Introduction
Atherosclerosis, the leading cause of cardiovascular
disease, is a chronic inflammatory disorder, during which the
dysfunction of endothelial cells is the first step (1–3).
Accumulating evidence has confirmed that endothelial cells have a
major role in the development of atherosclerosis and that
endothelial dysfunction and damage have an initial role (4,5).
Under pathological conditions, injured endothelial cells display
great plasticity by differentiating into healthy and functional
endothelial cells (6).
Atherosclerosis is a persistent inflammatory condition and vascular
endothelial cell apoptosis is considered the initiation factor of
the occurrence, development and pathogenesis of atherosclerosis
(7,8). Furthermore, endothelial-mesenchymal
transition (EndMT) helps endothelial cells to acquire a mesenchymal
phenotype (9). Oxidized
low-density lipoprotein (Ox-LDL) is a common factor in the
establishment of experimental models of atherosclerosis, which may
induce endothelial cell apoptosis and act as an essential risk
factor for the formation of atherosclerosis (10,11).
Sestrins (SESNs) are a family of highly conserved
stress-inducible proteins that regulate multiple cell homeostatic
mechanisms (12). A deficiency in
endogenous SESNs may lead to metabolic disorders, including insulin
resistance, fat accumulation, mitochondrial dysfunction and
oxidative damage (13). In
addition, the SESN family are antioxidant enzymes and
transcriptional targets of tumor suppressor protein p53 (14). As a primary member of the SESN
family, SESN1 functions as a vital mediator in multiple human
diseases, including human maxillary cancer, myoblast
differentiation and diabetes (15–17). Furthermore, SESN1 has been
reported to activate the adenosine monophosphate-activated protein
kinase catalytic subunit α1 (AMPK) signaling pathway and to
function as a suppressor of the mechanistic target of rapamycin
complex 1 kinase (18,19). Recent evidence has demonstrated
that SESN1 suppresses the inflammation of macrophages in a murine
atherosclerosis model (20). In
addition, Zhang et al (21) revealed that SESN1 is expressed, at
low levels, in endothelial cells subjected to mild shear stress.
However, whether SESN1 is involved in human umbilical vein
endothelial cell (HUVEC) injury induced by atherosclerosis has
remained largely elusive.
SESN1 has been indicated to reduce myocardial
hypertrophy and activate AMPK signaling (22). As metabolic sensors, AMPK and
sirtuin 1 (SIRT1) have been identified as master regulators of
metabolism (23). In a previous
study, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
analysis revealed that AMPK is able to regulate the expression of
downstream SIRT1 genes, while SIRT1 may inhibit the expression of
Ox-LDL receptor-1 (LOX1) (24).
It is well established that endothelial cell injury is induced by
Ox-LDL to drive the progression of atherosclerosis and LOX1 is the
primary OxLDL receptor of endothelial cells, which may be activated
by Ox-LDL, and it mediates cell damage in atherosclerosis (25). Therefore, blocking LOX1 expression
is considered to be a major strategy in the management of
atherosclerosis.
In the present study, the effects of SENS1 on the
inflammation, apoptosis and EndMT of HUVECs exposed to Ox-LDL were
investigated. In addition, the latent regulatory mechanisms among
SESN1, AMPK/SIRT1 signaling and LOX1 were identified. The present
results may highlight a novel target for the treatment of
atherosclerosis.
Materials and methods
Bioinformatics analysis
The Human Protein Atlas (https://www.proteinatlas.org/) was used to assess
SESN1 expression in HUVECs.
Cells and cell treatment
HUVECs (cat. no. KCB2012087YJ) obtained from the
Kunming Cell Bank of the Type Culture Collection of the Chinese
Academy of Sciences were maintained in Dulbecco's modified Eagle's
medium Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal
bovine serum (RWDLS) in a humidified atmosphere with 5%
CO2 at 37°C. For the subsequent experiments, various
concentrations (0, 25, 50, 75 and 100 µg/ml) of Ox-LDL (cat. no.
20605ES05; Yeasen Biotechnology Co., Ltd.) were used to pre-treat
the HUVECs for 24 h according to a previous study (26). In addition, 15 µM SIRT1 inhibitor,
nicotinamide (NAM; MilliporeSigma) (27) and 8 µM AMPK inhibitor (compound C;
MilliporeSigma) were respectively utilized to pre-treat the cells
for 30 min (28).
