Table II.Sequences of primers used for
site-directed mutagenesis. |
Table II.
Sequences of primers used for
site-directed mutagenesis.
| Primer name | Sequence,
5′-3′ | Used for clone
no. |
|---|
| AML_WT_11_T |
GAGTGGGTCTCAAGTATTcAGCATTTGGGTTCGCGG | 11 |
| AML_WT_11_B |
CCGCGAACCCAAATGCTgAATACTTGAGACCCACTC |
|
| AML_WT_14_T |
GAGTGGGTCTCAACGATTcAGGGGCGTGGTCATAGG | 14,22,44 |
| AML_WT_14_B |
CCTATGACCACGCCCCTgAATCGTTGAGACCCACTC |
|
| AML_WT_24_T |
GGAGTGGGTCTCAACTATTACTcAGCGGGGTAGTAG | 24,35 |
| AML_WT_24_B |
CTACTACCCCGCTgAGTAATAGTTGAGACCCACTCC |
|
| AML_WT_33_T |
GATTCAGAATCGTGGTcAGAGTACAGCGTACGCAGA | 33 |
| AML_WT_33_B |
TCTGCGTACGCTGTACTCTgACCACGATTCTGAATC |
|
Then, all selected scFv were cloned in frame into
SfiI-NotI restriction sites of the eukaryotic expression vector
pMS2 to express a bivalent antibody scFv-Fc (23).
Subsequently, 293T cells were transfected with the
insert-bearing pMS2 vector and transfected cells were selected
under zeocin pressure as described above. Finally, scFv-Fc fusion
proteins secreted into the supernatant of 293T were verified by
western blot and FACS analysis.
Western blot analysis of scFv-Fc
fusion proteins
For western blot analysis, a sodium dodecyl sulphate
polyacrylamide gel electrophoresis was conducted. Bolt™ 8% Bis-Tris
plus gels (Thermo Fisher Scientific; #NW00080BOX) were run with
NuPAGE® MOPS SDS Running Buffer (20X) (Life
Technologies; #NP0001) at 200 V, 50 min. The negative control was
supernatant of non-transfected 293T cells (NT). A scFv-Fc fusion
protein previously confirmed in another experiment served as
positive control. ScFv-Fc fusion proteins were usually purified
using Protein A/G UltraLink Resin (Thermo Fisher Scientific;
#53132) affinity chromatography. Then, purified scFv-Fc fusion
proteins were quantified using the BCA assay (Thermo Fisher
Scientific; #23227), commonly revealing concentrations in the range
of 0.15–0.25 mg/ml (23). Spectra
Multicolor Broad Range Protein Ladder (Thermo Fisher Scientific;
#26634) was added as marker of molecular weight. For each lane 20
µl sample volumes mixed with equivalent volumes of sample loading
buffer (NuPAGE™ LDS Sample Buffer (4X), Thermo Fisher Scientific;
#NP0007) were used. Then, a wet blot and a Coomassie brilliant blue
(G-250, Thermo Fisher Scientific; #20279) staining of the membrane
was applied. The horseradish peroxidase-labelled polyclonal rabbit
anti-mouse antibody (Dako; #P0260, diluted 1:5,000) was provided as
secondary antibody as previously described (23). For the detection of
chemiluminescence the Amersham™ ECL™ Prime Western Blotting
Detection Reagent (GE Healthcare; #12316992) was used. The analysis
was performed by ChemoStar Touch v.0.5.38 (Intas Science Imaging
Instruments GmbH).
Flow cytometric analysis of scFv-Fc
fusion proteins
For flow cytometry analysis, 2.5×105
Kasumi-1 cells were applied for each stain and incubated for 10 min
at RT in 10% AB serum (diluted with WB-FACS). Then, cells were
washed with 1 ml WB-FACS and centrifuged at 2,300 rpm for 5 min.
100 µl of the supernatant of 293T cells containing the scFv-Fc
fusion proteins was added and incubated for 30 min at 4°C. After
another washing step, 100 µl gam-Fitc solution was added (dilution
1:50 in WB-FACS), cells were resuspended and incubated under the
same conditions. Following a further wash step, cells were analyzed
using FACS. The negative control was non-transfected supernatant
(NT), while an scFv-Fc protein tested and validated in another
experiment was used as positive control. The same criteria used for
single phage analysis and polyclonal PD served as cut-off.
Measurement of internalization
activity by flow cytometry and confocal imaging
For each stain, 2×105 Kasumi-1 cells were
required. After washing 5 min with WB-FACS at 2,300 rpm, the cells
were blocked in 10% AB serum (diluted in WB-FACS) for 10 min at RT.
Subsequently, every step was conducted at 4°C or on ice until
internalization was intended. One washing step with WB-FACS was
performed at 4°C for each sample before resuspending with scFv-Fc
containing supernatants. Four different antibodies (AB 11, 22, 24
and 35) were tested for internalization (Fig. 4,Fig.
5,Fig. 6).
