Introduction
Although systemic inflammatory response syndrome
(SIRS) is commonly associated with pathogenic infection, certain
patients with SIRS do not suffer from such infection, thus
reflecting poor understanding of its pathophysiology (1). Therefore, other factors may also
serve a key role in the occurrence and development of SIRS.
Stimulating adaptive immune response pattern
recognition receptors (PRRs) is associated with development of
SIRS. PRRs are derived from pathogen-associated molecular patterns
(PAMPs) and damage-associated molecular patterns (DAMPs) of
exogenous and endogenous invaders, respectively (2). Among known DAMPs, mitochondrial
DAMPs (mtDAMPs), including mitochondrial DNA (mtDNA), N-formyl
peptides (NFPs), mitochondrial transcription factor (TFAM),
cardiolipin and ATP (3), have
attracted increasing attention from researchers. The aforementioned
molecules are considered to be danger signals released in response
to tissue injury, thus triggering an immune response similar to
that induced by pathogens (2).
Previous studies have shown that plasma levels of mtDAMPs may be
associated with clinical outcome of septic shock, major surgery or
severe trauma (4–6). In addition, it has been suggested
that mtDNA may be a more efficient biomarker than lactate
concentration/sequential organ failure assessment score in
predicting mortality rate in patients with sepsis following
emergency admission (7). However,
mtDNA has not been widely used in clinical practice to optimize
clinical treatment. Therefore, further research is urgently
required.
The present review aimed to summarize the effects
and mechanisms of mtDAMPs on activation of inflammatory responses
and development of SIRS. Although multiple types of mitochondrial
component and metabolites, such as mtDNA, NFPs, TFAM, transcription
factor A, ATP, cardiolipin, cytopigment C, succinate and
mitochondrial RNA serve as DAMPs (8), the present review focused on widely
studied mitochondria-derived DAMPs, such as mtDNA, NFPs and TFAM.
Additionally, the present review focused on the mechanisms
underlying the effects of the aforementioned DAMPs on inducing
sepsis-like responses and their potential clinical value.
Structure and function of mitochondria
Mitochondria, the ‘cellular energy factories’, are
organelles in eukaryotic cells, which comprise ~25% of the total
cytoplasm volume. Mitochondria are complex organelles due to their
unique evolutionary history and components, as well as their genome
(9). Additionally, mitochondria
are involved in a range of cell fate decisions, including energy
metabolism, reactive oxygen species (ROS) formation, calcium
homeostasis, cell proliferation and apoptosis (10). When mitochondrial homeostasis is
disrupted and the apoptosis pathway is activated under severe
stress conditions, including cytoskeleton alteration, extensive DNA
damage, endoplasmic reticulum (ER) and replication stress,
sustained ROS burst, calcium overload and mitotic defect,
mitochondrial outer membrane permeabilization is targeted by the
aforementioned stressors (11).
The activation of the pro-apoptotic pathway regulates the structure
of pro-apoptotic proteins BAX and Bcl2 homologous antagonist/killer
(12). In addition, it
permeabilizes the outer membrane of mitochondria to allow transport
of pro-apoptotic molecules from the inner membrane into the cytosol
to initiate a caspase cascade, resulting in rapid cell death
(13). It has also been reported
that the mitochondrial intermembrane space proteins, such as
cytochrome c, which are released into the cytoplasm
following increased membrane permeability, also mediate activation
of apoptotic proteases (14).
Displaced mtDAMPs have been identified following cell death or
mitochondrial dyshomeostasis in patients with trauma, intestinal
ischemia/reperfusion and lung injury (1,15–19). Therefore, it has been hypothesized
by clinicians that mitochondria serve a vital role in catalyzing
the pathophysiology of sterile inflammation following trauma
(1).
mtDAMPs
Mitochondria produce ATP via oxidative
phosphorylation; ROS are byproducts of this process (1). Therefore, mitochondria are the
primary source of ROS; ATP and ROS are also considered to be DAMPs.
Damage-induced mitochondrial secretion of ATP increases local
levels of ATP, thereby promoting macrophage-mediated death of
sepsis-causing bacteria via P2X7 and P2X4 receptors (20,21). Emerging evidence has suggested
that under physiological conditions, cells produce mitochondrial
components that are not actively secreted into the cytoplasm;
however, these components are released via cellular disruption
(18,22–24).
mtDNA
Human mtDNA is a 16,569-bp long superhelical
closed-loop double-stranded DNA molecule that encodes essential
protein subunits of the oxidative phosphorylation system, including
the electron transport chain (complex I–IV) and ATP synthase
(complex V), which drive oxidative phosphorylation and ATP
production. Apart from the nucleus, mitochondria are the only
source of DNA in cells (9,25).
Unlike nuclear DNA, mtDNA is more prone to injury and lacks repair
systems (26). It has been
suggested that mitochondria originated from gram-negative
(G−) bacteria. Following phagocytosis of G-bacteria by
eukaryotes, the bacteria may have formed a symbiotic association
with the host and gradually evolved into mitochondria (25). Due to this evolutionary homology,
mitochondria are similar to bacteria; both exhibit conserved
hypomethylated 5′-cytosine-phosphoguanine (CpG) gene sequences and
pro-inflammatory effects (9,27).
Following cell injury, these endogenous mitochondrial ‘enemies’ are
recognized by classical PRRs to induce immune responses, thereby
acting as a bridge between trauma, inflammation and SIRS (9).