Cell Counting Kit-8 (CCK-8) assay
Cell viability was evaluated by the CCK-8 reagent
(Beyotime Institute of Biotechnology) according to manufacturer's
protocol. The HUVECs were seeded into 96-well plates at the density
of 4×104 cells/well and cultured at 37°C overnight.
Following the indicated treatments, CCK-8 solution (10 µl) was
added to each well. Following incubation for 2 h at 37°C, cell
proliferation was detected using a microplate reader (Bio-Rad
Laboratories, Inc.) at an absorbance wavelength of 450 nm.
Reverse transcription-quantitative PCR
(RT-qPCR)
Isolation of total RNA was performed using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) in compliance with the manufacturer's instructions. The
temperature protocol for this step was as follows: 70°C for 5 min,
37°C for 5 min and 42°C for 1 h. Subsequently, complementary DNA
was synthesized using the PrimeScript RT reagent (Takara Bio,
Inc.). An ABI 7500 Fast Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with a SYBR Green Master Mix Kit
(Toyobo Co. Ltd.) were used for PCR analysis. The following
thermocycling conditions were used: Initial denaturation at 95°C
for 7 min; and 40 cycles of 95°C for 15 sec and 60°C for 30 sec;
and a final extension at 72°C for 30 sec. The primers sequences
were as follows: SESN1 forward, 5′-GCGACCAGGACGAGGAACTT-3′ and
reverse, 5′-TGCATCTGTGCGTCTTCACT-3′; tumor necrosis factor (TNF)-α
forward, 5′-CTGGGCAGGTCTACTTTGGG-3′ and reverse,
5′-CTGGAGGCCCCAGTTTGAAT-3′; interleukin (IL)-6 forward,
5′-TCCACAAGCGCCTTCGGTC-3′ and reverse, 5′-GGTCAGGGGTGGTTATTGCAT-3′;
IL-1β forward, 5′-GCTCGCCAGTGAAATGATGG-3′ and reverse,
5′-TCGTGCACATAAGCCTCGTT-3′; α smooth muscle actin (α-SMA) forward,
5′-AAAGCAAGTCCTCCAGCGTT-3′ and reverse, 5′-TTAGTCCCGGGGATAGGCAA-3′;
vimentin forward, 5′-AACTTAGGGGCGCTCTTGTC-3′ and reverse,
5′-ATTCAAGTCTCAGCGGGCTC-3′; CD31 (also known as platelet
endothelial cell adhesion molecule 1) forward,
5′-AGAGAGGCTGCTGTCATTGC-3′ and reverse, 5′-GGCCCCTCAGAAGACAACAT-3′;
von Willebrand factor (vWF) forward, 5′-CAACACCTGCATTTGCCGAA-3′ and
reverse, 5′-ATGCGGAGGTCACCTTTCAG-3′; GAPDH forward,
5′-AATGGGCAGCCGTTAGGAAA-3′ and reverse, 5′-GCGCCCAATACGACCAAATC-3′.
Relative gene expression was calculated using the 2−ΔΔCq
method using GAPDH as an internal control (29).
Western blot analysis
Total protein was extracted from the HUVECs using
lysis buffer (Beyotime Institute of Biotechnology). Total protein
was quantified using a bicinchoninic acid kit (Beyotime Institute
of Biotechnology). Subsequently, 40 µg of total protein was loaded
per lane on a 10% SDS-PAGE gel, electroblotted onto polyvinylidene
difluoride membranes (Thermo Fisher Scientific, Inc.) and blocked
using 5% skimmed milk (MilliporeSigma) for 1 h at room temperature.
The membranes were then incubated overnight at 4°C with the
following primary antibodies: Anti-SESN1 (1:1,000 dilution; cat.
no. ab134091), anti-inducible nitric oxide synthase (iNOS) (1:1,000
dilution; cat. no. ab178945), anti-total NF-κB p65 (t-NF-κB p65)
(1:1,000 dilution; cat. no. ab32536), anti-phosphorylated NF-κB p65
(p-NF-κB p65) (1:2,000 dilution; cat. no. ab86299), anti-BCL2
associated X, apoptosis regulator (Bax) (1:1,000 dilution; cat. no.
ab182733), anti-cleaved caspase-3 (1:400 dilution; cat. no.