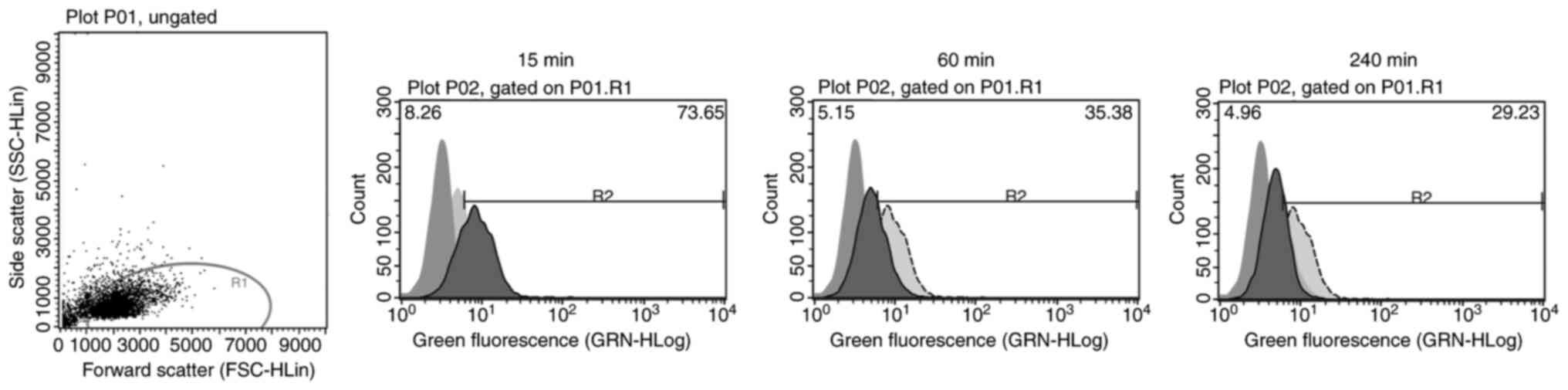
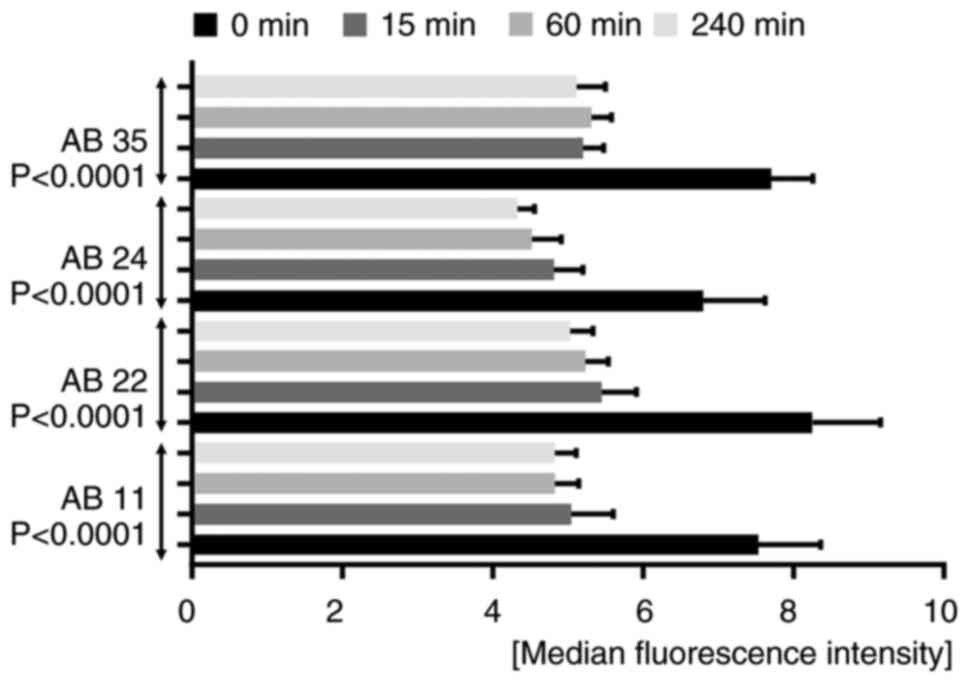 | Figure 4.Internalizing activity measured by
flow cytometry. A time-dependent bar chart presents the
fluorescence signal quenching of the tested antibodies AB 11, 22,
24 and 35. Bar graphs for each antibody indicate a decrease in
fluorescence signals the longer the internalization time. The
measurements were performed before internalization (0 min) and
after internalization times of 15, 60 and 240 min. All four
antibodies showed statistical significance using a repeated
measures one-way ANOVA with a Greenhouse-Geisser correction [AB 11,
F(1.851, 9.253)=75.65, P<0.0001; AB 22, F(1.133, 5.664)=131.3,
P<0.0001; AB 24, F(2.041, 10.21)=39.28, P<0.0001; AB 35,
F(1.296, 6.478)=102,7, P<0.0001]. AB, antibody. |
After resuspending, samples were incubated at 4°C
for 30 min. Following another washing step, the cells were
incubated for 15, 60 and 240 min at 37°C, 5% CO2, in
RPMI medium with 20% FBS in order to allow internalization as
described previously (23).
Internalization was stopped by adding ice-cold
WB-FACS and centrifugation for 5 min at 2,300 rpm, 4°C. Secondary
antibody gam-Fitc was used for detection by resuspending the cell
pellet in 100 µl of its solution (1:50 in WB-FACS) and incubating
the resulting mixture at 4°C for 30 min. Following a wash step in
ice-cold WB-FACS, the fluorescence signal was measured using flow
cytometry. As control, non-transfected supernatant (NT) plus
gam-Fitc was used. The experiment was performed three times in
duplicates.
Statistical analysis
Statistical analyses were carried out using GraphPad
Prism Software (Version 7.0; GraphPad Software). Results were
analyzed for statistical significance using repeated measures
one-way analysis of variance (ANOVA) with a Greenhouse-Geisser
correction. Significance was determined at P<0.005 (Fig. 4). The experiment was repeated three
times in duplicates.
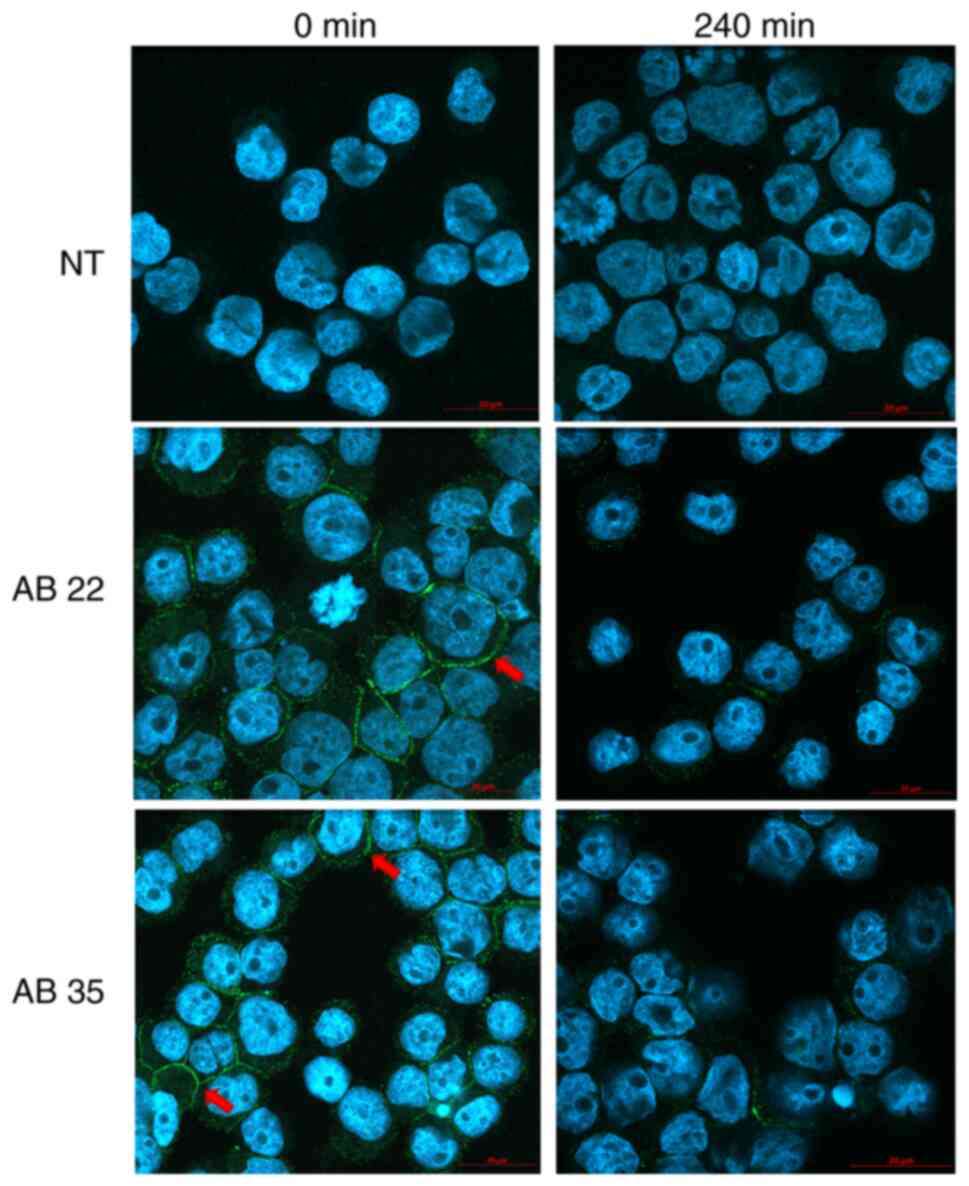
Confocal imaging preparation for
internalizing experiments
For confocal imaging, Kasumi-1 cells were stained as
described above. After staining, cells were resuspended in 1%
paraformaldehyde diluted in WB-FACS and centrifuged 5 min at 700
rpm in cytofunnels™ (Thermo Fisher Scientific; #13127963).