In critical patients with multiple organ dysfunction
syndrome (MODS) and sepsis, mitochondrial dysfunction may lead to
energetic and metabolic failure in white blood cells, thus altering
their function and attenuating the ability of the host to fight
infection (28). mtDNA is more
susceptible to damage compared with nuclear DNA since it lacks
introns and histones (29).
mtDNA damage affects the respiratory chain, enhances
oxidative stress and inflammatory responses, and induce apoptosis,
leading to cell dysfunction and tissue damage, which further
aggravate mitochondrial dysfunction in cells, thus forming a
feedback loop (30). Therefore,
mtDNA is considered to be a trigger that stimulates the innate
immune response (15). In 2013,
Nakahira et al (31)
published the first clinical trial in this area, including 200
patients from medical Intensive Care Units. The aforementioned
study showed that circulating mtDNA was significantly increased in
patients who died within 28 days of admission compared with those
who survived. In addition, mtDNA was positively associated with
mortality in patients who were hospitalized for up to 28 days.
Further studies confirmed these results (32–35). Additionally, other studies have
suggested that mtDNA serves a key role in the pathogenesis of
severe trauma, major abdominal surgery, acute lung injury
(ALI)/acute respiratory distress syndrome (ARDS),
ischemia/reperfusion (I/R) injury, inflammatory bowel disease,
rheumatoid arthritis, systemic lupus erythematosus (SLE),
myocarditis and myocardial infarction (1,15,19,36–38).
Effect of mtDNA on the occurrence and
development of SIRS
During mitochondrial stress, mitochondrial membrane
potential is decreased, leading to impaired membrane integrity
(39,40). These changes facilitate leakage of
mtDNA into the cytosol. mtDNA exhibits immunological potential and
is a key DAMP. Stimulators of interferon genes (STING) is produced
following activation of toll-like receptor 9 (TLR9),
nucleotide-binding oligomerization domain-like receptor family
pyrin domain-containing 3 (NLRP3), cyclic GMP-AMP synthase (cGAS)
and other signaling pathways, thereby promoting the occurrence and
development of SIRS by regulating the innate immune response and
contribute to inflammation initiation (25,41). The potential pathophysiological
mechanisms are illustrated in Fig.
1.
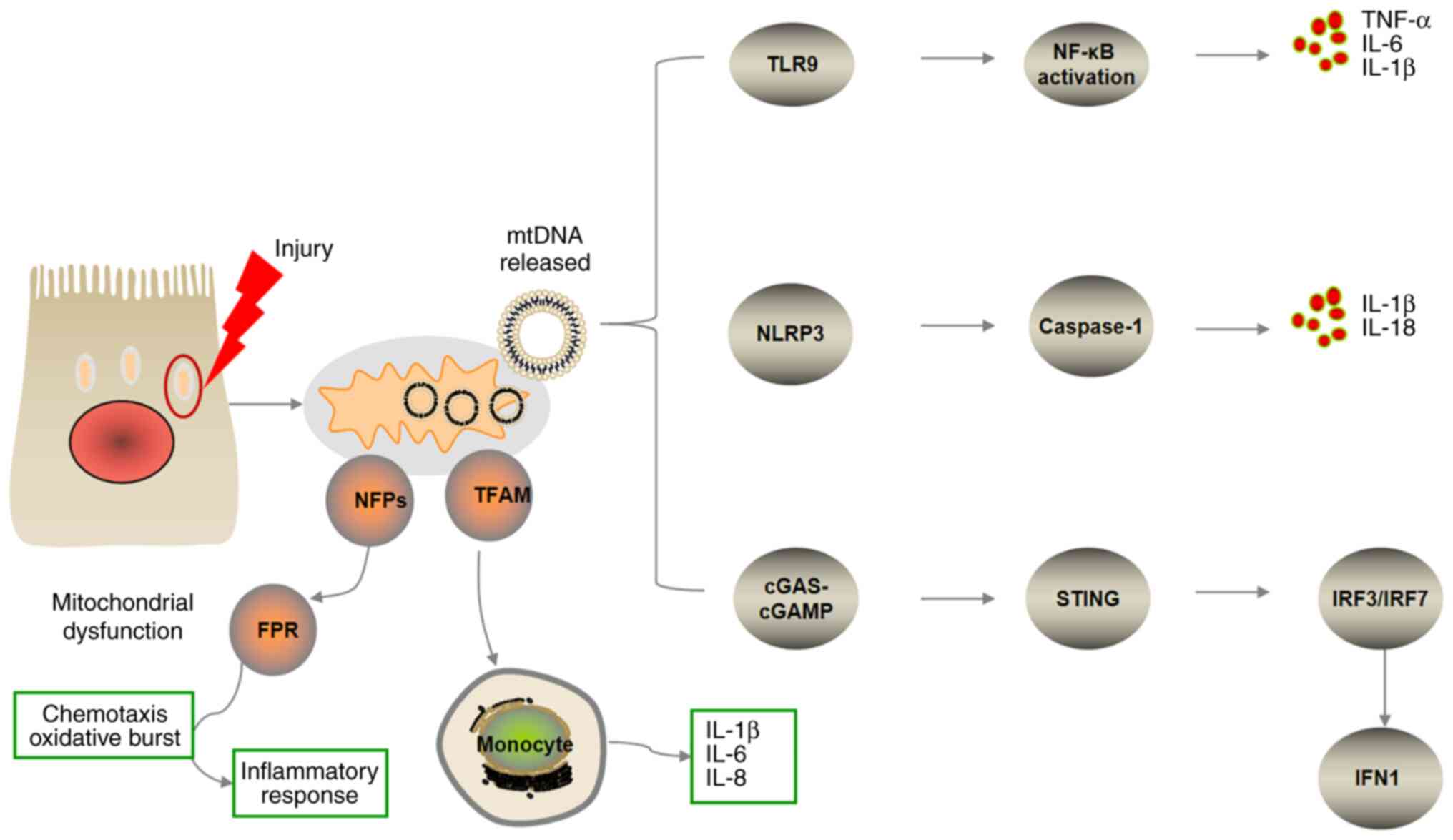 | Figure 1.Overview of pro-inflammatory
signaling pathways triggered by mitochondrial damage-associated
molecular patterns. Mitochondrial components released via cellular
disruption trigger systemic inflammatory response syndrome. mtDNA
triggers pro-inflammatory signaling pathways via endosomal
localized TLR9, cytosolic cGAS-STING or NLRP3 inflammasome. TLR9
binds mtDNA in the endosome, inducing NF-κB-dependent
pro-inflammatory signaling. mtDNA-dependent inflammasome activity
leads to caspase-1-dependent maturation or activation of IL-1 and
IL-8. cGAS recognizes mtDNA in the cytosol and activates
endoplasmic reticulum-localized STING to trigger an IFN response.