ab32042), anti-caspase-3 (1:5,000 dilution; cat. no. ab32351),
anti-poly(adenosine diphosphate ribose) polymerase (PARP) (1:1,000
dilution; cat. no. ab191217) anti-cleaved PARP (1:1,000 dilution;
cat. no. ab32064), anti-α-SMA (1:50 dilution; cat. no. ab150301),
anti-CD31 (1:1,000 dilution; cat. no. ab9498), anti-vimentin
(1:1,000 dilution; cat. no. ab92547), anti-vWF (1:1,000 dilution;
cat. no. ab287962), anti-p-AMPK (1:500 dilution; cat. no.
ab131357), anti-AMPK (1:500 dilution; cat. no. ab3759), anti-SIRT1
(1:1,000 dilution; cat. no. ab189494), anti-LOX1 (1:1,000 dilution;
cat. no. ab214427) and anti-β-actin (1:1,000 dilution; cat. no.
ab8227; all from Abcam) and anti-B cell lymphoma-2 (Bcl-2) (1:1,000
dilution; cat. no. 15071, Cell Signaling Technology, Inc.).
Following primary antibody incubation, the membranes were incubated
with horseradish peroxidase-conjugated secondary antibody (1:3,000
dilution; cat. no. 7074S; Cell Signaling Technology, Inc.) for 1 h
at room temperature and washed three times with PBS. Proteins bands
were visualized using enhanced chemiluminescence (Thermo Fisher
Scientific, Inc.) and ImageJ software (v6; National Institutes of
Health) was used to analyze the protein bands.
Cell transfection
To overexpress SESN1, transfection of the pcDNA3.1
vector (Shanghai GenePharma, Co., Ltd.) containing the SESN1 gene
(Ov-SESN1; 4 µg) or the empty vector plasmid (Ov-NC; 4 µg) was
performed using Lipofectamine 2000® (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C in accordance with the
manufacturer's protocol. After 48 h, the transfected cells were
collected for use in subsequent experiments.
Test for inflammatory factors
contents
The cell culture medium was collected and the
supernatant was obtained via centrifugation. The ELISA sandwich
method was employed to evaluate the concentrations of TNF-α (cat.
no. F02810), IL-6 (cat. no. F01310) and IL-1β (cat. no. F01220) in
culture medium supernatant in accordance with the manufacturer's
protocols. These kits were acquired from Shanghai Xitang
Biotechnology. The optical density at 450 nm was measured with a
microplate reader (Bio-Rad Laboratories, Inc.).
Terminal deoxynucleotidyl
transferase-mediated nick-end labeling (TUNEL) staining
A TUNEL staining kit (Beyotime Institute of
Biotechnology) was utilized according to the manufacturer's
instructions. In brief, following incubation with 4%
paraformaldehyde at 4°C for 20 min, the cells were treated with
0.5% Triton X-100. They were then incubated with 50 µl TUNEL
reaction buffer for 1 h at 37°C. Cell nuclei were stained with 2
µg/ml DAPI solution for 10 min at room temperature in the dark.
Images were acquired under a fluorescence microscope (Olympus
Corporation) and cells were counted in five randomly selected
microscopic fields.
Immunofluorescence staining
HUVECs were first cultured on sterilized coverslips.
Subsequently, the transfected HUVECs were fixed with 4%
paraformaldehyde for 20 min at room temperature, permeabilized with
0.5% Triton X-100 and blocked with 1% bovine serum albumin
(MilliporeSigma) for 1 h at room temperature. The cells were then
incubated with primary antibodies against CD31 (1:1,000 dilution;
cat. no. ab9498; Abcam) or α-SMA (1:50 dilution; cat. no. ab150301;
Abcam) overnight at 4°C. Subsequently, cells were incubated with
Alexa Fluor® 488-conjugated secondary antibody (1:400
dilution; cat. no. A11008; Molecular Probes; Thermo Fisher
Scientific, Inc.) for 1 h at room temperature. After washing with
PBS three times, DAPI was used to stain the coverslips for 5 min at
room temperature. The results were imaged using a fluorescence
microscope (Olympus Corporation).