Subsequently, VECTASHIELD® mounting medium with DAPI
(4′,6-diamidino-2-phenylindole, Vector Laboratories Inc.; #H-1200)
was added and cytospins were analyzed in a confocal microscope
(LSM800, Carl Zeiss AG). Analysis of scans was carried out using
the Zen 2.3 software (Carl Zeiss AG) (Fig. 6).
Results
Standardization and optimization of
cell-based panning protocols
Cell-based phage display panning procedures using
primary tumor cells are the optimal methods for the discovery of
antibodies against tumor-associated surface antigens because target
antigens are displayed in their natural configuration and with
post-translational modifications (27). Several protocols for whole cell
panning using leukemic cells have been reported, but these describe
a variety of screening strategies (19,21,22,28–31).
To examine these differences, existing articles are briefly
summarized and compared based on their technical approaches to
cell-based panning procedures.
First, the number of selection rounds (SR) varied
between two (21,22) and five (29). Second, panning material applied
during the SRs differed substantially regarding cell counts and
types of cells: Only one laboratory used patients' primary cells
during the entire selection procedure (29), although these cells originated from
a patient with chronic myelogenous leukemia (CML) in blast crisis.
Two articles described partial use of primary AML blasts during
cell panning in combination with AML cell lines and a mixture of
AML/CML-derived cell lines, respectively (22). Third, a subtractive panning strategy
by including a depletion step (DS) was described in multiple
publications (19,21,22),
but these DS varied concerning handling of the healthy donors'
cells: Peripheral blood cells (PBC) were either acquired by buffy
coat (21,22) or as membrane fragments after DGC
(19).
In summary, in view of the variety of existing
technical approaches, we standardized the number of selection
rounds by restricting them to exactly three. Moreover, we used AML
patients' cells throughout the whole cell-based panning process and
found that initial blast cell counts played an important role in
determining the outcome of phage antibody selection. Finally, we
demonstrated the impact of optimizing the diversity of depletion
antigens and were able to isolate four promising, AML blast-binding
and internalizing antibodies.
Positive Selection: Initial AML blast
counts are associated with polyclonal enrichment of specific phage
antibodies
Regarding the selection process of patient 0, no
enrichment of specific binders during polyclonal PD was observed in
FACS analysis. Patient 0 had blast percentages of 52% after DGC and
was not treated via CD34 MACS, but rather used as control in
comparisons with CD34 sorted selection procedures (Fig. 1).
In contrast to patient 0, all other selection
strategies showed polyclonal enrichment of specific phage antibody
binders ranging from 93.4 % (patient D) to 10.1% (patient B)
(Fig. 2). Of these, only patient
D's cells were not magnetically sorted because of their blast
portion of 96%, which made CD34+ cell enrichment
unnecessary (Fig. 1). Patient C
exhibited blast percentages of 76% after DGC and was therefore only
sorted during the first SR (Fig.
1).
Because the first SR proved to be the most important
round for selecting binders (16,32),
the first SR always underwent CD34 enrichment. Using flow cytometry
analysis as described below, we identified that the purity of
patient C's enriched blasts during the first SR was 99%. Patient A
and B were sorted during all three SRs and reached average purities
of 95 and 96%, respectively, in FACS analysis. To the best of our
knowledge, this is the first report using CD34-enriched blast cells
for cell- based panning procedures.
Negative selection: Diversity of
depletion antigens influences outcomes of cell- based selection
procedures
Patient 0's depletion step (DS) consisted of DGC, a
procedure, which excludes granulocytes and erythrocytes and
preserves mononuclear cells only, e.g., lymphocytes and monocytes
(33). Thus, less antigen diversity
(AD) was available during this DS than in the DS of patient B, C
and D (Fig. 1, here an increase in
antigen diversity is shown by an increase in size of the grey ramp
in the background).
Although patient A had the same DS as patient 0,
patient A showed polyclonal enrichment of AML blast-binding phage
particles (Fig. 2). The sole
difference between both selection procedures was that patient A was
CD34-sorted for panning. In contrast to patient 0 and A, the
depletion procedures of patient B, C and D were carried out by
human, erythrocyte-lysed peripheral blood cells. Hence, nothing but
erythrocytes were excluded. Consequently, AD was higher in their DS
compared with the negative selection of patient 0 and A (Fig. 1).
However, the highest AD was observed during patient
D's selection procedure because of its additional DS with enriched
monocytes (Fig. 1). Here, only one
clone was selected which bound AML blasts, but all portions of
healthy blood cells as well (Fig.
1). Consequently, this clone had overgrown all other more
specific phages during the selection strategy and thus, was not
pursued further (Fig. 3, clone no.
9).
