Introduction
Tubular atrophy/interstitial fibrosis (TA/IF), the
histological characteristic of kidney allograft destruction over
time, is a major cause of late allograft loss (1,2). TA/IF
is a chronic, progressive, non-specific and irreversible
histopathological entity that occurs in the early
post-transplantation period (3),
and is associated with significant morbidity and mortality in
patients (4). Currently, the major
challenge faced is the incomplete understanding of the factors that
promote the development of TA/IF. Therefore, it is of great
significance to examine the identifiable causes of chronic
allograft TA/IF and to develop cause-specific treatment
strategies.
Inflammation is a physiological defense mechanism
against adverse stimuli, such as tissue damage and infection
(5). A timely and powerful
inflammatory response can effectively resist these harmful stimuli,
while weak and continuous inflammation will aggravate the situation
(6). Kidney allograft survival in
patients with inflammation detected in the fibrotic areas of
indication biopsies has been reported to be significantly worse
compared with patients with non-inflammatory interstitial fibrosis
(7–9), which suggested the close association
between inflammation and TA/IF. Therefore, we hypothesized that
effective inhibition of chronic inflammation may suppress
TA/IF.
Receptor-interacting protein 3 (RIP3) is a cytosolic
serine/threonine kinase that consists of an active kinase domain at
the amino terminus (10), and it
serves an important role in the process of necroptosis, a
programmed form of necrotic cell death (11,12).
RIP3 associates with RIP1 to form a signaling complex known as the
necrosome, and then mixed lineage kinase domain-like protein (MLKL)
causes a change in its conformation, leading to its translocation
to the plasma membrane and subsequent membrane disruption (13,14).
Necroptosis is a highly inflammatory type of cell death caused as a
result of the release of intracellular immunogenic contents, which
stimulates innate immune cells and subsequently inflammation
(15). However, there are few
reports regarding the role of necroptosis in kidney
transplantation.
Reactive oxygen species (ROS) have long been
considered as a driving force for necroptosis (16). For example, it has been reported
that TNF can induce mitochondrial ROS, and ROS can enhance
necrosome formation (17,18). It has been also been revealed that
RIP1 can sense ROS via the modification of three crucial cysteine
residues, and its autophosphorylation on S161 is subsequently
induced. This phosphorylation event allows for the efficient
recruitment of RIP3 to RIP1 to form a functional necrosome
(19); however, research examining
the relationship between ROS and RIP3 has been inconclusive.
In the present study, the differences in expression
of RIP3 in patients with chronic TA/IF and patients who had a good
recovery after renal transplant were tested. Then, normal renal
tubular epithelial cells HK-2 were used to establish a cellular
model with RIP3 overexpression in order to examine the effect of
RIP3 on renal epithelial cells after transplantation. Once the
importance of RIP3 is determined, novel treatment strategies could
be developed that suppress inflammation to improve TA/IF.
Materials and methods
Tissue samples
Patients who underwent kidney transplant and did not
have chronic diseases such as diabetes, hypertension or fatty liver
disease were enrolled in the present experiment between March 2018
and October 2019. Puncture specimens from all patients with kidney
transplant were obtained from the Department of Renal
Transplantation, Ningbo Urology and Nephrology Hospital (Ningbo,
China). A total of 45 puncture specimens were collected, of which
16 had good recovery [normal transplant (NT) group] and the other
29 samples had chronic TA/IF. The study protocol was approved by
the Ethical Committee of Ningbo Urology and Nephrology Hospital
(approval no. YZRY2017120034), and informed written consent was
obtained from all the subjects. The clinicopathological
characteristics of the included patients are summarized in Table I.
 | Table I.Clinicopathological characteristics
of the patients included in the present study. |
Table I.
Clinicopathological characteristics
of the patients included in the present study.
| Clinicopathological
characteristics | Normal group | TA/IF group |
|---|
| Total number,
n | 16 | 29 |
| Age, years, mean ±
SEM | 43±8 | 45±12 |
| Sex |
|
|
| Male,
n | 9 | 18 |
| Female,
n | 7 | 11 |
| Puncture time,
monthsa |
|
|
|
<12 | 2 | 3 |
|
≥12 | 14 | 26 |
| Creatinine, µmol/l,
mean ± SEM | 97±42 | 212±98 |
| Hemoglobin, g/l,
mean ± SEM | 95±14 | 102±19 |
Cell culture
The cell line, HK-2, used in the present study was
obtained from the American Type Culture Collection, and cells were
cultured in DMEM (HyClone; Cytiva), supplemented with 10%
heat-inactivated FBS (Shanghai ExCell Biology, Inc.). Cells were
maintained at 37°C with 5% CO2 in a humid
environment.
Generation of stable cell lines
RIP3-expressing lentivirus with green fluorescent
protein and puromycin resistance markers were purchased from
Shanghai GeneChem Co., Ltd. RIP3-expressing lentivirus packaging
used three plasmids: GV492 plasmid carrying RIP3 CDS region, helper
plasmids pHelper 1.0 and pHelper 2.0 (all Shanghai GeneChem Co.,
Ltd.). A 3rd generation lentivirus packaging system was used. A
total of 20 GV492, 15 pHelper 1.0 and 10 µg pHelper 2.0 were mixed
with 25 µl transfection reagent (cat. no. LPK001; Shanghai GeneChem
Co., Ltd.), adjusted to a total volume of 1 ml and added to 293
cells (American Type Culture Collection) after standing for 15 min.
The 293 cells were cultured in a 37°C, 5% CO2 incubator
for 6 h. Next, fresh medium containing 10% serum was added before
culturing for 48–72 h. Supernatant was collected, centrifuged at
4,000 × g for 10 min at 4°C to remove cell debris, then filtered
with a 0.45-µM filter. Stable cell lines were established according
to the manufacturer's protocol. Briefly, 7.5×104 HK-2
cells were seeded onto a 6-well plate and transfected with
lentivirus vectors (MOI=10), and the cell culture medium was
replaced with complete medium after 24 h. Lentivirus-transfected
HK-2 cells were selected using complete medium containing 4 µg/ml
puromycin after 72 h. At 1 week after screening, the cells were
used for subsequent experiments. Subsequently, the cells were
maintained with 4 µg/ml puromycin. RIP3 overexpression was
confirmed via western blotting. Cells overexpressing RIP3 were
referred to as the RIP3-OE group. Cells with control lentivirus
were used as the negative control (NC) group.
Cell treatment
To inhibit MLKL, HK-2 cells were treated with 5 µM
necrosulfonamide (NSA; cat. no. 480073; Sigma-Aldrich; Merck KGaA)
and cultured in a 37°C cell incubator for 3 h, then the cells were
collected for subsequent experiments. For NF-κB inhibitor (BAY
11-7085) treatment, HK-2 cells were treated with 5 µM BAY 11-7085
(cat. no. HY-10257; MedChemExpress) and cultured in a 37°C cell
incubator for 3 h, then the cells were collected for subsequent
experiments.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from tissue and cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RT was conducted using the PrimeScript RT Master Mix kit
according to the manufacturer's protocol. (Takara Biotechnology
Co., Ltd.). cDNAs were amplified via RT-qPCR using SYBR Green PCR
Master mix (Roche Diagnostics) on a LightCycler® 480
system (Roche Diagnostics) as follows: 95°C for 15 sec, 60°C for 30
sec and 72°C for 30 sec, 45 cycles in total. The relative
expression levels were normalized against the expression level of
the endogenous control, GAPDH using the 2−ΔΔCq method
(20). The PCR primers are
presented in Table II.
| Table II.Oligonucleotide sequences of the
primers used for reverse transcription-quantitative PCR. |
Table II.