Statistical analysis
Values are expressed as the mean ± standard
deviation and analyzed using GraphPad Prism software (version 8.0;
GraphPad Software, Inc.). All experiments were performed as three
independent replicates. An unpaired Student's t-test was used to
compare differences between two groups, while differences between
more than two groups were analyzed using a one-way ANOVA followed
by Tukey's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
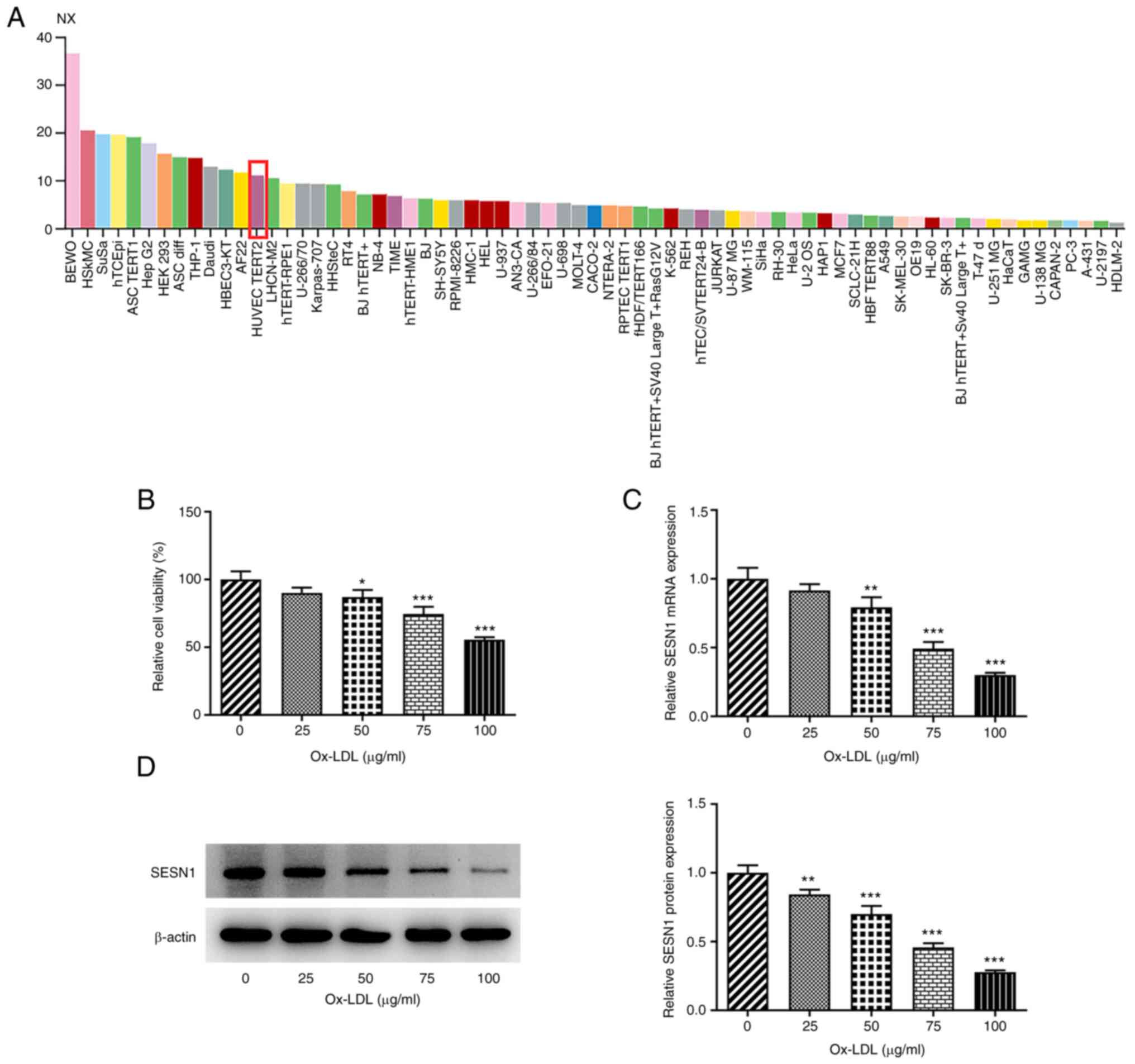
SESN1 expression is downregulated in
Ox-LDL-stimulated HUVECs
To evaluate the role of SESN1 in HUVECs, SESN1
expression was first examined in HUVECs. Using the Human Protein
Atlas, SESN1 was indicated to be highly expressed in HUVECs
(Fig. 1A). Subsequently, various
concentrations (0, 25, 50, 75 and 100 µg/ml) of Ox-LDL were
utilized to stimulate the HUVECs and the viability was examined
using a CCK-8 assay. The results suggested that the viability of
HUVECs was decreased upon exposure to Ox-LDL in a
concentration-dependent manner (Fig.