NFPs are released into the blood circulation to activate the
chemokine FPR, which recruits immune cells and promotes
inflammatory responses. TFAM serves as a pro-inflammatory cell
signaling molecule and is recognized by monocytes, leading to
enhanced secretion of pro-inflammatory factors, such as IL-1β, −6
and −8. mtDNA, mitochondrial DNA; FPR, formyl peptide receptor;
TFAM, mitochondrial transcription factor; NFP, N-formyl peptide;
TLR9, toll-like receptor; NLRP3, NOD-like receptor 3; cGAS, cyclic
GMP-AMP synthase; cGAMP, cyclic GMP-AMP; STING, stimulator of
interferon genes; IRF, interferon regulatory factor; IFN,
interferon. |
mtDNA-binding TLR9 activates the
downstream pathway of inflammation
The discovery of membrane-bound TLR family members
indicated that a number of PAMPs, including lipid, lipoprotein,
protein, glycan and nucleic acid, initiate innate immune responses.
TLR9 is the most common recognition receptor of mtDNA. TLR9 is
primarily expressed in macrophages, dendritic cells and B
lymphocytes, and recognizes the DNA CpG motif in bacteria and
viruses (42). Similar to
bacterial DNA, mtDNA is hypomethylated on the CpG motif (43), making it a potent activator of
TLR9. In infection-mediated mitochondrial injury, the body clears
damaged mitochondria and mtDNA via mitophagy, the underlying
mechanisms of which have been summarized in a previous review
article (44). However, when
mitophagy is inhibited, released mtDNA can be recognised by nucleic
acid recognition receptors in the cytoplasm (45). For example, TLR9 identifies mtDNA
via the CpG motif. However, CpG-independent TLR9 activation may
also occur. Activation of TLR9 leads to activation of NF-κB via the
myeloid differentiation factor 88 (MyD88)-dependent (classical)
pathway, which induces expression of downstream pro-inflammatory
genes, especially those of early cytokines, such as TNF-α and IL-6
(45).
Lin et al (46) showed that cyclic stretch (CS) of
lungs promotes mitochondrial injury in a mechanical ventilation rat
model, resulting in release of mtDNA. mtDNA, as a DAMP, is
recognized by TLR9 to activate the TLR9/MyD88/NF-κB signaling
pathway, exacerbating inflammation and lung injury (46). Jing et al (47) studied ventilator-induced lung
injury also using a CS cell culture model and suggested that
PTEN-induced putative kinase 1 (PINK1)-dependent mitophagy and
associated TLR9 activation is a key factor in stretch-induced cell
injury. Knockdown of PINK1, which is involved in regulating
mitophagy, has also been shown to promote mitochondrial
dysfunction, defective mitophagy and more severe lung injury
(48). By contrast, PINK1
overexpression may mitigate stretching-induced inflammation and
injury. Similar effects have been observed following TLR9
overexpression to induce expression of MyD88 and NF-κB/p65.
Furthermore, MyD88 silencing protects lung epithelial cells from
traction injury and downregulates NF-κB/p65. These findings
suggested that PINK1-dependent autophagy and TLR9 activation are
key factors in stretching-induced cell damage. Release of mtDNA
could activate TLR9, which induces the MyD88/NF-κB pathway, leading
to lung injury (47). Inhibiting
the release of mtDNA and activation of TLR9 may be a therapeutic
approach for preventing lung inflammation and injury.
Activation of mtDNA and NLRP3
inflammasome
The NLRP3 inflammasome, a member of the NLR family,
is a macromolecular complex composed of NLRP3, caspase-1 and
apoptosis-associated speck-like protein (40). Oxidative mtDNA binds to and
activates NLRP3 inflammasomes, thereby leading to secretion of
caspase-1-dependent pro-inflammatory cytokines, such as IL-1β and
IL-18, resulting in enhanced inflammatory cell death (pyroptosis)
(9,49). During pyroptosis, the cell bursts
and releases its contents, such as DNA fragments, into the
intercellular stroma (49).
Emerging evidence has suggested that mtDNA serves a
key role in activating the NLRP3 inflammasome. Sok et al
(50) revealed that inhibiting
release of oxidative mtDNA decreased generation of mitochondrial
ROS and inhibited activation of NLRP3 inflammasomes. Severe fever
with thrombocytopenia syndrome caused by viral infection can also
cause the release of oxidative mtDNA and activate the NLRP3
inflammasome, leading to intensive inflammation (51). A previous study demonstrated that
inhibiting synthesis and production of oxidized mtDNA could
alleviate the severity of ARDS (52). Wu et al (53) performed burn and delayed
resuscitation experiments in rats and showed that delayed
resuscitation could cause liver injury and oxidative stress. ROS
can cause liver injury via destroying mitochondrial integrity and
activating the mtDNA/NLRP3 axis. However, pre-intervention of
mitochondria-targeted antioxidants could protect the structure and
function of mitochondria and inhibit the release of mtDNA. These
findings indicated that mtDNA may serve a key role in occurrence
and development of systemic inflammation and organ dysfunction via
activating the NLRP3 inflammasome (53). Therefore, protecting mitochondrial
function and inhibiting mtDNA release to the cytosol may improve
clinical symptoms of patients with SIRS/MODS).