Oligonucleotide sequences of the
primers used for reverse transcription-quantitative PCR.
| Gene | Primer sequences
(5′→3′) |
|---|
| RIP3 | F:
CTGAGTGGCTAAACAAACTGAATC |
|
| R:
AGGTAGGGCTGGGCATCTG |
| IL-8 | F:
ACTCCAAACCTTTCCACCCC |
|
| R:
TTCTCAGCCCTCTTCAAAAACT |
| IL-1β | F:
CAGAAGTACCTGAGCTCGCC |
|
| R:
AGATTCGTAGCTGGATGCCG |
| IL-33 | F:
TTATGAAGCTCCGCTCTGGC |
|
| R:
CCAAAGGCAAAGCACTCCAC |
| NLRP3 | F:
AGAACTTTCTGTGTGGACCGA |
|
| R:
AGCCCTTCTGGGGAGGATAG |
| BMF | F:
TGGAAACAATACCGCACCGT |
|
| R:
ACTCGATTGGGAAGGAGGGA |
| BNIP3 | F:
CTGGAGTCTGACTTGGTTCGT |
|
| R:
CCACCCCAGGATCTAACAGC |
| GAPDH | F:
AATGGGCAGCCGTTAGGAAA |
|
| R:
GCGCCCAATACGACCAAATC |
Western blot analysis
RIPA kit (cat. no. R0010; Beijing Solarbio Science
& Technology Co., Ltd.) was used to extract cellular protein.
Protein concentrations were determined with BCA kit (cat. no.
P0010; Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Proteins (30 µg per lane) were separated
via 10% SDS-PAGE, transferred to a polyvinylidene fluoride membrane
(Bio-Rad Laboratories, Inc.) and blocked with TBS with 0.1%
Tween-20 (TBST) containing 5% non-fat dry milk at room temperature
for 90 min. Anti-RIP3 (1:1,000; cat. no. ab56164; Abcam),
anti-phosphorylated (p)-RIP3 (1:1,000; cat. no. ab209384; Abcam),
anti-MLKL (1:1,000; cat. no. ab183770; Abcam), anti-p-MLKL
(1:1,000; cat. no. ab187091; Abcam), anti-RIP1 (1:1,000; cat. no.
4926; Cell Signaling Technology, Inc.), anti-p-RIP1 (1:1,000; cat.
no. 44590; Cell Signaling Technology, Inc.), anti-NF-κB (1:2,000;
cat. no. 06-418; EMD Millipore) and anti-GAPDH (1:1,000; cat. no.
ab181602; Abcam) antibodies were diluted in TBST containing 3%
non-fat dry milk and incubated with the membranes overnight at 4°C.
Membranes were then washed with TBST and incubated for 1 h at room
temperature with HRP-conjugated secondary antibody (1:5,000; cat.
no. sc2030; Santa Cruz Biotechnology, Inc.). Subsequently,
membranes were washed with TBST and developed using an ECL-Plus
reagent (Cytiva). ImageJ software version 1.48 (National Institutes
of Health) was used to evaluate the gray value of the western
blots.
Measuring ROS
In order to detect the level of ROS and exclude the
interference of GFP, HK-2 cells transfected with a RIP3-expressing
lentivirus that did not contain GFP but had puromycin resistance
was also constructed as aforementioned. RIP3-expressing lentivirus
packaging used three plasmids: GV341 plasmid carrying RIP3 CDS
region, helper plasmids pHelper 1.0 and pHelper 2.0 (all Shanghai
GeneChem Co., Ltd.) A total of 3×105 cells suspended in
2 ml fresh media were plated in each well of a 6-well plate and
incubated overnight. Then, the cells were treated with or without
10 µM N-acetylcysteine (NAC; Beyotime Institute of Biotechnology)
at 37°C for 6 h. The cells were incubated with 10 µmol/l
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) at 37°C for 30
min to assess the ROS-mediated oxidation of DCFH-DA to the
fluorescent compound DCF. The images showing the green fluorescence
of DCF in the cells were acquired using a Nikon Ti-U fluorescence
microscope (Nikon Corporation). Next, the cells were harvested, and
the pellets were suspended in 1 ml PBS. Samples were analyzed at an
excitation wavelength of 480 nm and an emission wavelength of 525
nm using a FACScan flow cytometer (Becton, Dickinson and Company).
Data were analyzed with FlowJo software version 10 (Becton,
Dickinson and Company).
Necroptosis assay determining lactate
dehydrogenase (LDH) release
RIP3-OE and NC cells (1×104/well) were
seeded onto a 96-well microplate and cultured for 24 h. The LDH
cytotoxicity assay kit (cat. no. C0017; Beyotime Institute of
Biotechnology) was used to detect cell death after the
overexpression of RIP3, according to the manufacturer's
instructions. Briefly, cells (~1×105 cell/well) were
added into a 96-well cell culture plate (the cell density did not
exceed 80–90% when it was tested), and then each culture well was
divided into the following groups: i) Cell-free culture medium
wells (background blank control wells); ii) control cell wells
without treatment (sample control wells); iii) cell wells that were
not treated for subsequent lysis (sample maximum enzyme activity
control wells); and iv) cell wells with treatment (experimental
sample wells). Then, 1 h before the scheduled detection time, the
cell culture plate was removed and the LDH release reagent (10% of
the original culture fluid volume) provided in the kit was added to
the ‘sample maximum enzyme activity control well’, mixed several
times and incubated for 30 min. Subsequently, the cell culture
plate with a multi-well plate was centrifuged at 400 × g, 4°C for 5
min. A total of 120 µl supernatant was removed from each well and
added to the corresponding wells of a new 96-well plate, and then
sample determination was conducted. The absorbance value of each
sample was read at 490 nm (iMark™ Microplate Reader; Bio-Rad
Laboratories, Inc.).
Statistical analysis
The data are presented as the mean ± SEM of three
independent experiments (n=3). GraphPad Prism 5 software (GraphPad
Software, Inc.) was used for analysis. All data in this study
conformed to a normal distribution. First, a Shapiro-Wilk test was
used to analyze whether the data followed a normal distribution.
Then, one-way ANOVA was performed followed by post hoc Bonferroni's
correction to compare the differences between the groups. To
compare differences between two groups, an F-test was first used to
compare variances, following which unpaired t-test was used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Overexpression of RIP3 can induce
necroptosis in renal tubular epithelial cells
To detect the expression level of RIP3 in patients
with renal transplant, a total of 45 puncture specimens were
collected and RT-qPCR was performed using samples from the NT and
chronic TA/IF groups. Compared with the NT group, the patients with
chronic TA/AF were found to have upregulated RIP3 expression
(Fig. 1A).