1B). In addition, RT-qPCR and western blot analysis
demonstrated that SESN1 expression at both the mRNA and protein
level was decreased as the concentration of Ox-LDL increased; when
the Ox-LDL concentration was 100 µg/ml, SESN1 expression was the
lowest (Fig. 1C and D).
Therefore, 100 µg/ml Ox-LDL was used in the subsequent experiments.
Taken together, the results demonstrated that SESN1 exhibits low
expression in Ox-LDL-stimulated HUVECs.
SESN1 overexpression mitigates the
decrease in the viability and inflammation of HUVECs stimulated
with Ox-LDL
For the purpose of assessing the function of SESN1
in HUVECs, SESN1 was first overexpressed by transfection with the
Ov-SESN1 plasmid and the transfection efficiency was examined using
RT-qPCR and western blot analysis (Fig. 2A and B). Subsequently, CCK-8
assays demonstrated that SESN1 overexpression led to an increase in
cell viability following stimulation with Ox-LDL (Fig. 2C). Furthermore, RT-qPCR revealed
that the enhanced mRNA levels of inflammatory factors, including
TNF-α, IL-6 and IL-1β, in the HUVECs stimulated with Ox-LDL were
decreased following the overexpression of SESN1 (Fig. 2D-F). Consistently, Ox-LDL
stimulation led to significant increases in the contents of TNF-α,
IL-6 and IL-1β compared to the control group, which were restored
by overexpression of SESN1 (Fig.
2G). Furthermore, western blot analysis indicated that