Activation of the cGAS/STING signaling
pathway by mtDNA
STING was initially identified as a key immune
molecule that detects nuclear or cytoplasmic DNA fragments from
pathogen-infected cells and triggers defensive immune responses
(54). cGAS and STING are
expressed in different types of cell, including cancer, immune and
non-immune cells (55). However,
increasing evidence has suggested that activation of the STING
pathway can lead to both tissue inflammation and damage (55,56). mtDNA can promote the onset of
inflammatory signaling responses via activating the
cGAS/STING/interferon regulatory factor 3 (IRF3) pathway (57,58). The cGAS/STING complex activated by
mtDNA may provide novel insights on the mechanisms of sepsis and
may further emphasize the key role of mtDNA in sepsis (57). The specific signaling pathway is
illustrated in Fig. 1. Cyclic
GMP-AMP (cGAMP) is generated following cGAS binding to DNA.
Secondary messenger cGAMP binds to STING in the endoplasmic
reticulum (ER) membrane. After binding, STING changes its
conformation and is activated. Activated STING is transferred from
the ER to the ER/Golgi intermediate organ and Golgi apparatus.
During this process, the carboxyl terminus of STING recruits and
activates TANK binding kinase 1 via phosphorylating transcription
factor IRF3. Phosphorylated IRF3 forms a dimer and translocates
into the nucleus, where it initiates the type I interferon response
(57). The type I interferon
response activates innate and adaptive immunity in a pleiotropic
manner (59).
In vivo experiments using a burn-induced ALI
model revealed that plasma levels of mtDNA were increased, and cGAS
and STING were both upregulated in lung tissue and neutrophil
infiltration was enhanced following burn injury (60). These results indicated that
increased plasma mtDNA-mediated activation of the cGAS-STING
pathway may induce ALI and neutrophil infiltration in rats. Hu
et al (58) showed that
inhibition of mtDNA release attenuated sepsis-induced inflammatory
responses and intestinal injury; therefore, it was hypothesized
that inhibition of the mtDNA/cGAS/STING signaling pathway could
protect against sepsis-induced organ damage and intestinal barrier
dysfunction. Liu et al (61) revealed that levels of cyclic mtDNA
and STING activation were enhanced in patients with severe ALI. In
addition, STING activation may serve a key role in mtDNA-mediated
lung injury, which promotes inflammatory storm and is involved in
autophagy via decreasing lysosomal acidification in an
IFN-dependent manner (61). STING
overactivation has also been reported to be associated with the
pathogenesis of SLE, neurological degeneration and sepsis (27). Consistent with a previous study
(48), Sliter et al
(62) demonstrated that PINK1, a
key gene in mitophagy that remove damaged mitochondria from cells,
could inhibit STING-mediated inflammatory responses, thus providing
a potential novel model for studing mitophagy, and attenuating the
inflammatory response.
NFPs
NFP is a potent chemotactic polypeptide synthesized
in mitochondria (23). In the
absence of cellular damage, bacteria-like NFPs are isolated within
the mitochondria; however, during severe trauma and cell death,
NFPs are released into the blood circulation to activate the
chemokine formyl peptide receptor (FPR), which recruits immune
cells and promotes inflammatory responses (63). FPR comprises conserved
G-protein-coupled receptors that serve a key role in host defense
and inflammatory responses (64).
FPR1, FPR2/lipoxygenase A4 receptor and FPR3 have been identified
in humans (1). These receptors
are expressed in multiple types of cell, with the highest
expression levels of FPR1 and FPR2 observed in neutrophils, and
those of FPR3 in monocytes/macrophages (64). A number of structurally and
chemically different ligands (such as microbial origins peptides,
endogenous peptides and synthetic small molecules) activate FPRs
(64), while NFP is the only
common ligand for all three receptors in humans (65).
Wenceslau et al (66) showed that mitochondrial FPs
(mtFPs) caused inflammation and vascular dysfunction via FPRs, and
accelerated the development of sepsis. Another study revealed that
mtFPs could cause sepsis-like syndrome, heart failure, heatstroke,
vascular leakage, thrombosis and lung injury in a rat model,
suggesting that NFPs may be a bridge between trauma, SIRS and
cardiovascular failure (67).
Another clinical study on traumatic hemorrhagic shock-induced lung
injury demonstrated that trauma-induced release of NFPs could
promote infiltration of neutrophils into the lung, and aggravate
SIRS and sepsis (16). This may
be because mtFPs-induced FPR activation could cause sepsis-like
symptoms, leading to ALI. Dorward et al (68) found increased bronchoalveolar
lavage fluid and serum levels of mtFPs in patients with ARDS. FPR1
has been reported to be involved in neutrophil recruitment and
alveolar leakage following sterile injury. Previous in vivo
and in vitro studies revealed that FPR1 could also be
activated in a neutrophil activation-dependent manner, suggesting
that pluripotency of FPR1-induced by mtFPs may be the mechanism
underlying their inflammatory effect (24,36). Therefore FPR1 may be a potential
therapeutic target for treating aseptic ALI.
TFAM
TFAM is an abundant mitochondrial protein; each
mtDNA molecule binds to ~1,000 TFAM molecules. The tight binding of
TFAM with mtDNA stabilizes the structure of mtDNA and protects mt
function (29,69). When mitochondria are damaged,
mtDNA and TFAM are both released into the cytoplasm (17,19). TFAM serves as a pro-inflammatory
cell signaling molecule and is recognized by monocytes, leading to
enhanced secretion of pro-inflammatory factors such as IL-1β, IL-6
and IL-8 (70). Hepokoski et
al (17) showed that
pulmonary I/R-mediated lung injury was associated with accumulation
of extracellular mtDNA and TFAM, whereas circulating TFAM promoted
infiltration of neutrophils in the lung. In addition, West et
al (71) indicated that TFAM
may serve a key role in maintaining the stability of mtDNA;
therefore, when TFAM levels are low, the stability of mtDNA is
decreased. This was also confirmed by van der Slikke et al
(30); this previous study
demonstrated that in sepsis-induced acute kidney damage, the degree
of mitochondrial damage was inversely proportional to the
expression of TFAM. In conclusion, TFAM may serve a key dual role
in the activation of inflammation-associated signaling pathways.