In conclusion, medium levels of AD during the
depletion strategies of patient B and C entailed the most specific
AML-binding phages as confirmed by monoclonal analysis and
sequencing, respectively.
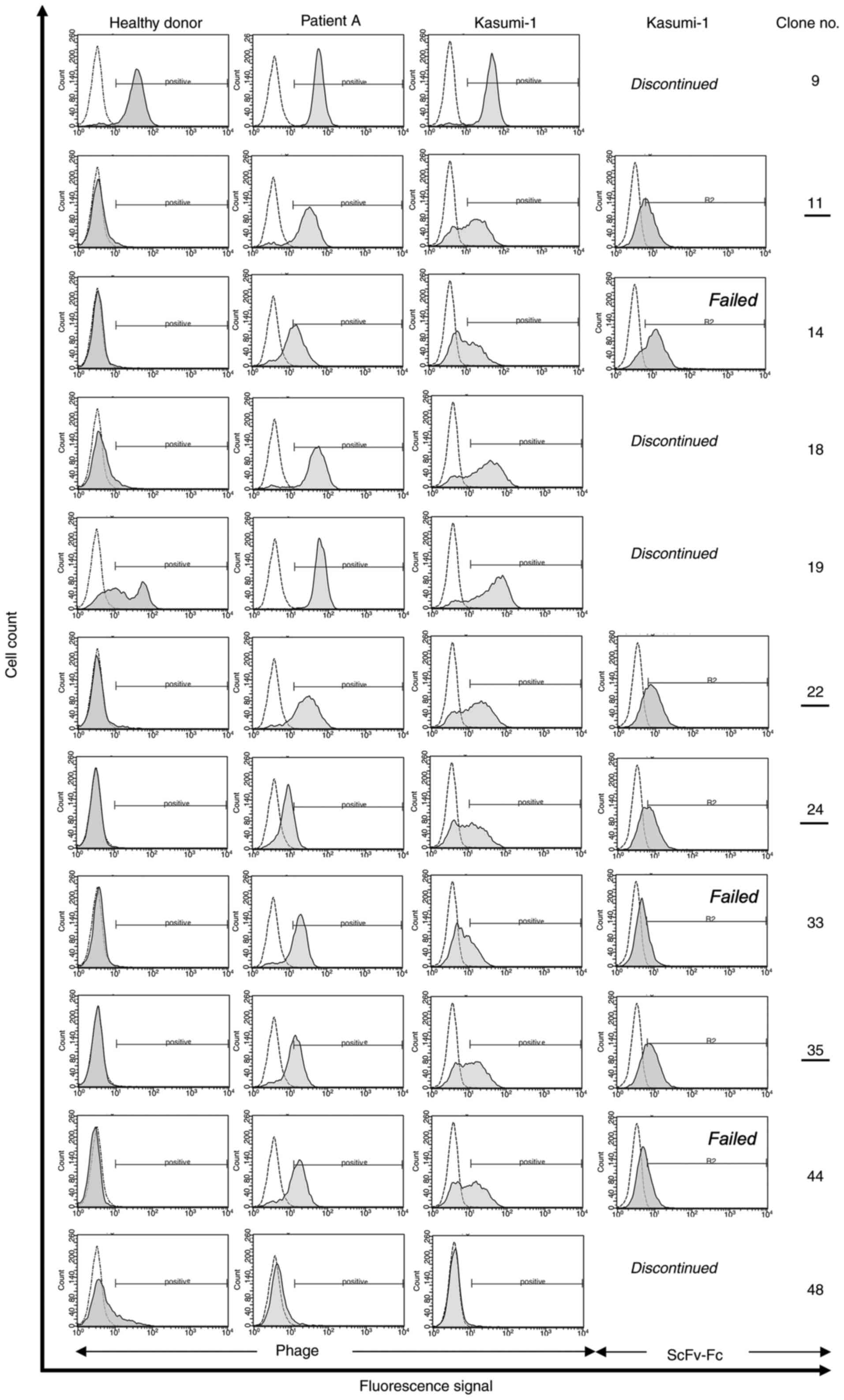
Panning-enriched antibody clones
contain consensus sequences
After confirmation of polyclonal enrichment of phage
particles, randomly selected single antibody clones from the third
SR of the successful selection methods were tested for monoclonal
binding activity (Fig. 1). To do
so, 352 samples were investigated for each patient individually (A,
B, C and D) by flow cytometry. FACS-positive binders, (8 out of 352
for patient A, 25 out of 352 for patient B, 10 out of 352 for
patient C, and 10 out of 352 for patient D) were DNA sequenced (∑
53 sequenced phages; Fig. 1).
Comparisons of these sequences and alignments with publicly
available sequence databases (NCBI, http://blast.ncbi.nlm.nih.gov/Blast.cgi) revealed a
total of 11 unique antibody clones.
Based on their similarity, three out of these 11
clones contained shared sequence identities, especially in the CDR2
and CDR3 sequences of their heavy chain. Interestingly, clones with
shared sequence identities were isolated from different depletion
and selection strategies and thus, were called consensus phages.
Seven of the 11 tested phage antibodies were positive for AML
blasts (Fig. 3), but negative for
healthy blood cells. Subsequently, all blast-specific candidates
were expressed as soluble scFv-Fc fusion protein antibodies (AB 11,
14, 22, 24, 33, 35 and 44) and further investigated regarding their
binding and internalizing activity. Two of these candidates were
former consensus phages (AB 33, AB 35).
In addition, all seven clones contained an amber
termination codon (TAG) in the DNA sequence of every CDR2 of their
immunoglobulin heavy chain, which was changed by site- directed
mutagenesis to CAG (Glutamine, Q) (Table I).
Moreover, homologies of heavy chain sequences were
noticed between AB 24 and 35, as well as between AB 14, 22 and 44.
Intriguingly, AB 24 and 35 differed only in the CDR3 portion of
their immunoglobulin light chain (Table
I).
Effective tumor cell-binding activity
against primary AML blasts and the human AML cell line
Kasumi-1
Specific binding activity of selected antibody
clones was analyzed on Kasumi-1 cells and primary AML blasts using
scFv-presenting phage particles as well as soluble fusion protein
(Fig. 3). After functional protein
expression, clones AB 33 and 44 failed to meet our binding criteria
and were therefore not pursued. In addition, AB 14 showed
unspecific binding activity on 293T cells and was therefore also
not pursued.
Cellular internalization of the
scFv-Fc fusion proteins
Internalizing activity was observed for antibody
clones AB 11, 22, 24 and 35 via flow cytometry and confocal imaging
using Kasumi-1 cells (Fig.