overexpression of SESN1 attenuated the iNOS and p/t-NF-κB p65
protein levels triggered by Ox-LDL (Fig. 2H). In summary, these results
demonstrated that SESN1 exerted protective effects against the
Ox-LDL-induced decrease in the viability and inflammation of
HUVECs.
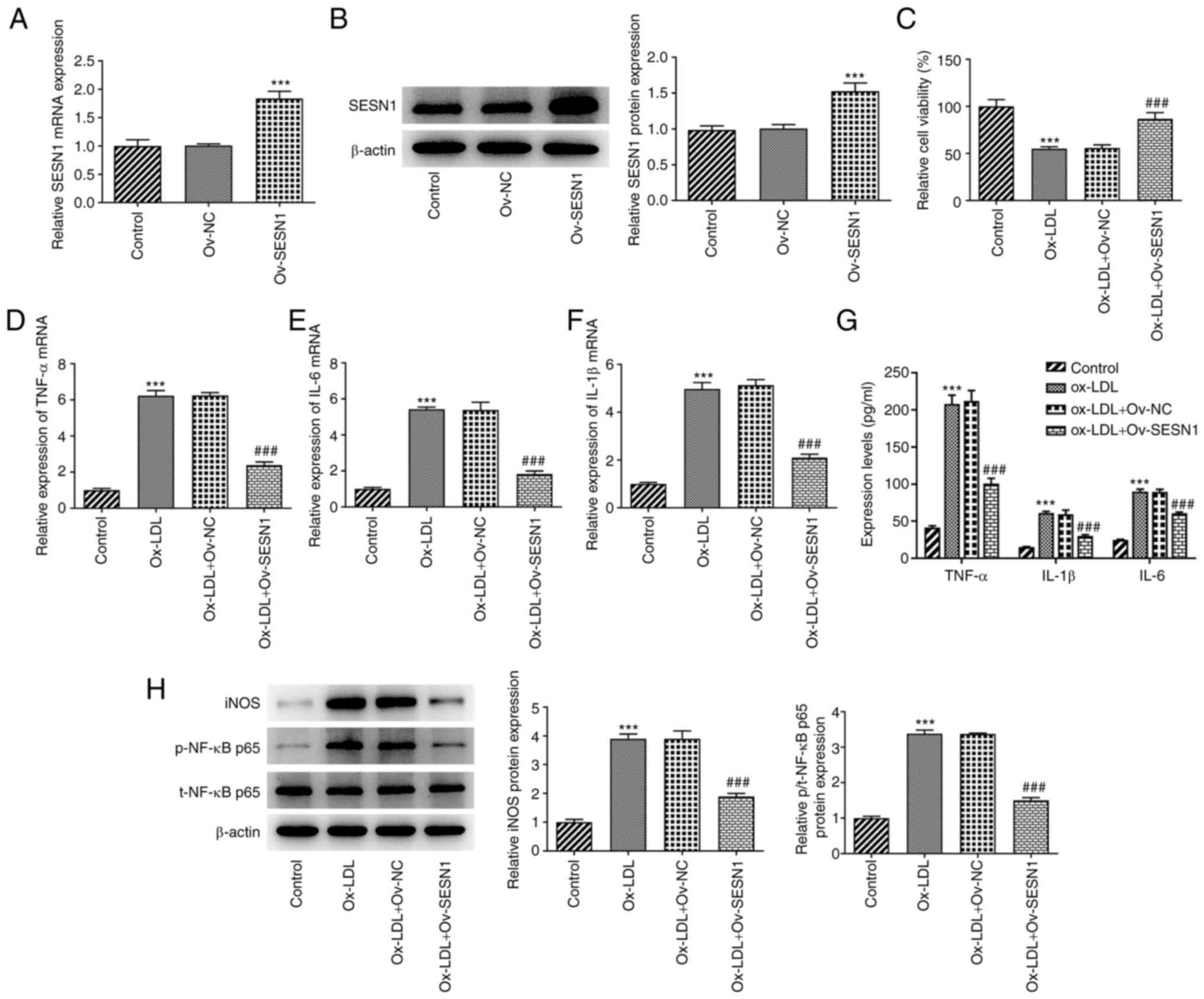 | Figure 2.SESN1 overexpression mitigates the
decrease in the viability and inflammatory response of human
umbilical vein endothelial cells stimulated with Ox-LDL. (A and B)
SESN1 overexpression efficiency was examined using (A) RT-qPCR and
(B) western blot analysis. ***P<0.001 vs. Ov-NC. (C) Cell
Counting Kit-8 assays were used to assess cell proliferation. (D-F)
RT-qPCR was to analyze the mRNA levels of (D) TNF-α, (E) IL-6 and
(F) IL-1β. (G) The contents of TNF-α, IL-6 and IL-1β in cell
culture supernatant were assessed with ELISA kits. (H) Western blot
analysis was used to examine the protein levels of iNOS, p-NF-κB
p65 and t-NF-κB p65. ***P<0.001 vs. control;
###P<0.001 vs. Ox-LDL + Ov-NC. RT-qPCR, reverse
transcription-quantitative PCR; SESN1, sestrin 1; Ox-LDL, oxidized
low-density lipoprotein; Ov, overexpression; NC, negative control;
iNOS, inducible nitric oxide synthase; p-, phosphorylated protein;
t-, total protein. |
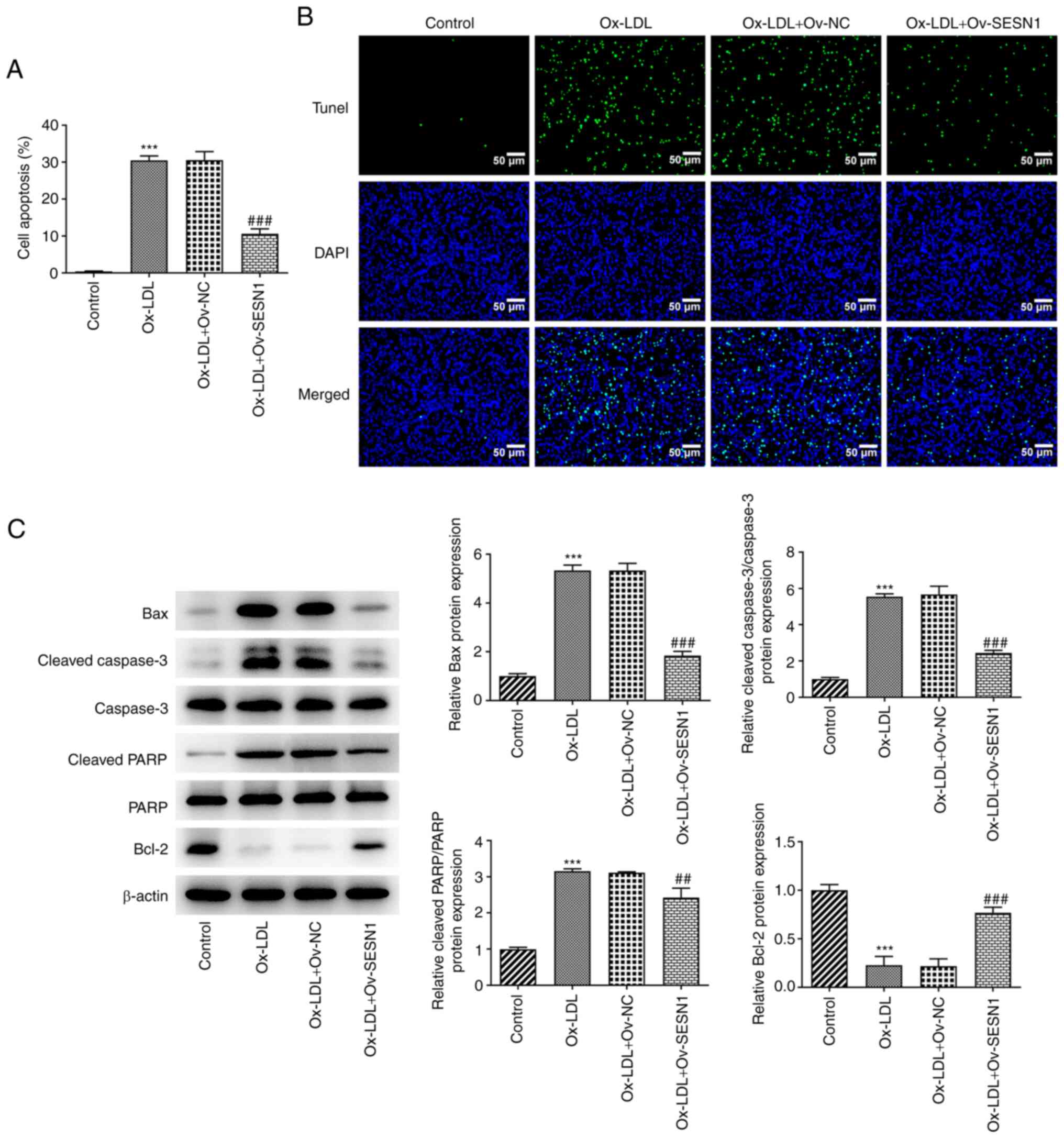
SESN1 overexpression suppresses the
apoptosis of Ox-LDL-stimulated HUVECs