Firstly, TFAM could stabilize the structure and function of mtDNA
(70), and secondly, as a DAMP,
TFAM could also enhance inflammatory responses, similar to other
DAMPs, and cause injury to vital organs, such as the lung and
kidney (17,70). However, the synergistic effect of
TFAM and mtDNA on inflammatory responses requires further
investigation.
mtDAMPs and intestinal barrier
dysfunction
Intestinal mucosal epithelial cells exhibit a strong
metabolism and rapidly proliferate, renewing every 4–5 days
(72); therefore, the rate of
mitochondrial energy metabolism and the number of mitochondria are
increased in these cells compared with other cells (73). Additionally, intestinal epithelium
has been reported to be prone to hypoxia; therefore, intestinal I/R
can promote both intestinal mucosal epithelial cell and
mitochondrial injury, resulting in increased circulating mtDNA
levels (74). A previous study
showed that oxidized mtDNA could trigger a powerful inflammatory
response (15). The
aforementioned findings suggested that mtDNA may not only serve as
a marker of intestinal I/R injury, but could also be involved in
inflammation and cell death.
Just as mtDNA is thought to trigger the innate
immune response, the gut is also considered to promote SIRS and
multiple organ dysfunction (75).
Consistent with previous studies on intestinal I/R injury (15,76), Hu et al (15) demonstrated that mtDNA was
associated with increased secretion of inflammatory cytokines and
intestinal barrier injury. However, intervention with
mitochondrial-targeted antioxidant MitoQ could protect the
intestinal barrier during I/R (15,76). It has been reported that several
inflammatory cytokines, such as TNF-α, IFN-γ and IL, are involved
in regulation of intestinal tight junction integrity (77). TNF-α is a key factor in elevating
gut permeability via occludin (OCLN); OCLN overexpression has been
shown to mitigate cytokine-mediated increased gut permeability
(78). Furthermore, anti-TNF
therapy could improve epithelial barrier function (79). Based on the aforementioned
studies, it was hypothesized that mtDAMPs and mtDAMP-mediated
inflammatory responses could be associated with intestinal barrier
function. When the intestinal barrier is damaged, bacterial
translocation occurs and products of microorganisms, such as
alimentary antigens, may enter the bloodstream, thus intensifying
SIRS and promoting the formation of a feedback loop that
facilitates development of fatal sepsis (75,80).
Conclusion
Injury-released mtDAMPs serve a key role in the
occurrence and development of systemic inflammatory response
(1). The inflammatory response
exerts a protective effect on the body; however, excessive
inflammatory response damages organ function, leading to the
occurrence and development of sepsis, MODS and death (81). In addition, mtDAMP-mediated
activation of inflammatory signaling pathways can aggravates
mitochondrial damage, resulting in release of more mtDAMPs and
intestinal barrier dysfunction. The aforementioned processes
contribute to the development of SIRS when accompanied by displaced
intestinal flora and release of harmful metabolites (15,58). Further studies on mtDAMPs are
required to determine whether mtDNA serves as a biomarker for
predicting disease severity or mortality, and to determine the
mechanism underlying DAMP-induced SIRS pathogenesis and development
during mitochondrial injury. Additionally, the effect of drugs on
protecting mitochondrial function or antagonizing mtDAMP-associated
receptors to interrupt this pathophysiological process should be
investigated to support the potential role of mtDAMPs as a
significant target for preventing and treating sepsis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Clinical Research Fund
Project of Zhejiang Medical Association (grant no. 2019ZYC-A182)
and the Taizhou Municipal Science and Technology Bureau (grant no.
1902ky37).
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
TF designed the review and collected the literature.
CK and WS drafted the manuscript. All authors reviewed the
literature. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DAMP
|
damage-associated molecular
pattern
|
|
mt
|
mitochondrial
|
|
NFP
|
N-formyl peptide
|
|
FPR
|
formyl peptide receptor
|
|
SIRS
|
systemic inflammatory response
syndrome
|
|
PRR
|
pattern recognition receptor
|
|
PAMP
|
pathogen-associated molecular
pattern
|
|
TFAM
|
mitochondrial transcription factor
|
|
ROS
|
reactive oxygen species
|
|
CpG
|
5′-cytosine-phosphoguanine
|
|
MODS
|
multiple organ dysfunction
syndrome
|
|
ALI
|
acute lung injury
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
I/R
|
ischemia/reperfusion
|
|
SLE
|
systemic lupus erythematosus
|
|
cGAS
|
cyclic GMP-AMP synthase
|
|
STING
|
stimulators of interferon genes
|
|
TLR9
|
toll-like receptor 9
|
|
NLRP3
|
nucleotide-binding oligomerization
domain-like receptor family pyrin domain-containing 3
|
|
MyD88
|
myeloid differentiation factor 88
|
|
CS
|
cyclic stretch
|
|
PINK1
|
PTEN-induced putative kinase 1
|
|
IRF3
|
interferon regulatory factor 3
|
|
cGAMP
|
cyclic GMP-AMP
|
|
ER
|
endoplasmic reticulum
|
References
|
1
|
Itagaki K, Riça I, Konecna B, Kim HI, Park
J, Kaczmarek E and Hauser CJ: Role of mitochondria-derived danger
signals released after injury in systemic inflammation and sepsis.
Antioxid Redox Signal. 35:1273–1290. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kang J, Kim S, Cho H and Lee S: DAMPs
activating innate immune responses in sepsis. Ageing Res Rev.