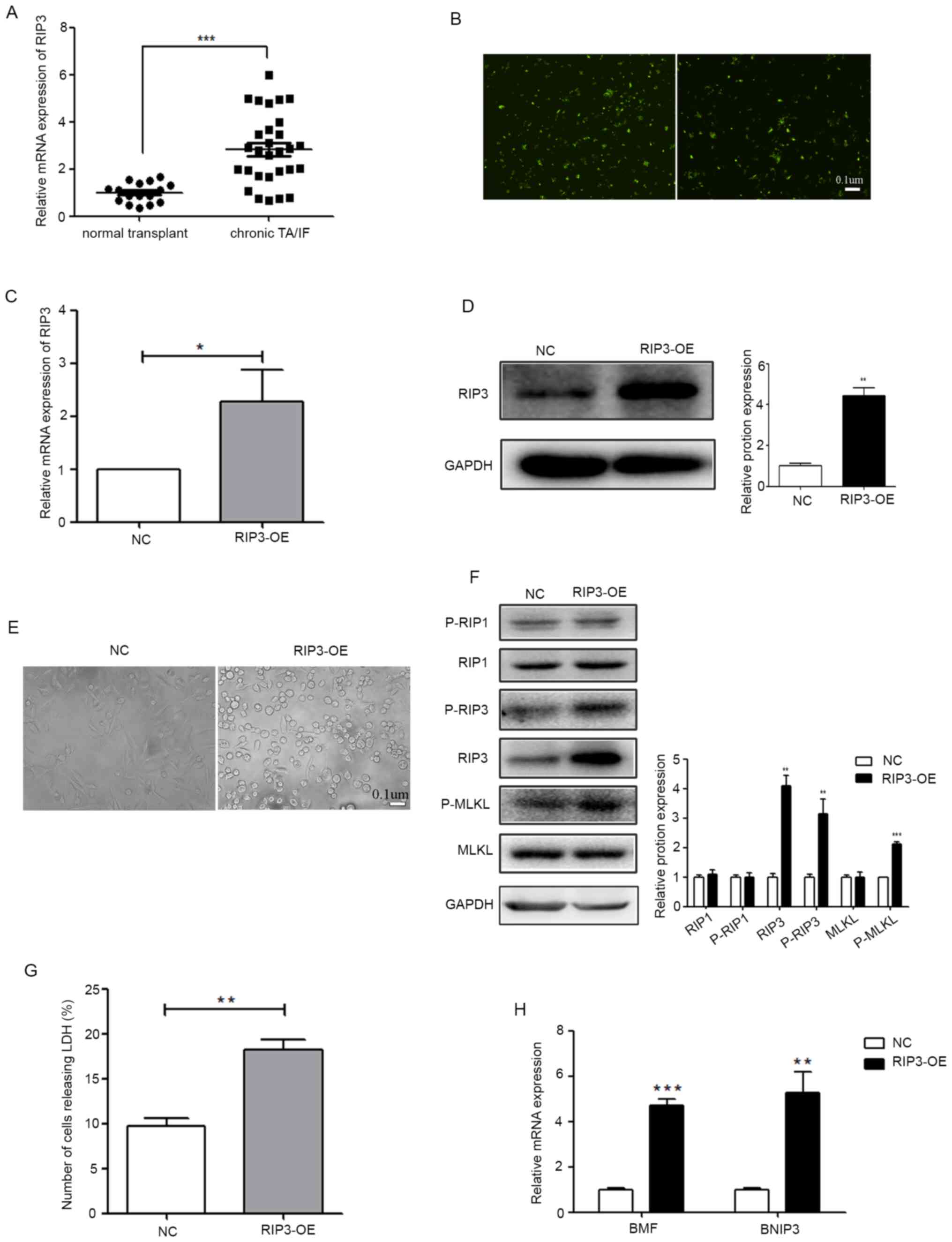 | Figure 1.RIP3 overexpression can induce
necroptosis in renal tubular epithelial cells. (A) mRNA expression
level of RIP3 was analyzed in 16 patients with NT and in 29
patients with chronic TA/IF using RT-qPCR. ***P<0.001 vs. NT
group. (B) A lentivirus vector-mediated RIP3 overexpressing stable
HK-2 cell line was established and the transfection efficiency of
the virus was determined (scale bar, 0.1 µm). (C) RT-qPCR was
performed to detect the expression level of RIP3 in HK-2 cells. (D)
Western blotting was performed to examine the expression level of
RIP3. The lentivirus RIP3-OE vector significantly increased the
expression level of RIP3 in HK-2 cells. (E) The influence of RIP3
on the morphological changes of HK-2 cells (scale bar, 0.1 µm). (F)
Protein expression levels of RIP1, RIP3 and MLKL and their
phosphorylation levels were examined via western blotting in HK-2
cells. (G) LDH release assay results identified that the level of
cell death was increased in the RIP3-OE group compared with the NC.
(H) RT-qPCR was performed to detect the expression levels of BMF
and BNIP3 in HK-2 cells. *P<0.05, **P<0.01, ***P<0.001 vs.
NC. RT-qPCR, reverse transcription-quantitative PCR; RIP,
receptor-interacting protein; NT, normal transplant; TA/IF, tubular
atrophy and/or interstitial fibrosis; OE, overexpression group; NC,
negative control; MLKL, mixed lineage kinase domain-like protein;
BMF, Bcl-2-modifying factor; BNIP3, Bcl-2-interacting protein 3;
LDH, lactate dehydrogenase; p-, phosphorylated. |
To further evaluate the relationship between RIP3
and TA/AF, a lentivirus vector-mediated RIP3 overexpressing stable
HK-2 cell line was established and the transfection efficiency of
the virus is presented in Fig. 1B.
RT-qPCR and western blotting were then performed. Compared with the
NC group, the expression of RIP3 was significantly higher in the
RIP3-OE group (Fig. 1C and D).
Since RIP3 is a key regulatory gene of necroptosis (11,12),
the changes in cell morphology were analyzed via microscopy
(Fig. 1E). Cells began to round out
and cytoplasmic vacuolation appeared following RIP3-OE. The
phosphorylation level of RIP3, as well as two key genes RIP1 and
MLKL located upstream and downstream, were examined in the RIP3-OE
group. As presented in Fig. 1F, the
phosphorylation levels of RIP3 and MLKL were significantly
increased, while the phosphorylation level of RIP1 was not
changed.
Differing from apoptotic cell death, necroptosis
does not result in chromatin condensation, shrinkage of the cell
body or a fragmented genome, but is characterized by the damaged
integrity of the cytoplasm membrane, which can be confirmed by LDH
leakage (21,22). The results demonstrated that LDH
leakage, which can indicate the degree of necroptosis, was
significantly increased in the RIP3-OE group compared with the NC
(Fig. 1G). At the molecular level,
necroptotic cells usually have higher expression levels of the
genes that are associated with the RIP cascades, such as
Bcl-2-modifying factor (BMF) and Bcl-2-interacting protein 3
(BNIP3) (21,23). Therefore, the expression levels of
BMF and BNIP3 were detected in the RIP3-OE group, and it was found
that both were increased (Fig. 1H).
These results indicated that RIP3 overexpression can induce
necroptosis in renal tubular epithelial cells.