4,Fig. 5,Fig. 6). All four antibodies showed a
reduction of fluorescence signals by increasing internalization
time. This reduction showed statistical significance (P<0.0001)
for each tested antibody [AB 11 (n=6), F(1.851, 9.253)=75.65,
P<0.0001; AB 22 (n=6), F(1.133, 5.664)=131.3, P<0.0001; AB 24
(n=6), F(2.041, 10.21)=39.28, P<0.0001; AB 35 (n=6), F(1.296,
6.478)=102,7, P<0.0001; Fig. 4].
Confocal imaging confirmed and visualized the internalizing
activity of all tested antibodies (Fig.
6).
Discussion
The fundamental rationale for the implementation of
antibody-based targeted therapies in cancer relies on their
sophisticated ability to selectively tackle cancerous populations
while leaving healthy cells alone. However, the identification of
highly selective cell surface biomarkers capable of discriminating
between benign and malignant cells is an enormous experimental
challenge. Standard selection procedures performed by
immobilization of recombinant target proteins or cell membrane
fractions on plastic surfaces may not be sufficient to meet this
challenge because of significantly altering protein conformations
(34), so that isolated antibodies
may not bind the native conformation of targeted antigens
efficiently. Thus, experimental strategies, which account for
three-dimensional structures of cell surface antigens, seem more
promising.
In this context, we implemented intact primary AML
blasts as complex source of targetable antigens for the selection
of specific antibodies against acute myeloid leukemia, thus
preserving the native conformational structure of membrane antigens
(35). However, primary tumor
cell-based panning techniques are often hampered by low abundance
of blast-specific antigens, which discriminate healthy from
tumorous cells against a background of irrelevant antigens
(36).
Additionally, a major difficulty in working with
primary blasts instead of e.g., human cell lines is their variable
ratio in patients' bloods, which seldom reach 100% of all cells.
Thus, we implemented a blast enrichment step in consideration of
patients' initial blast percentages, making PD selection procedures
feasible for patients regardless their blast counts. Moreover, we
established novel depletion strategies to exclude irrelevant and
healthy antigens from positive selection steps and, thereby, showed
that the diversity of depletion antigens has a major impact on
individual selection outcomes (Fig.
1).
An essential prerequisite for phage display success
is significant enrichment of specific phage particles during
consecutive biopanning rounds. Up to four selection rounds are
typically performed to allow strong accumulation of
antigen-specific phages without losing phage diversity (37). In line with this approach, we
carried out three subsequent rounds of panning in our
experiments.
Relative to the considerations above, we determined
that initial counts of at least 75% primary blasts after DGC are
critical for successful whole cell-based panning using primary
tumor cells. For blast counts below this threshold, we recommend a
blast enrichment step before panning. By contrast, blast counts
above 90% did not benefit from enrichment in our experiments.
For enrichment, we established a strategy to achieve
highly concentrated CD34+ blasts. CD34 is well-known as marker of
hematopoietic stem cells (HSC) and hematopoietic progenitor cells
(24). It is expressed in roughly
40% of all AML cases (38). By
contrast, CD33, the myeloid antigen marker of the commercially
available antibody conjugate gemtuzumab-ozogamicin (GO) (39), is expressed in ~75% of AMLs in
adults, but has response rates up to 30% only, suggesting a
heterogeneous expression of CD33 by AML cells (40). Interestingly, it was the most
primitive progenitor compartment (as indicated by CD34 expression)
which showed reduced response rates to GO (41) as well as overall reduced surface
expression of CD33 in vitro (40). Thus, targeting CD34+ leukemic cells
is a promising candidate approach for the complementation of
CD33-based antibody therapies in AML.
To the best of our knowledge, the CD34 enrichment
approach described here is the first report of the individual
generation of novel antibodies for AML patients regardless of their
initial blast counts. The relevance of this step was demonstrated
in patient 0's PD procedure, which did not include an enrichment
step despite initial blast counts below the cut-off value and
subsequently failed to achieve accumulation of specific phage
particles (Fig. 2). Future studies
will be needed to determine whether other surface antigens, e.g.,
the transmembrane protein receptor CD117 (42), can also be implemented for our
suggested blast enrichment procedure.
For identification of novel blast-specific binders,
the phage antibody library Tomlinson J was initially depleted
against equivalent non-cancerous blood cells (43). This negative selection (depletion
step) was introduced prior to positive selection to remove binders
directed against unspecific antigens, which are expressed on the
surface of both benign and malignant cells. It has been reported
that insufficient depletion can reduce the success of the
biopanning procedure (15).
However, our literature research showed that depletion is not
performed using a standardized procedure or not at all, indicating
that optimization of the depletion step could be an effective
method to improve panning results. Therefore, we investigated the
impact of three depletion strategies on antigen diversity (AD) and,
thus, overall selection success. The following strategies (in
ascending order of antigen diversity) were analyzed: density
gradient centrifugation (DGC), lysis of erythrocytes (LE) and lysis
of erythrocytes plus enrichment of monocytes by adherence (MA).
The quantity of different antigens was determined by
the addition or overrepresentation of different cell populations.
Thus, DGC retained only PBMCs, i.e., lymphocytes and monocytes, LE
contained the populations specified above plus granulocytes and MA
included the same cell fractions as LE, but additionally enriched
monocytes, a minor cell fraction in blood. This monocytic
enrichment was conducted based on reports of similar antigens on
myelomonocytic leukemia (FAB M4) and monoblastic/monocytic leukemia
(FAB M5) cells (44). We initially
hypothesized that the enrichment of blast-specific antibodies is
correlated to antigen diversity. However, our experiments did not
support this hypothesis as the strategy with the highest diversity
depletion antigens (patient D) retained just one single phage,
which also bound all populations of healthy donor cells (Fig. 3, clone 9). According to Smith and
Petrenko (16), stringency is a
possible reason for the failure of high abundancies of DA when the
attempt to force enrichment of target-specific phage is made by
presenting a large diversity of depletion antigens, especially
during the first SR, where merely 100 phage antibodies per unique
clone are present (16). This
approach can cause target-specific phages to be lost in the
background of unspecific interactions between phages, cells and
antigens (16).
On the other hand, shown by the experiments on
material from patient 0 and patient A (Fig. 1), inadequate DA levels can result in
an abundance of unabsorbed, but unspecific phages (15). The PD procedure using material from
patient 0 did not result in any specific phage and patient A
supplied only one.
In conclusion, we recommend using medium levels of
antigen diversity provided by lysis of erythrocytes containing
normal blood compositions of cellular antigens to retain most
specific phage antibodies.