24:54–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khwaja B, Thankam FG and Agrawal DK:
Mitochondrial DAMPs and altered mitochondrial dynamics in OxLDL
burden in atherosclerosis. Mol Cell Biochem. 476:1915–1928. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schneck E, Edinger F, Hecker M, Sommer N,
Pak O, Weissmann N, Hecker A, Reichert M, Markmann M, Sander M and
Koch C: Blood levels of free-circulating mitochondrial DNA in
septic shock and postsurgical systemic inflammation and its
influence on coagulation: A secondary analysis of a prospective
observational study. J Clin Med. 9:20562020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiménez-Sousa MA, Tamayo E,
Guzmán-Fulgencio M, Heredia M, Fernández-Rodríguez A, Gómez E,
Almansa R, Gómez-Herreras JI, García-Álvarez M, Gutiérrez-Junco S,
et al: Mitochondrial DNA haplogroups are associated with severe
sepsis and mortality in patients who underwent major surgery. J
Infect. 70:20–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, Wang
G, Gu G and Li J: Elevated levels of plasma mitochondrial DNA are
associated with clinical outcome in intra-abdominal infections
caused by severe trauma. Surg Infect (Larchmt). 18:610–618. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kung CT, Hsiao SY, Tsai TC, Su CM, Chang
WN, Huang CR, Wang HC, Lin WC, Chang HW, Lin YJ, et al: Plasma
nuclear and mitochondrial DNA levels as predictors of outcome in
severe sepsis patients in the emergency room. J Transl Med.
10:1302012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li S, Hu Q, Huang J, Wu X and Ren J:
Mitochondria-derived damage-associated molecular patterns in
sepsis: From bench to bedside. Oxid Med Cell Longev.
2019:69148492019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
West AP and Shadel GS: Mitochondrial DNA
in innate immune responses and inflammatory pathology. Nat Rev
Immunol. 17:363–375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryoo IG and Kwak MK: Regulatory crosstalk
between the oxidative stress-related transcription factor
Nfe2l2/Nrf2 and mitochondria. Toxicol Appl Pharmacol. 359:24–33.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pihán P, Carreras-Sureda A and Hetz C:
BCL-2 family: Integrating stress responses at the ER to control
cell demise. Cell Death Differ. 24:1478–1487. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Picca A, Calvani R, Coelho-Junior HJ and
Marzetti E: Cell death and inflammation: The role of mitochondria
in health and disease. Cells. 10:5372021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bock FJ and Tait SWG: Mitochondria as
multifaceted regulators of cell death. Nat Rev Mol Cell Biol.
21:85–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Campbell KJ and Tait SWG: Targeting BCL-2
regulated apoptosis in cancer. Open Biol. 8:1800022018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu Q, Ren H, Ren J, Liu Q, Wu J, Wu X, Li
G, Wang G, Gu G, Guo K, et al: Released mitochondrial DNA following
intestinal ischemia reperfusion induces the inflammatory response
and gut barrier dysfunction. Sci Rep. 8:73502018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wenceslau CF, Szasz T, McCarthy CG, Baban
B, NeSmith E and Webb RC: Mitochondrial N-formyl peptides cause
airway contraction and lung neutrophil infiltration via formyl
peptide receptor activation. Pulm Pharmacol Ther. 37:49–56. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hepokoski M, Wang J, Li K, Li Y, Gupta P,
Mai T, Moshensky A, Alotaibi M, Crotty Alexander LE, Malhotra A and
Singh P: Altered lung metabolism and mitochondrial DAMPs in lung
injury due to acute kidney injury. Am J Physiol Lung Cell Mol
Physiol. 320:L821–L831. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McIlroy DJ, Bigland M, White AE, Hardy BM,
Lott N, Smith DW and Balogh ZJ: Cell necrosis-independent sustained
mitochondrial and nuclear DNA release following trauma surgery. J
Trauma Acute Care Surg. 78:282–288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pencovich N, Nevo N, Weiser R, Bonder E,
Bogoch Y and Nachmany I: Postoperative rise of circulating
mitochondrial DNA is associated with inflammatory response in
patients following pancreaticoduodenectomy. Eur Surg Res. 62:18–24.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Csóka B, Németh ZH, Szabó I, Davies DL,
Varga ZV, Pálóczi J, Falzoni S, Di Virgilio F, Muramatsu R,
Yamashita T, et al: Macrophage P2X4 receptors augment bacterial
killing and protect against sepsis. JCI Insight. 3:e994312018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Csóka B, Németh ZH, Törő G, Idzko M, Zech
A, Koscsó B, Spolarics Z, Antonioli L, Cseri K, Erdélyi K, et al:
Extracellular ATP protects against sepsis through macrophage P2X7
purinergic receptors by enhancing intracellular bacterial killing.
FASEB J. 29:3626–3637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Konecna B, Park J, Kwon WY, Vlkova B,
Zhang Q, Huang W, Kim HI, Yaffe MB, Otterbein LE, Itagaki K and
Hauser CJ: Monocyte exocytosis of mitochondrial danger-associated
molecular patterns in sepsis suppresses neutrophil chemotaxis. J
Trauma Acute Care Surg. 90:46–53. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaczmarek E, Hauser CJ, Kwon WY, Riça I,
Chen L, Sandler N, Otterbein LE, Campbell Y, Cook CH, Yaffe MB, et
al: A subset of five human mitochondrial formyl peptides mimics
bacterial peptides and functionally deactivates human neutrophils.
J Trauma Acute Care. 85:936–943. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fang C, Wei X and Wei Y: Mitochondrial DNA
in the regulation of innate immune responses. Protein Cell.