The detection of cell apoptosis was performed using
a TUNEL assay. The results revealed that the Ox-LDL-induced
apoptosis of HUVECs was inhibited by SESN1 (Fig. 3A and B). In addition, western blot
analysis was used to examine the protein levels of Bax, cleaved
caspase-3, caspase-3, cleaved PARP, PARP and Bcl-2, all of which
are apoptosis-related factors. It was noted that following
stimulation with Ox-LDL, the Bax, cleaved caspase-3/caspase-3 and
cleaved PARP/PARP protein levels were increased, while the Bcl-2
protein level was decreased, indicating that Ox-LDL induced
apoptosis of HUVECs. However, when SESN1 was overexpressed, HUVEC
apoptosis was impeded (Fig. 3C).
These results suggested that SESN1 exerted a suppressive effect
against HUVEC apoptosis following stimulation with Ox-LDL.
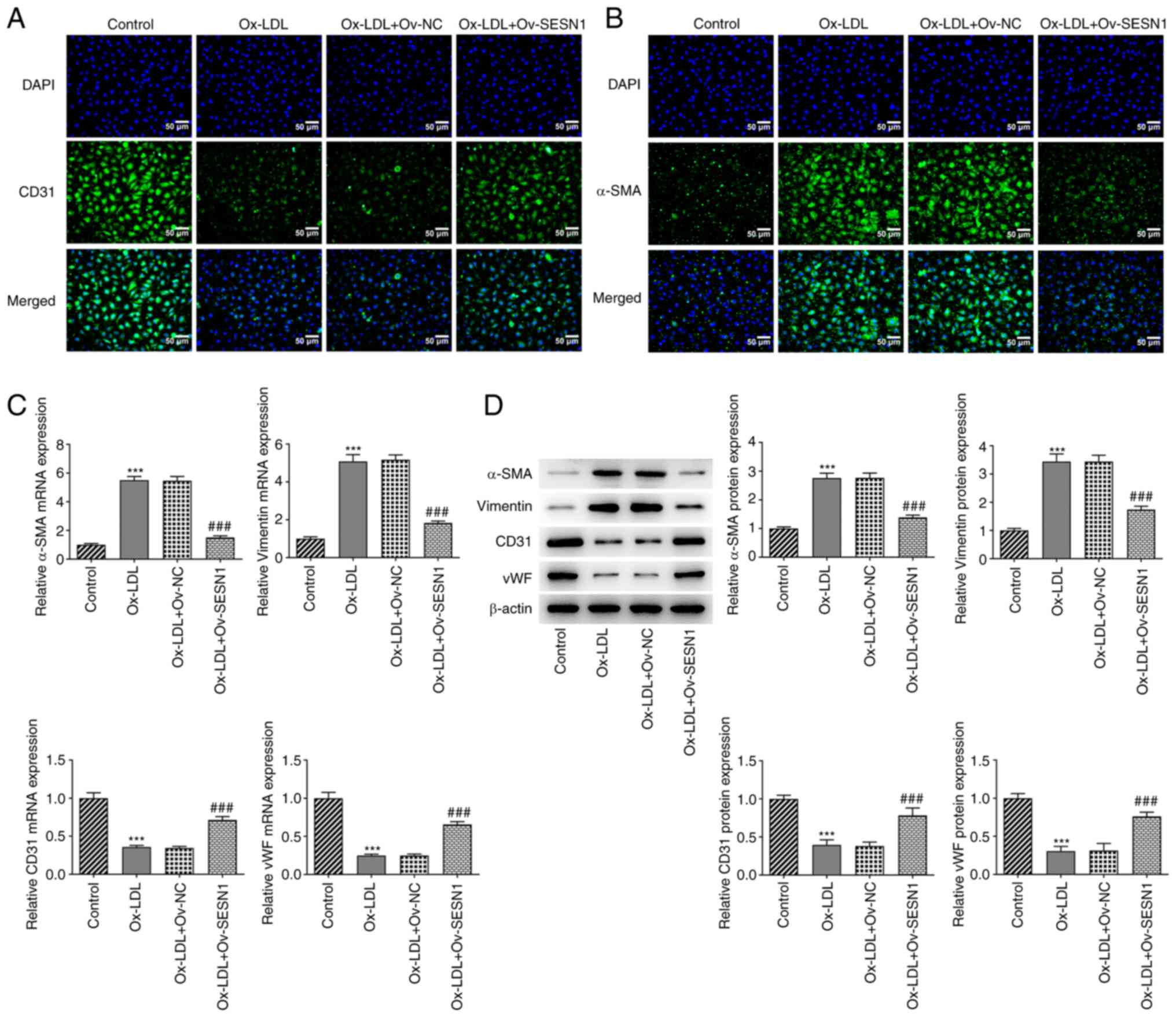
SESN1 attenuates the Ox-LDL-induced
EndMT of HUVECs
Using immunofluorescence staining, it was
demonstrated that Ox-LDL decreased the expression of the
endothelial marker CD31 (Fig. 4A)
and increased that of the mesenchymal marker α-SMA (Fig. 4B) in HUVECs; these effects were
mitigated by SESN1 overexpression. In addition, the increased
expression of mesenchymal markers (α-SMA and vimentin) and the
decreased expression of endothelial markers (CD31 and vWF) in
Ox-LDL-stimulated HUVECs were both reversed by SESN1 overexpression