Overexpression of RIP3 causes
inflammation in HK-2 cells
Kidney allograft survival in patients with
inflammation detected in the fibrotic areas of indication biopsies
has been reported to be significantly worse compared with patients
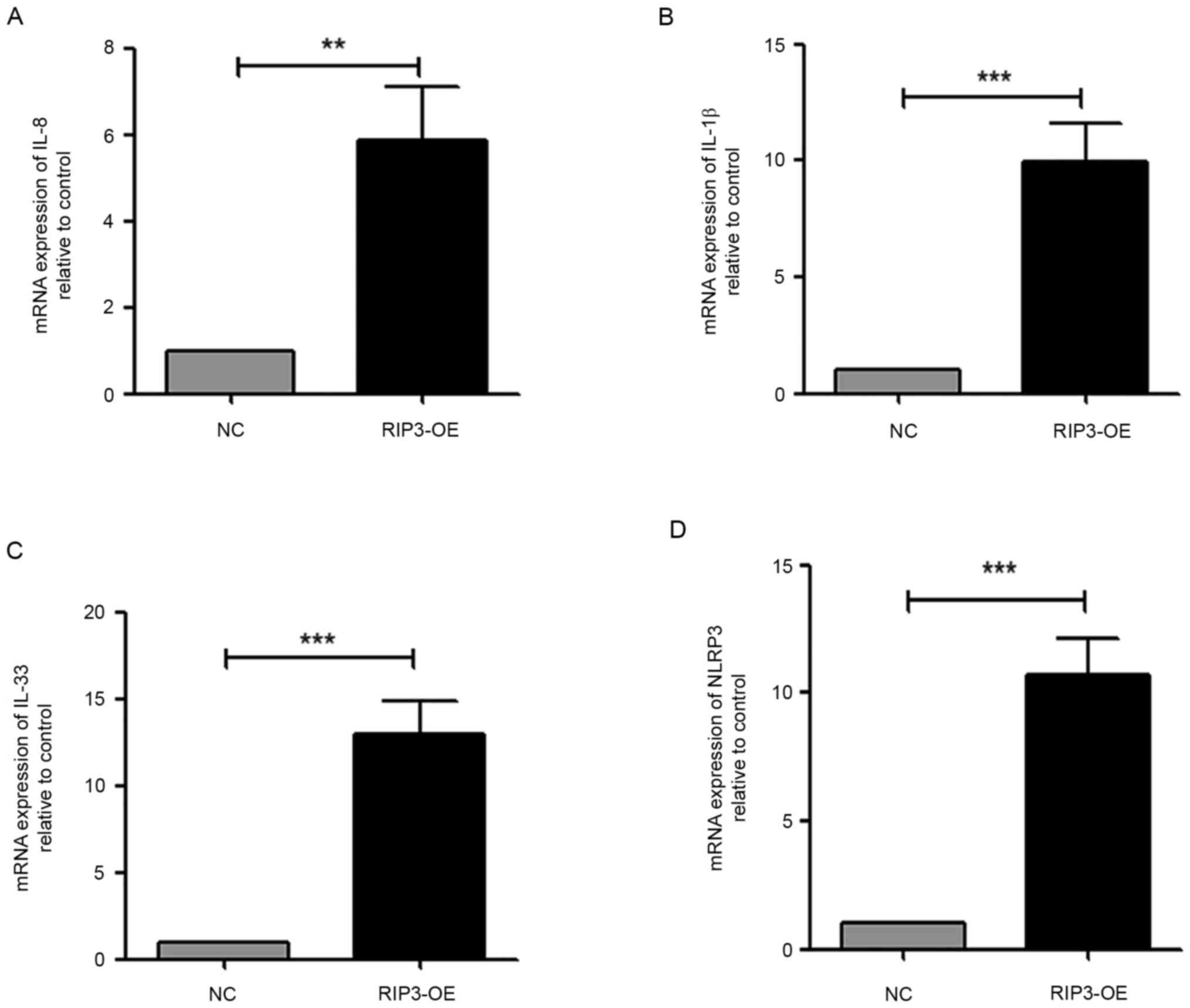
with non-inflammatory interstitial fibrosis (7). Thus, the expression levels of the
pro-inflammatory cytokines IL-1β, IL-8 and the IL-33 were measured.
The results demonstrated a significant increase in the transcript
levels of all cytokines in HK-2 cells in the RIP3-OE group
(Fig. 2A-C). Moreover, the
expression of NLR family pyrin domain containing 3 (NLRP3), which
is a well-characterized inflammasome (24), was examined. It was found that the
NLRP3 transcript was significantly increased in the RIP3-OE group
compared with the NC group (Fig.
2D). Taken together, these data suggested that the
overexpression of RIP3 caused inflammation in HK-2 cells.
Necroptosis induced by RIP3 is
involved in inflammation in HK-2 cells
Necrotic cell death is a highly inflammatory type of
cell death due to the release of intracellular immunogenic contents
that stimulate innate immune cells and subsequently inflammation
(15). In order to verify whether
PIR3-induced necroptosis was the cause of inflammation, NSA was
used to detect inflammatory-related indicators. First, it was
identified that NSA could effectively inhibit the phosphorylation
level of MLKL (Fig. 3A), and
significantly reduce the number of necroptotic cells (Fig. 3B). Furthermore, after NSA treatment,
the expression levels of IL-1β, IL-8, IL-33 and NLRP3 were
significantly lower compared with those of the RIP3-OE group
(Fig. 3C-F). These data revealed
that necroptosis induced by RIP3 was involved in inflammation in
HK-2 cells.
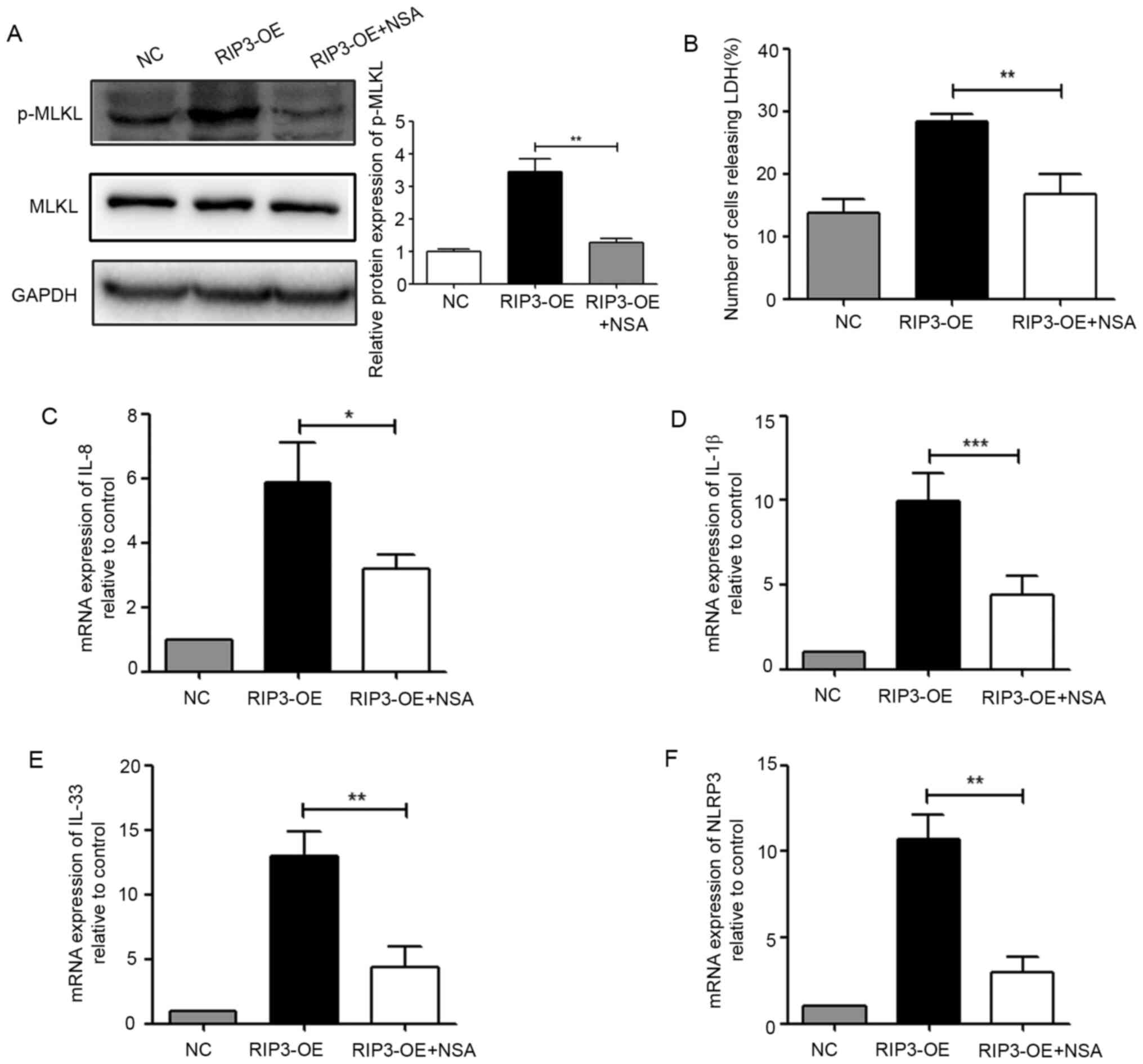 | Figure 3.Necroptosis induced by RIP3 is
involved in inflammation in HK-2 cells. (A) Western blot analysis
of p-MLKL expression in HK-2 cells after treatment with the MLKL
inhibitor NSA (5 µM). (B) LDH release was detected in HK-2 cells
after treatment with the MLKL inhibitor NSA (5 µM). Reverse
transcription-quantitative PCR analysis of the mRNA expression
levels of (C) IL-8, (D) IL-1β, (E) IL-33 and (F) NLRP3 in HK-2
cells after treatment with the MLKL inhibitor NSA (5 µM).