7:11–16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu Z, Sainz AG and Shadel GS:
Mitochondrial DNA: Cellular genotoxic stress sentinel. Trends
Biochem Sci. 46:812–821. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riley JS and Tait SW: Mitochondrial DNA in
inflammation and immunity. EMBO Rep. 21:e497992020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Garrabou G, Morén C, López S, Tobías E,
Cardellach F, Miró O and Casademont J: The effects of sepsis on
mitochondria. J Infect Dis. 205:392–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonekamp NA and Larsson NG: SnapShot:
Mitochondrial nucleoid. Cell. 172:388–388.e1. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van der Slikke EC, Star BS, van Meurs M,
Henning RH, Moser J and Bouma HR: Sepsis is associated with
mitochondrial DNA damage and a reduced mitochondrial mass in the
kidney of patients with sepsis-AKI. Crit Care. 25:362021.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakahira K, Kyung SY, Rogers AJ, Gazourian
L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, et
al: Circulating mitochondrial DNA in patients in the ICU as a
marker of mortality: Derivation and validation. PLoS Med.
10:e10015772013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Busani S, De Biasi S, Nasi M, Paolini A,
Venturelli S, Tosi M, Girardis M and Cossarizza A: Increased plasma
levels of mitochondrial DNA and normal inflammasome gene expression
in monocytes characterize patients with septic shock due to
multidrug resistant bacteria. Front Immunol. 11:7682020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang WZ, Hoffman KL, Schiffer KT,
Oromendia C, Rice MC, Barjaktarevic I, Peters SP, Putcha N, Bowler
RP, Wells JM, et al: Association of plasma mitochondrial DNA with
COPD severity and progression in the SPIROMICS cohort. Respir Res.
22:1262021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faust HE, Reilly JP, Anderson BJ, Ittner
CAG, Forker CM, Zhang P, Weaver BA, Holena DN, Lanken PN, Christie
JD, et al: Plasma mitochondrial DNA levels are associated with ARDS
in trauma and sepsis patients. Chest. 157:67–76. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McIlroy DJ, Minahan K, Keely S, Lott N,
Hansbro P, Smith DW and Balogh ZJ: Reduced deoxyribonuclease enzyme
activity in response to high postinjury mitochondrial DNA
concentration provides a therapeutic target for systemic
inflammatory response syndrome. J Trauma Acute Care Surg.
85:354–358. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Boyapati RK, Dorward DA, Tamborska A,
Kalla R, Ventham NT, Doherty MK, Whitfield PD, Gray M, Loane J,
Rossi AG, et al: Mitochondrial DNA is a pro-inflammatory
damage-associated molecular pattern released during active IBD.
Inflamm Bowel Dis. 24:2113–2122. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bliksøen M, Mariero LH, Torp MK, Baysa A,
Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G and Stensløkken
KO: Extracellular mtDNA activates NF-κB via toll-like receptor 9
and induces cell death in cardiomyocytes. Basic Res Cardiol.
111:422016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Simmons JD, Lee Y, Mulekar S, Kuck JL,
Brevard SB, Gonzalez RP, Gillespie MN and Richards WO: Elevated
levels of plasma mitochondrial DNA DAMPs are linked to clinical
outcome in severely injured human subjects. Ann Surg. 258:591–598.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Harrington JS, Choi AM and Nakahira K:
Mitochondrial DNA in Sepsis. Curr Opin Crit Care. 23:284–290. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Q, Zhang D, Hu D, Zhou X and Zhou Y:
The role of mitochondria in NLRP3 inflammasome activation. Mol
Immunol. 103:115–124. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou L and Tan L: Role of mitochondrial
DNA in acute lung injury/acute respiratory distress syndrome
induced by sepsis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue.
32:253–256. 2020.(In Chinese). PubMed/NCBI
|
|
42
|
Shepard CR: TLR9 in MAFLD and NASH: At the
intersection of inflammation and metabolism. Front Endocrinol
(Lausanne). 11:6136392021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Medeiros TC and Graef M: Autophagy
determines mtDNA copy number dynamics during starvation. Autophagy.
15:178–179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pickles S, Vigié P and Youle RJ: Mitophagy
and quality control mechanisms in mitochondrial maintenance. Curr
Biol. 28:R170–R185. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang JZ, Liu Z, Liu J, Ren JX and Sun TS:
Mitochondrial DNA induces inflammation and increases TLR9/NF-κB
expression in lung tissue. Int J Mol Med. 33:817–824. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin JY, Jing R, Lin F, Ge WY, Dai HJ and
Pan L: High tidal volume induces mitochondria damage and releases
mitochondrial DNA to aggravate the ventilator-induced lung injury.
Front Immunol. 9:14772018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jing R, Hu ZK, Lin F, He S, Zhang SS, Ge
WY, Dai HJ, Du XK, Lin JY and Pan LH: Mitophagy-mediated mtDNA
release aggravates stretching-induced inflammation and lung
epithelial cell injury via the TLR9/MyD88/NF-κB pathway. Front Cell
Dev Biol. 8:8192020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bueno M, Lai YC, Romero Y, Brands J, St
Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, et
al: PINK1 deficiency impairs mitochondrial homeostasis and promotes
lung fibrosis. J Clin Invest. 125:521–538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sok SPM, Ori D, Wada A, Okude H, Kawasaki
T, Momota M, Nagoor NH and Kawai T: 1′-Acetoxychavicol acetate
inhibits NLRP3-dependent inflammasome activation via mitochondrial
ROS suppression. Int Immunol. 33:373–386. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li S, Li H, Zhang YL, Xin QL, Guan ZQ,
Chen X, Zhang XA, Li XK, Xiao GF, Lozach PY, et al: SFTSV infection
induces BAK/BAX-dependent mitochondrial DNA release to trigger
NLRP3 inflammasome activation. Cell Rep. 30:4370–4385.e7. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xian H, Liu Y, Rundberg Nilsson A,
Gatchalian R, Crother TR, Tourtellotte WG, Zhang Y, Aleman-Muench
GR, Lewis G, et al: Metformin inhibition of mitochondrial ATP and
DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary
inflammation. Immunity. 54:1463–1477. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu Y, Hao C, Liu X, Han G, Yin J, Zou Z,
Zhou J and Xu C: MitoQ protects against liver injury induced by
severe burn plus delayed resuscitation by suppressing the
mtDNA-NLRP3 axis. Int Immunopharmacol. 80:1061892020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishikawa H and Barber GN: STING is an
endoplasmic reticulum adaptor that facilitates innate immune
signalling. Nature. 455:674–678. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bryant JD, Lei Y, VanPortfliet JJ, Winters
AD and West AP: Assessing mitochondrial DNA release into the
cytosol and subsequent activation of innate immune-related pathways
in mammalian cells. Curr Protoc. 2:e3722022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li
Y, Azares AR, Zhang M, Guo J, Ghaghada KB, et al: Critical role of
cytosolic DNA and its sensing adaptor STING in aortic degeneration,
dissection, and rupture. Circulation. 141:42–66. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wan D, Jiang W and Hao J: Research
advances in how the cGAS-STING pathway controls the cellular
inflammatory response. Front Immunol. 11:6152020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J,
Zheng J, Huang J, Slade DA, Wu X and Ren J: STING-mediated
intestinal barrier dysfunction contributes to lethal sepsis.