To determine the number of different antibody clones
that bound specifically to AML blast cells, 53 FACS-positive clones
were characterized by DNA sequencing and aligned. This analysis
revealed 11 unique sequences and a significant enrichment of
consensus sequences in the variable regions of the heavy and light
chains. To categorize these findings, the synthesis process of the
applied PD library has to be considered, as the naïve human
semi-synthetic Tomlinson J library is mutated only in the
complementarity determining region 2 (CDR2) and CDR3 of the
variable parts of the light chain (VL) as well as the heavy chain
(VH) (45). Consequently, both
CDR1s are maintained throughout the whole PD library. Consequently,
certain degrees of sequence limitations and, thus, restrictions
regarding the diversity of phage antibodies become obvious. For
example, Tomlinson J seems to be prone to the selection of amber
codon bearing phages (46–48), roughly ~45% caused by its random
synthesis process utilizing an NNK triplet codon bias (i.e., N=A,
T, G, or C and K=T or G) (49).
Moreover, we found identical sequences of VH between
phage antibodies (PH) 14, PH 22, and 44 as well as sequence
identities between PH 24 and 35, whereas the last two phage
antibodies differed only regarding their CDR3 of VL (Table I). Such sequence identities seemed
to occur frequently during PD selection strategies performed by the
Tomlinson J library. For example, Fitting et al (19) also used the Tomlinson library and
retained two antibodies containing the same VL, but different VH.
Moreover, Huang et al (50)
were confronted with identical VH sequences of in total five
antibody clones by using the Tomlinson PD library. Huang et
al also reported identical sequences in two clones with the
exception of CDR3 of VL (50).
Interestingly, in our study, three unique phage
antibodies were selected in independent selection procedures of
different AML patients (Fig. 1,
indicated by the shared sequences in box brackets). Thus, we termed
them consensus phages (PH 9, PH 33, PH 35).
Several explanation approaches can be taken for the
phenomenon of consensus phages: First, loss of diversity can happen
during the amplification process between selection rounds, leading
to so-called propagation-related phage particles (37,51,52).
These particles are biologically enriched for reasons including
intrinsic advantages regarding the assembly process of phages
bearing special fusion proteins (53), improved binding of phages to the
bacterial pili (15), and mutations
in regulatory regions of phage genes (52,54).
Consequently, Vodnik et al (51) described this phenomenon as almost
intrinsic to phage display technology.
Second, consensus phages may bind contents of the
experimental setting i.e., buffers, blocking reagents, plastic
tubes, et cetera (52,55) and, therefore, be subject to
selection-procedure-related enrichment (51).
Third, the target itself may favor the enrichment of
consensus phages (16).
In our experiments, all three explanations may have
contributed as combination of target-induced selection pressure,
comparable experimental conditions and uniform bacterial production
conditions.
Moreover, Sasso et al (56) performed ten selection procedures
with comparable in vitro conditions and reported several
clones commonly enriched in all ten selection procedures.
Nevertheless, these clones were deemed unspecific without further
investigation (56).
By contrast, we explored the potential of our
consensus phage PH 35 further and showed its blast-binding ability
after conversion into an scFv-Fc antibody format as well as its
internalizing activity (Figs. 3,
4 and 6). Additionally, clones 11, 22, and 24
showed internalizing activity; showing that they are potential
candidates for antibody-drug conjugate in AML immunotherapy
(57).
The first limitation of this study is the lack of
sufficient patient material for antibody selection and therefore,
we were not able to perform all selection strategies with the same
patient. Second, our implemented CD34+ enrichment step
is dependent on the CD34 antigen expression of primary blast cells.
However, only 40% of AML cells express this antigen on their cell
surface. Thus, future studies will be needed to determine whether
other surface antigens, e.g., CD117, can also be implemented for
the blast enrichment procedure.
Third, the semi-synthetic Tomlinson library is based
on one single human framework (VH-3 and Vκ-1) and thus, is
restricted by the selected V-genes. When using a complete naïve
antibody library that includes all human frameworks, there is a
much greater chance for isolation of high-potent antibodies.
In summary, we presented a CD34-based enrichment
strategy, which improves the standard cell-based panning procedures
for the generation of AML blast-specific internalizing antibodies,
thus potentially providing AML patients with access to personalized
antibodies irrespective of their initial blast counts.
In addition, we explored depletion strategies with
increasing levels of depletion antigens regarding their impact on
PD selection outcomes and found that medium levels of depletion
antigens were most effective to isolate promising phage
antibodies.
Finally, we discovered sequence similarities between
our selected phage antibodies, indicating a common observation when
using the semi-synthetic Tomlinson library.
After conversion of the isolated candidate scFv
antibody fragments into scFv-Fc fusion proteins we found that four
of these internalized successfully into cells of the AML-derived
cell line Kasumi-1, which makes them potential candidates for
immunotherapeutic agents.
Acknowledgements
Not applicable.
Funding
The Justus Liebig University Trainee Program for
young medical scientists funded this work.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
TW conceived the clinical and experimental concept,
performed the lab experiments and analyzed the data. TW performed
the literature research and prepared the manuscript. MKT and TW
revised the manuscript. SP supported all lab experiments and
critically verified the correct execution. SG, MR, WB, UG, ATS and
NR evaluated and discussed all lab results thoroughly. MR and WB
supported the clinical setting and enrollment of patients with AML.
SG and MKT provided lab material, discussed and interpretated the
study results and provided their expertise for the successful
outcome of this study. MKT, SP, WB and TW confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All patients with AML gave written informed
consent, and the Clinical Ethics Committee of the Justus Liebig
University (Giessen, Germany) approved all experimental work
(approval no. 140/16). Furthermore, AB serum was received from
patients after written informed consent, which was also approved by
the Clinical Ethics Committee of the Justus Liebig University
Giessen (approval no. 05/00).
Patient consent for publication
All patients gave written informed consent.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AD
|
antigen diversity
|
|
anti-M13
|
antibody against phage proteins
|
|
DGC
|
density gradient centrifugation
|
|
DS
|
depletion step
|
|
LE
|
lysis of erythrocytes
|
|
MA
|
enrichment of monocytes by
adherence
|
|
NT
|
supernatant of non-transfected 293T
cells
|
|
PD
|
phage display
|
|
PS
|
panning step
|
|
RPMI
|
RPMI 1640 GlutaMAX™ medium
|
|
RT
|
room temperature
|
|
scFv
|
single chain fragment variable
|
|
scFv-Fc
|
scFv cloned to a fragment
crystallizable of an IgG2a mouse antibody
|
|
SR
|
selection round
|
|
WB-FACS
|
washing buffer for
fluorescence-activated cell-sorting experiments
|
References
|
1
|
Medinger M, Lengerke C and Passweg J:
Novel therapeutic options in acute myeloid leukemia. Leuk Res Rep.