*P<0.05, **P<0.01, ***P<0.001 vs. RIP3-OE group. NLRP3,
NLR family pyrin domain containing 3; RIP3, receptor-interacting
protein 3; MLKL, mixed lineage kinase domain-like protein; p-,
phosphorylated; LDH, lactate dehydrogenase; NSA, necrosulfonamide;
OE, overexpression group; NC, negative control. |
ROS production is a mediator of
necroptosis-induced inflammation in HK-2 cells
Necrosomes have been reported to impair mitochondria
energy metabolism by disturbing ROS homeostasis, leading to ATP
depletion, mitochondrial depolarization and eventually cell death
(25). It is well-known that ROS
are important mediators to induce oxidative stress, which is a
common mechanism of injury in chronic allograft TA/IF (26). Therefore, it was suggested that ROS
production may be a mediator of necroptosis-induced inflammation.
To confirm this, the fluorescent dye DCFH-DA was utilized to detect
ROS levels. From the fluorescence image in Fig. 4A, it was observed that ROS levels
were increased in the RIP3-OE group. Moreover, flow cytometry was
used for the semi-quantitative analysis of ROS production, and
consistent results are presented in Fig. 4B. These results indicated that
overexpression of RIP3 caused increased ROS levels in HK-2
cells.
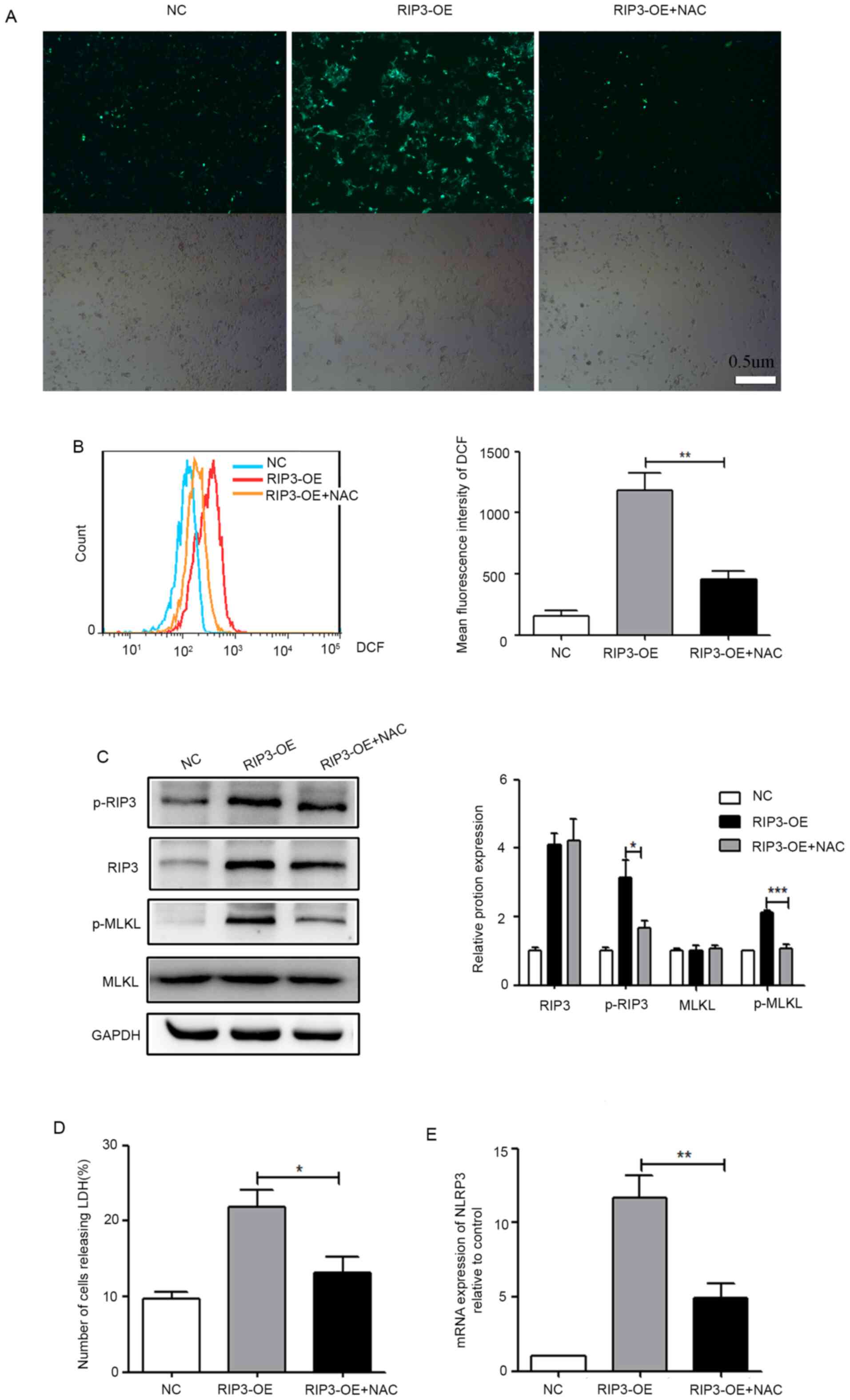 | Figure 4.ROS production is a mediator of
necroptosis-induced inflammation in HK-2 cells. (A) HK-2 cells were
pretreated with/without 10 mM NAC for 6 h, then fluorescence
microscopy of cells was conducted following 10 µM DCFH-DA staining
for 30 min (scale bar, 0.5 µm). (B) Flow cytometry was used for the
semi-quantitative analysis of (A). (C) Western blot analysis of
p-RIP3 and p-MLKL expression in HK-2 cells after treatment with NAC
to decrease ROS levels. (D) LDH release was detected in HK-2 cells
after treatment with NAC. (E) Reverse transcription-quantitative
PCR analysis of the mRNA expression level of NLRP3 in HK-2 cells
after treatment with NAC. *P<0.05, **P<0.01, ***P<0.001
vs. RIP3-OE group. NLRP3, NLR family pyrin domain containing 3;
RIP3, receptor-interacting protein 3; MLKL, mixed lineage kinase
domain-like protein; p-, phosphorylated; ROS, reactive oxygen
species; LDH, lactate dehydrogenase; NAC, N-acetylcysteine;
DCFH-DA, 2,7-dichlorodihydrofluorescein diacetate; OE,
overexpression group; NC, negative control. |
To address whether ROS production was a mediator of
necroptosis-induced inflammation, the effects of the antioxidant
NAC on the necroptosis induced by the overexpression of RIP3 were
evaluated. As shown in Fig. 4A and
B, the generation of ROS in the RIP3-OE + NAC group was
significantly suppressed. At the same time, it was found that, with
the decrease of ROS, the phosphorylation levels of RIP3 and MLKL
(Fig. 4C) and the release of LDH
(Fig. 4D) were significantly
reduced in the RIP3-OE + NAC group, which indicated that
necroptosis was inhibited. Furthermore, the expression of NLRP3 was
significantly reduced in the RIP3-OE + NAC group (Fig. 4E), indicating that inflammation had
been markedly improved with the reduction of ROS. Collectively,
these experimental results suggested that ROS production was a
mediator of necroptosis-induced inflammation.