Ebiomedicine. 41:497–508. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vringer E and Tait SW: Mitochondria and
inflammation: Cell death heats up. Front Cell Dev Biol. 7:1002019.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Comish PB, Liu M, Huebinger R, Carlson D,
Kang R and Tang D: The cGAS-STING pathway connects mitochondrial
damage to inflammation in burn-induced acute lung injury in rat.
Burns. 48:168–175. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y
and Ren J: Circulating mitochondrial DNA-triggered autophagy
dysfunction via STING underlies sepsis-related acute lung injury.
Cell Death Dis. 12:6732021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sliter DA, Martinez J, Hao L, Chen X, Sun
N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al: Parkin
and PINK1 mitigate STING-induced inflammation. Nature. 561:258–262.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Banoth B and Cassel SL: Mitochondria in
innate immune signaling. Transl Res. 202:52–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ye RD, Boulay F, Wang JM, Dahlgren C,
Gerard C, Parmentier M, Serhan CN and Murphy PM: International
Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for
the formyl peptide receptor (FPR) family. Pharmacol Rev.
61:119–161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
He HQ and Ye RD: The formyl peptide
receptors: Diversity of ligands and mechanism for recognition.
Molecules. 22:4552017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wenceslau CF, McCarthy CG, Goulopoulou S,
Szasz T, NeSmith EG and Webb RC: Mitochondrial-derived N-formyl
peptides: Novel links between trauma, vascular collapse and sepsis.
Med Hypotheses. 81:532–535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wenceslau CF, McCarthy CG, Szasz T,
Goulopoulou S and Webb RC: Mitochondrial N-formyl peptides induce
cardiovascular collapse and sepsis-like syndrome. Am J Physiol
Heart Circ Physiol. 308:H768–H777. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dorward DA, Lucas CD, Doherty MK, Chapman
GB, Scholefield EJ, Conway Morris A, Felton JM, Kipari T, Humphries
DC, Robb CT, et al: Novel role for endogenous mitochondrial
formylated peptide-driven formyl peptide receptor 1 signalling in
acute respiratory distress syndrome. Thorax. 72:928–936. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ueda S, Shimasaki M, Ichiseki T, Hirata H,
Kawahara N and Ueda Y: Mitochondrial transcription factor A added
to osteocytes in a stressed environment has a cytoprotective
effect. Int J Med Sci. 17:1293–1299. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Schindler SM, Frank MG, Annis JL, Maier SF
and Klegeris A: Pattern recognition receptors mediate
pro-inflammatory effects of extracellular mitochondrial
transcription factor A (TFAM). Mol Cell Neurosci. 89:71–79. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
van der Flier LG and Clevers H: Stem
cells, self-renewal, and differentiation in the intestinal
epithelium. Annu Rev Physiol. 71:241–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rath E, Moschetta A and Haller D:
Mitochondrial function-gatekeeper of intestinal epithelial cell
homeostasis. Nat Rev Gastroenterol Hepatol. 15:497–516. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang X, Wu J, Liu Q, Li X, Li S, Chen J,
Hong Z, Wu X, Zhao Y and Ren J: mtDNA-STING pathway promotes
necroptosis-dependent enterocyte injury in intestinal ischemia
reperfusion. Cell Death Dis. 11:10502020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Druml W: Intestinal cross-talk: The gut as
motor of multiple organ failure. Med Klin Intensivmed Notfmed.
113:470–477. 2018.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, Gu
G, Ren H, Hong Z and Li J: The mitochondrially targeted antioxidant
MitoQ protects the intestinal barrier by ameliorating mitochondrial
DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis.
9:4032018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chelakkot C, Ghim J and Ryu SH: Mechanisms
regulating intestinal barrier integrity and its pathological
implications. Exp Mol Med. 50:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Marchiando AM, Shen L, Graham WV, Weber
CR, Schwarz BT, Austin JN II, Raleigh DR, Guan Y, Watson AJ,
Montrose MH and Turner JR: Caveolin-1-dependent occludin
endocytosis is required for TNF-induced tight junction regulation
in vivo. J Cell Biol. 189:111–126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Odenwald MA and Turner JR: The intestinal
epithelial barrier: A therapeutic target? Nat Rev Gastroenterol
Hepatol. 14:9–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Assimakopoulos SF, Triantos C, Thomopoulos
K, Fligou F, Maroulis I, Marangos M and Gogos CA: Gut-origin sepsis
in the critically ill patient: Pathophysiology and treatment.
Infection. 46:751–760. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhou M, Aziz M and Wang P:
Damage-associated molecular patterns as double-edged swords in
sepsis. Antioxid Redox Signal. 35:1308–1323. 2021. View Article : Google Scholar : PubMed/NCBI
|