6:39–49. 2016.PubMed/NCBI
|
|
2
|
Southam CM, Craver LF, Dargeon HW and
Burchenal JH: A study of the natural history of acute leukemia with
special reference to the duration of the disease and the occurrence
of remissions. Cancer. 4:39–59. 1951. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Juliusson G, Antunovic P, Derolf A,
Lehmann S, Mollgard L, Stockelberg D, Tidefelt U, Wahlin A and
Höglund M: Age and acute myeloid leukemia: Real world data on
decision to treat and outcomes from the Swedish acute leukemia
registry. Blood. 30:4179–4187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berger DP, Mertelsmann R and Duyster J;
Tumorzentrum Freiburg- CCCF (eds). Das Rote Buch, : Hämatologie und
internistische Onkologie. überarbeitete und erweiterte Auflage.
Landsberg am Lech: ecomed Medizin. p14002017.
|
|
5
|
Patel JP, Gönen M, Figueroa ME, Fernandez
H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S,
Aminova O, et al: Prognostic relevance of integrated genetic
profiling in acute myeloid leukemia. N Engl J Med. 22:1079–1089.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Papaemmanuil E, Gerstung M, Bullinger L,
Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F,
Bolli N, et al: Genomic classification and prognosis in acute
myeloid leukemia. N Engl J Med. 9:2209–2221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network, ;
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
30:2059–2074. 2013.PubMed/NCBI
|
|
8
|
Talati C and Sweet K: Recently approved
therapies in acute myeloid leukemia: A complex treatment landscape.
Leuk Res. 73:58–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dugan J and Pollyea D: Enasidenib for the
treatment of acute myeloid leukemia. Expert Rev Clin Pharmacol.
3:755–760. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pollyea DA: New drugs for acute myeloid
leukemia inspired by genomics and when to use them. Hematology Am
Soc Hematol Educ Program. 30:45–50. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Egan PC and Reagan JL: The return of
gemtuzumab ozogamicin: A humanized anti-CD33 monoclonal
antibody-drug conjugate for the treatment of newly diagnosed acute
myeloid leukemia. Onco Targets Ther. 11:8265–8272. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McCafferty J, Griffiths AD, Winter G and
Chiswell DJ: Phage antibodies: Filamentous phage displaying
antibody variable domains. Nature. 348:552–524. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Breitling F, Dübel S, Seehaus T,
Klewinghaus I and Little M: A surface expression vector for
antibody screening. Gene. 15:147–153. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Frei JC and Lai JR: Protein and antibody
engineering by phage display. Methods Enzymol. 5806:45–87. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barbas III CF, Burton DR, Scott JK and
Silverman GJ: Phage Display: A Laboratory Manual. Cold Spring
Harbor Laboratory; New York, NY: 2004, PubMed/NCBI
|
|
16
|
Smith GP and Petrenko VA: Phage display.
Chem Rev. 97:391–410. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tur MK, Rothe A, Huhn M, Goerres U, Klimka
A, Stöcker M, Engert A, Fischer R, Finner R and Barth S: A novel
approach for immunization, screening and characterization of
selected scFv libraries using membrane fractions of tumor cells.
Int J Mol Med. 11:523–527. 2003.PubMed/NCBI
|
|
18
|
Ten Haaf A, Gattenlöhner S and Tur MK:
Antibody selection on FFPE tissue slides. Methods Mol Biol.
1701:381–391. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fitting J, Blume T, Ten Haaf A, Blau W,
Gattenlöhner S, Tur MK and Barth S: Phage display-based generation
of novel internalizing antibody fragments for immunotoxin-based
treatment of acute myeloid leukemia. MAbs. 7:390–402. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ljungars A, Svensson C, Carlsson A,
Birgersson E, Tornberg UC, Frendéus B, Ohlin M and Mattsson M: Deep
mining of complex antibody phage pools generated by cell panning
enables discovery of rare antibodies binding new targets and
epitopes. Front Pharmacol. 10:8472019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bakker ABH, van den Oudenrijn S, Bakker
AQ, Feller N, van Meijer M, Bia JA, Jongeneelen MA, Visser TJ, Bijl
N, Geuijen CA, et al: C-type lectin-like molecule-1: A novel
myeloid cell surface marker associated with acute myeloid leukemia.
Cancer Res. 15:8443–8450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Geuijen CAW, Bijl N, Smit RCM, Cox F,
Throsby M, Visser TJ, Jongeneelen MA, Bakker AB, Kruisbeek AM,
Goudsmit J and de Kruif J: A proteomic approach to tumour target
identification using phage display, affinity purification and mass
spectrometry. Eur J Cancer. 41:178–187. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
ten Haaf A, Pscherer S, Fries K, Barth S,
Gattenlöhner S and Tur MK: Phage display-based on-slide selection
of tumor-specific antibodies on formalin-fixed paraffin-embedded
human tissue biopsies. Immunol Lett. 166:65–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sidney LE, Branch MJ, Dunphy SE, Dua HS
and Hopkinson A: Concise review: Evidence for CD34 as a common
marker for diverse progenitors. Stem Cells. 32:1380–1289. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leach RM, Drummond M, Doig A, McKay P,
Jackson B and Bain BJ: Practical Flow Cytometry in Haematology: 100
worked examples. John Wiley and Sons, Inc.; Hoboken, NJ: 2015,
View Article : Google Scholar
|
|
26
|
Fitting J, Killian D, Junghanss C,
Willenbrock S, Escobar HM, Lange S, Nolte I, Barth S and Tur MK:
Generation of recombinant antibody fragments that target canine
dendritic cells by phage display technology. Vet Comp Oncol.
9:183–195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sánchez-Martín D, Sørensen MD, Lykkemark
S, Sanz L, Kristensen P, Ruoslahti E and Álvarez-Vallina L:
Selection strategies for anticancer antibody discovery: Searching
off the beaten path. Trends Biotechnol. 33:292–301. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jäger S, Jahnke A, Wilmes T, Adebahr S,
Vögtle FN, Delima-Hahn E, Pfeifer D, Berg T, Lübbert M and Trepel
M: Leukemia-targeting ligands isolated from phage-display peptide
libraries. Leukemia. 21:411–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galili N, Devemy E and Raza A: Isolation
of specific and biologically active peptides that bind cells from
patients with acute myeloid leukemia (AML). J Hematol Oncol.