Necroptosis induces inflammation via a
ROS-dependent NF-κB pathway
NF-κB consists of a family of transcription factors
that serve a central role in the expression levels of
inflammatory-related factors (27).
Therefore, the current study further investigated the relationship
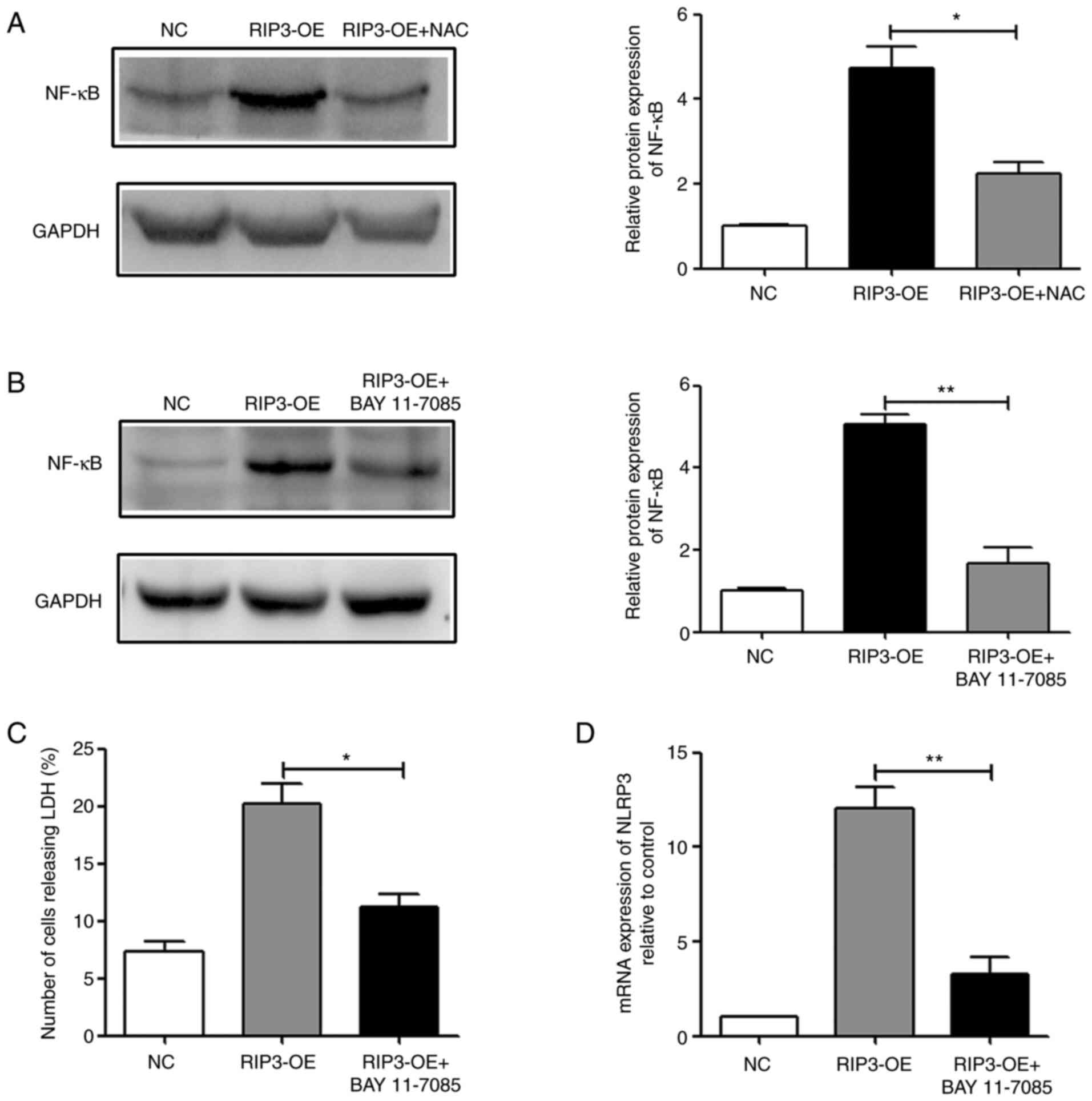
between necroptosis, NF-κB and inflammation. In the RIP3-OE group,
it was found that overexpression of RIP3 could activate NF-κB, and
reducing the levels of ROS in the RIP3-OE + NAC group could
partially reverse this effect (Fig.
5A). Therefore, it was suggested that overexpression of RIP3
could activate the NF-κB signaling pathway via ROS.
Next, BAY 11-7085 was used to treat RIP3-OE HK-2
cells. The results demonstrated that treatment with BAY 11-7085
could effectively suppress the activity of NF-κB (Fig. 5B). Then, necroptosis and
inflammation were examined, and it was identified that in the
RIP3-OE + BAY 11-7085 group, LDH release and NLRP3 expression
levels were significantly reduced compared with the RIP3-OE group
(Fig. 5C and D). Thus, indicating
that inhibition of NF-κB activity could effectively reduce the
occurrence of necroptosis and the appearance of inflammation caused
by RIP3 overexpression. Taken together, the results suggested that
necroptosis induced inflammation via a ROS-dependent NF-κB
pathway.
Discussion
During clinical renal transplantation, multiple
renal allografts are lost in the long-term due to chronic renal
dysfunction associated with the development of TA/IF (1,2).
Inflammation within the areas of interstitial fibrosis and tubular
atrophy are associated with accelerated TA/IF, arterial
fibrointimal hyperplasia and chronic glomerulopathy, as well as
with reduced renal function (7,28).
However, the underlying mechanisms and relationships between
inflammation and TA/IF require further investigation. In the
present study, patients with chronic TA/IF were found to have
upregulated RIP3 expression compared with patients who had good
recovery after renal transplant. Therefore, normal renal tubular
epithelial cells HK-2 were used to establish a cellular model with
RIP3 overexpression in order to investigate the effect of RIP3 on
renal epithelial cells after transplantation.
RIP3 is a cytosolic serine/threonine kinase that
consists of an active kinase domain at the amino terminus. RIP3
functions as an essential adaptor for necroptosis, which is a type
of regulated necrosis controlled by RIP3 and its downstream
effector MLKL (11). Thus, in the
current study, after the overexpression of RIP3, relevant
indicators of necroptosis were detected, and it was found that the
phosphorylation levels of RIP3 and MLKL were significantly
increased, and LDH release, which is a key feature of necroptosis,
was elevated. These findings indicated that the overexpression of
RIP3 can induce cell necroptosis.
During necroptosis, cells can release
damage-associated molecular patterns molecules that can initiate an
inflammatory response in the absence of infection, which suggests
that necroptosis is pro-inflammatory (29). Therefore, the present study detected
the expression levels of the pro-inflammatory cytokines IL-1β, IL-8
and IL-33, and the well-characterized inflammasome NLRP3, after the
overexpression of RIP3. The current study identified a significant
increase in the aforementioned indicators, suggesting that
overexpression of RIP3 caused inflammation in HK-2 cells. In order
to further verify that the inflammatory response caused by the
overexpression of RIP3 was induced via necroptosis, the present
study used the inhibitor NSA to block the activity of the final
executive molecule of necroptosis, MLKL. It was found that after
necroptosis was inhibited, the inflammatory response was also
markedly suppressed.
Next, the mechanism via which RIP3 induces
necroptosis and ultimately leads to inflammation was determined.
Reactive species, which mainly include ROS, are products generated
as a consequence of metabolic reactions in the mitochondria of
eukaryotic cells (30). It is
well-known that ROS are important mediators of cellular damage, and
lipid peroxidation is the most important cause of ROS-induced
oxidative stress (31). The harmful
role of ROS in transplanted organs has been revealed both
experimentally and clinically in kidney transplantation (26). Of note, the present study
demonstrated that, after RIP3 overexpression, the production of ROS
was increased significantly. After removing the excessive ROS using
NAC, both necroptosis and inflammation could be restored to a large
extent. These findings indicated that ROS was closely associated
with necroptosis and inflammation.
NF-κB is a transcription factor that regulates the
expression levels of multiple genes (32), and governs various cellular
functions, including inflammation and necroptotic signaling
(33). In addition, NF-κB has been
proposed to be a sensor for oxidative stress that can be activated
by ROS (34). The present study
first detected that overexpression of RIP3 could effectively
activate the NF-κB signaling pathway, and this activation could be
markedly inhibited by the ROS scavenger NAC, which indicated that
ROS are important mediators in the NF-κB pathway. Subsequently, the
cells were treated with NF-κB inhibitors, and it was identified
that necroptosis and the inflammatory response induced by RIP3 were
also inhibited. Collectively, these results suggested that
necroptosis induced inflammation via a ROS-dependent NF-κB
pathway.
Although the current study identified the importance
of the necroptosis signaling pathway at the cellular level, there
are some limitations of this study. First, an animal model of
kidney transplantation with TA/IF was not established for in
vivo studies, and corresponding cells from animals will be
obtained for further research. Once the findings have been
confirmed both in vivo and in vitro, RIP3 may be a
therapeutic target against inflammation in TA/IF.
In recent years drug repositioning has emerged as a
promising alternative to develop drug-like compounds. Accumulating
evidence has shown that multi-targeting kinase inhibitors used for
the treatment of cancer also display anti-necroptotic activity. For
example, dabrafenib and vemurafenib have been reported to block
RIP3 (35). Moreover, sorafenib has
been revealed to block the kinase activity of both RIP1 and RIP3
(36), while ponatinib directly
targets RIP3 (37,38). These therapeutic drugs that have
been used clinically to treat cancer provide a foundation for the
subsequent development of therapies for TA/IF.
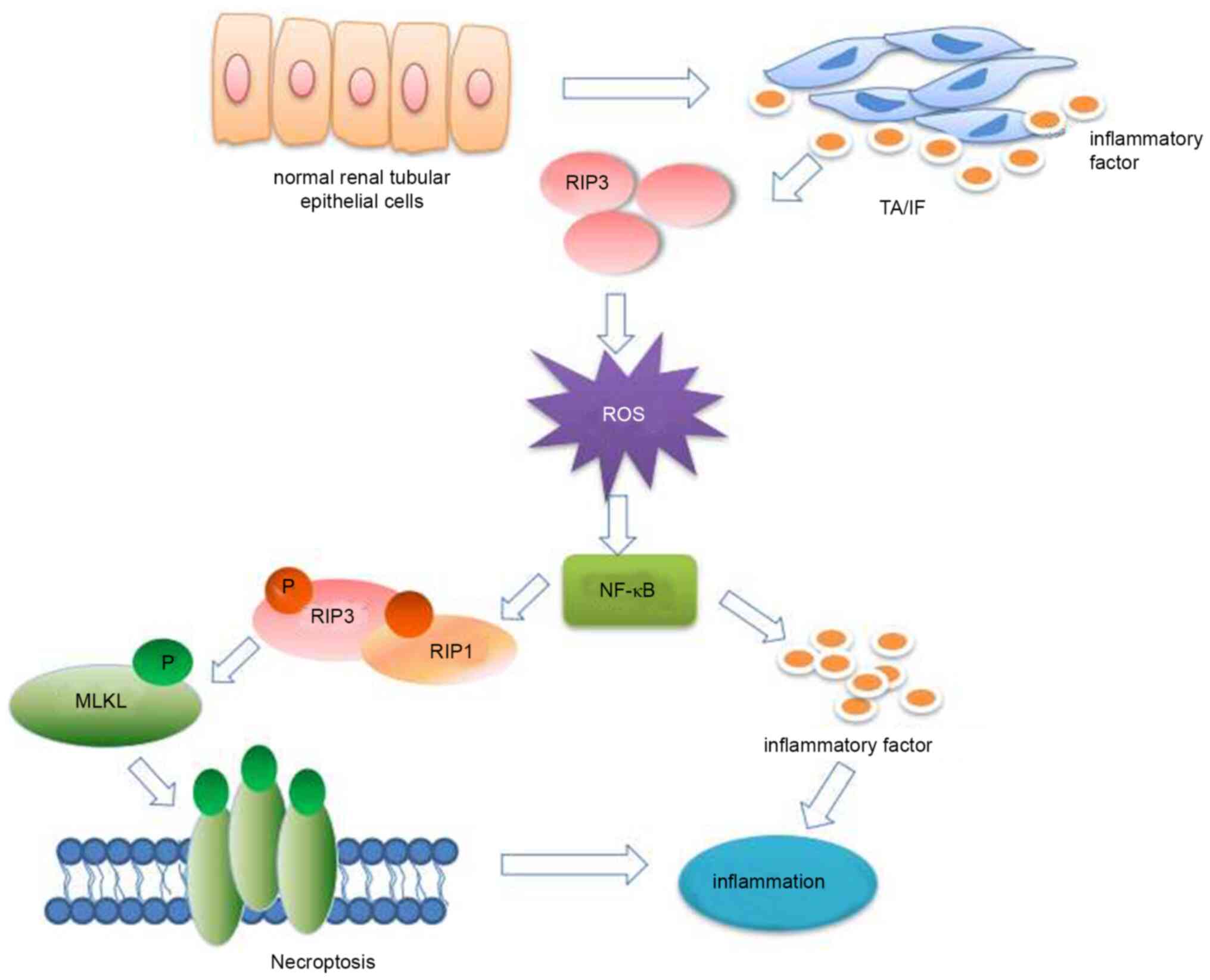
In conclusion, the present study demonstrated that
RIP3 expression was upregulated in patients with chronic TA/IF.