10:82008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karjalainen K, Jaalouk DE, Bueso-Ramos CE,
Zurita AJ, Kuniyasu A, Eckhardt BL, Marini FC, Lichtiger B, O'Brien
S, Kantarjian HM, et al: Targeting neuropilin-1 in human leukemia
and lymphoma. Blood. 20:920–927. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nodehi SM, Repp R, Kellner C, Bräutigam J,
Staudinger M, Schub N, Peipp M, Gramatzki M and Humpe A: Enhanced
ADCC activity of affinity maturated and Fc-engineered
mini-antibodies directed against the AML stem cell antigen CD96.
PLoS One. 7:e424262012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hoen PA, Jirka SM, ten Broeke BR, Schultes
EA, Aguilera B, Pang KH, Heemskerk H, Aartsma-Rus A, van Ommen GJ
and den Dunnen JT: Phage display screening without repetitious
selection rounds. Anal Biochem. 421:622–631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luttmann W, Bratke K, Küpper M and Myrtek
D: Der Experimentator: Immunologie. Springer; Heidelberg: 2014,
View Article : Google Scholar
|
|
34
|
Ngai PK, Ackermann F, Wendt H, Savoca R
and Bosshard HR: Protein A antibody-capture ELISA (PACE): An ELISA
format to avoid denaturation of surface-adsorbed antigens. J
Immunol Methods. 158:267–276. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nikfarjam S, Tohidkia MR, Mehdipour T,
Soleimani R, Rahimi AA and Nouri M: Successful application of whole
cell panning for isolation of phage antibody fragments specific to
differentiated gastric cancer cells. Adv Pharm Bull. 9:624–631.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jones ML, Alfaleh MA, Kumble S, Zhang S,
Osborne GW, Yeh M, Arora N, Hou JJ, Howard CB, Chin DY and Mahler
SM: Targeting membrane proteins for antibody discovery using phage
display. Sci Rep. 6:262402016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Derda R, Tang S, Li SC, Ng S, Matochko W
and Jafari MR: Diversity of phage-displayed libraries of peptides
during panning and amplification. Molecules. 21:1776–1803. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Naeim F, Nagesh Rao P, Song SX and Phan
RT: Principles of Immunophenotyping. Atlas of Hematopathology.
Elsevier; pp. 29–56. 2018, View Article : Google Scholar
|
|
39
|
Ricart AD: Antibody-drug conjugates of
calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab
ozogamicin. Clin Cancer Res. 15:6417–6427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vercauteren SM, Zapf R and Sutherland HJ:
Primitive AML progenitors from most CD34(+) patients lack CD33
expression but progenitors from many CD34(−) AML patients express
CD33. Cytotherapy. 9:194–204. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsui H, Takeshita A, Naito K, Shinjo K,
Shigeno K, Maekawa M, Yamakawa Y, Tanimoto M, Kobayashi M, Ohnishi
K and Ohno R: Reduced effect of gemtuzumab ozogamicin (CMA-676) on
P-glycoprotein and/or CD34-positive leukemia cells and its
restoration by multidrug resistance modifiers. Leukemia.
16:813–819. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wells SJ, Bray RA, Stempora LL and Farhi
DC: CD117/CD34 expression in leukemic blasts. Am J Clin Pathol.
1:192–195. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Alfaleh MA, Jones ML, Howard CB and Mahler
SM: Strategies for selecting membrane protein-specific antibodies
using phage display with cell-based panning. Antibodies (Basel).
5:102017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu Y, McKenna RW, Wilson KS, Karandikar
NJ, Schultz RA and Kroft SH: Immunophenotypic identification of
acute myeloid leukemia with monocytic differentiation. Leukemia.
20:1321–1324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Reader RH, Workman RG, Maddison BC and
Gough KC: Advances in the production and batch reformatting of
phage antibody libraries. Mol Biotechnol. 61:801–815. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pokorny NJ, Boulter-Bitzer JI, Hall JC,
Trevors JT and Lee H: Inhibition of Cryptosporidium parvum
infection of a mammalian cell culture by recombinant scFv
antibodies. Antonie Van Leeuwenhoek. 94:353–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu S, Ke A and Doudna JA: A fast and
efficient procedure to produce scFvs specific for large
macromolecular complexes. J Immunol Methods. 318:95–101. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Boulter-Bitzer JI, Lee H and Trevors JT:
Single-chain variable fragment antibodies selected by phage display
against the sporozoite surface antigen P23 of cryptosporidium
parvum. J Parasitol. 95:75–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marcus WD, Lindsay SM and Sierks MR:
Identification and repair of positive binding antibodies containing
randomly generated amber codons from synthetic phage display
libraries. Biotechnol Prog. 2:919–922. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang W, Samanta M, Crawford SE, Estes MK,
Neill FH, Atmar RL and Palzkill T: Identification of human
single-chain antibodies with broad reactivity for noroviruses.
Protein Eng Des Sel. 27:339–349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vodnik M, Zager U, Strukelj B and Lunder
M: Phage display: Selecting straws instead of a needle from a
haystack. Molecules. 19:790–817. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thomas WD, Golomb M and Smith GP:
Corruption of phage display libraries by target-unrelated clones:
Diagnosis and countermeasures. Anal Biochem. 407:237–240. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuzmicheva GA, Jayanna PK, Sorokulova IB
and Petrenko VA: Diversity and censoring of landscape phage
libraries. Protein Eng Des Sel. 22:9–18. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Brammer LA, Bolduc B, Kass JL, Felice KM,
Noren CJ and Hall MF: A target-unrelated peptide in an M13 phage
display library traced to an advantageous mutation in the gene II
ribosome-binding site. Anal Biochem. 373:88–98. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Menendez A and Scott JK: The nature of
target-unrelated peptides recovered in the screening of
phage-displayed random peptide libraries with antibodies. Anal
Biochem. 336:145–157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sasso E, D'Avino C, Passariello M, D'Alise
AM, Siciliano D, Esposito ML, Froechlich G, Cortese R, Scarselli E,
Zambrano N, et al: Massive parallel screening of phage libraries
for the generation of repertoires of human immunomodulatory
monoclonal antibodies. MAbs. 11:1–13. 2018. View Article : Google Scholar
|
|
57
|
Harper J, Mao S, Strout P and Kamal A:
Selecting an optimal antibody for antibody-drug conjugate therapy:
Internalization and intracellular localization. Methods Mol Biol.
1045:41–49, 2013.0. View Article : Google Scholar : PubMed/NCBI
|