This overexpression of RIP3 can produce excessive ROS and activate
the NF-κB signaling pathway to induce necroptosis, and ultimately
lead to inflammation (Fig. 6).
These potential mechanisms suggest that RIP3 may be a therapeutic
target against inflammation in TA/IF.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ and GW conceived the present study. JW and LC
designed the experiments. JW, DW, LT, ZX and WC carried out the
experiments and collected the data. JW and LC wrote and edited the
manuscript. SZ, GW and JW confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethical
Committee of Ningbo Urology and Nephrology Hospital (Ningbo,
China). Written informed consent was obtained from all
patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Colvin RB: Chronic allograft nephropathy.
N Engl J Med. 349:2288–2290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nankivell BJ, Borrows RJ, Fung CL,
O'Connell PJ, Allen RDM and Chapman JR: The natural history of
chronic allograft nephropathy. N Engl J Med. 349:2326–2333. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pascual M, Theruvath T, Kawai T,
Tolkoff-Rubin N and Cosimi AB: Strategies to improve long-term
outcomes after renal transplantation. N Engl J Med. 346:580–590.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gaston RS, Fieberg A, Helgeson ES,
Eversull J, Hunsicker L, Kasiske BL, Leduc R, Rush D and Matas AJ;
DeKAF Investigators*, : Late Graft loss after kidney
transplantation: Is ‘death with function’ really death with a
functioning allograft? Transplantation. 104:1483–1490. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koyama Y and Brenner DA: Liver
inflammation and fibrosis. J Clin Invest. 127:55–64. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mack M: Inflammation and fibrosis. Matrix
Biol. 68-69:106–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mannon RB, Matas AJ, Grande J, Leduc R,
Connett J, Kasiske B, Cecka JM, Gaston RS, Cosio F, Gourishankar S,
et al: Inflammation in areas of tubular atrophy in kidney allograft
biopsies: A potent predictor of allograft failure. Am J Transplant.
10:2066–2073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sellarés J, de Freitas DG, Mengel M, Sis
B, Hidalgo LG, Matas AJ, Kaplan B and Halloran PF: Inflammation
lesions in kidney transplant biopsies: Association with survival is
due to the underlying diseases. Am J Transplant. 11:489–499. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Naesens M, Kuypers DR, De Vusser K,
Evenepoel P, Claes K, Bammens B, Meijers B, Sprangers B, Pirenne J,
Monbaliu D, et al: The histology of kidney transplant failure: A
long-term follow-up study. Transplantation. 98:427–435. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng S, Yang Y, Mei Y, Ma L, Zhu D, Hoti
N, Castanares M and Wu M: Cleavage of RIP3 inactivates its
caspase-independent apoptosis pathway by removal of kinase domain.
Cell Signal. 19:2056–2067. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galluzzi L and Kroemer G: Necroptosis: A
specialized pathway of programmed necrosis. Cell. 135:1161–1163.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H,
Song Y, Chen X, Shao J and Han J: Distinct roles of RIP1-RIP3
hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis.
Cell Death Differ. 21:1709–1720. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Sun L, Su L, Rizo J, Liu L, Wang
LF, Wang FS and Wang X: Mixed lineage kinase domain-like protein
MLKL causes necrotic membrane disruption upon phosphorylation by
RIP3. Mol Cell. 54:133–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Li W, Ren J, Huang D, He WT, Song
Y, Yang C, Li W, Zheng X, Chen P and Han J: Translocation of mixed
lineage kinase domain-like protein to plasma membrane leads to
necrotic cell death. Cell Res. 24:105–121. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schulze-Osthoff K, Bakker AC,
Vanhaesebroeck B, Beyaert R, Jacob WA and Fiers W: Cytotoxic
activity of tumor necrosis factor is mediated by early damage of
mitochondrial functions. Evidence for the involvement of
mitochondrial radical generation. J Biol Chem. 267:5317–5323. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goossens V, Grooten J, De Vos K and Fiers
W: Direct evidence for tumor necrosis factor-induced mitochondrial
reactive oxygen intermediates and their involvement in
cytotoxicity. Proc Natl Acad Sci USA. 92:8115–8119. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fulda S: Regulation of necroptosis
signaling and cell death by reactive oxygen species. Biol Chem.
397:657–660. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ,
Chen X, Cai Q, Yang ZH, Huang D, Wu R and Han J: RIP1
autophosphorylation is promoted by mitochondrial ROS and is
essential for RIP3 recruitment into necrosome. Nat Commun.
8:143292017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen T: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li N, He Y, Wang L, Mo C, Zhang J, Zhang
W, Li J, Liao Z, Tang X and Xiao H: D-galactose induces necroptotic
cell death in neuroblastoma cell lines. J Cell Biochem.
112:3834–3844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hitomi J, Christofferson DE, Ng A, Yao J,
Degterev A, Xavier RJ and Yuan J: Identification of a molecular
signaling network that regulates a cellular necrotic cell death
pathway. Cell. 135:1311–1323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vanden Berghe T, Linkermann A,
Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis:
The expanding network of non-apoptotic cell death pathways. Nat Rev
Mol Cell Biol. 15:135–147. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perrea DN, Moulakakis KG, Poulakou MV,
Vlachos IS, Papachristodoulou A and Kostakis AI: Correlation
between oxidative stress and immunosuppressive therapy in renal
transplant recipients with an uneventful postoperative course and
stable renal function. Int Urol Nephrol. 38:343–348. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thoma A and Lightfoot AP: NF-κB and
inflammatory cytokine signalling: Role in skeletal muscle atrophy.
Adv Exp Med Biol. 1088:267–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farris AB and Colvin RB: Renal
interstitial fibrosis: Mechanisms and evaluation. Curr Opin Nephrol
Hypertens. 21:289–300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weinlich R, Oberst A, Beere HM and Green
DR: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang T, Zhang X and Li JJ: The role of
NF-kappaB in the regulation of cell stress responses. nt
Immunopharmacol. 2:1509–1520. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moriwaki K and Chan FK: The inflammatory
signal adaptor RIPK3: Functions beyond necroptosis. Int Rev Cell
Mol Biol. 328:253–275. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li N and Karin M: Is NF-kappaB the sensor
of oxidative stress? FASEB J. 13:1137–1143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su
Y, Shen YY, Chen Y, Xiong B, Yang CH, et al: The B-Raf(V600E)
inhibitor dabrafenib selectively inhibits RIP3 and alleviates
acetaminophen-induced liver injury. Cell Death Dis. 5:e12782014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martens S, Jeong M, Tonnus W, Feldmann F,
Hofmans S, Goossens V, Takahashi N, Bräsen JH, Lee EW, Van der
Veken P, et al: Sorafenib tosylate inhibits directly necrosome
complex formation and protects in mouse models of inflammation and
tissue injury. Cell Death Dis. 8:e29042017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fauster A, Rebsamen M, Huber KVM,
Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M,
Gridling M, Parapatics K, et al: A cellular screen identifies
ponatinib and pazopanib as inhibitors of necroptosis. Cell Death
Dis. 6:e17672015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Morioka S, Broglie P, Omori E, Ikeda Y,
Takaesu G, Matsumoto K and Ninomiya-Tsuji J: TAK1 kinase switches
cell fate from apoptosis to necrosis following TNF stimulation. J
Cell Biol. 204:607–623. 2014. View Article : Google Scholar : PubMed/NCBI
|