(Fig. 4C and D). Thus, SESN1
prevented the Ox-LDL-induced EndMT of HUVECs.
SESN1 mediates AMPK/SIRT1/LOX1
signaling in Ox-LDL-stimulated HUVECs
It is well established that SESN1 is capable of
stimulating the AMPK signaling pathway (16). Using KEGG pathway enrichment
analysis, it was indicated that SIRT1 was a critical regulator of
AMPK activation and it inhibited the expression of LOX1, which has
been identified as a major receptor for Ox-LDL in endothelial cells
(30,31). Hence, whether SESN1 was associated
with the activation of AMPK/SIRT1 signaling to suppress LOX1, thus
participating in the inflammation, apoptosis and EndMT of HUVECs,
was next assessed. Western blot analysis revealed that Ox-LDL
stimulation decreased the protein levels of p-AMPK/AMPK and SIRT1,
whereas it increased the protein levels of LOX1. However, these
effects were reversed by SESN1 overexpression (Fig. 5A). Of note, the addition of the
AMPK inhibitor compound C notably downregulated p-AMPK/AMPK and
SIRT1 expression and upregulated LOX1 expression when compared with
the Ox-LDL + Ov-SESN1 group (Fig.
5B). In addition, the SIRT1 inhibitor NAM decreased SIRT1
expression and increased LOX1 expression. However, the inhibitors
had no effect on p-AMPK/AMPK expression. In conclusion, SESN1
activated AMPK/SIRT1 signaling to suppress LOX1 in HUVECs following
stimulation with Ox-LDL.
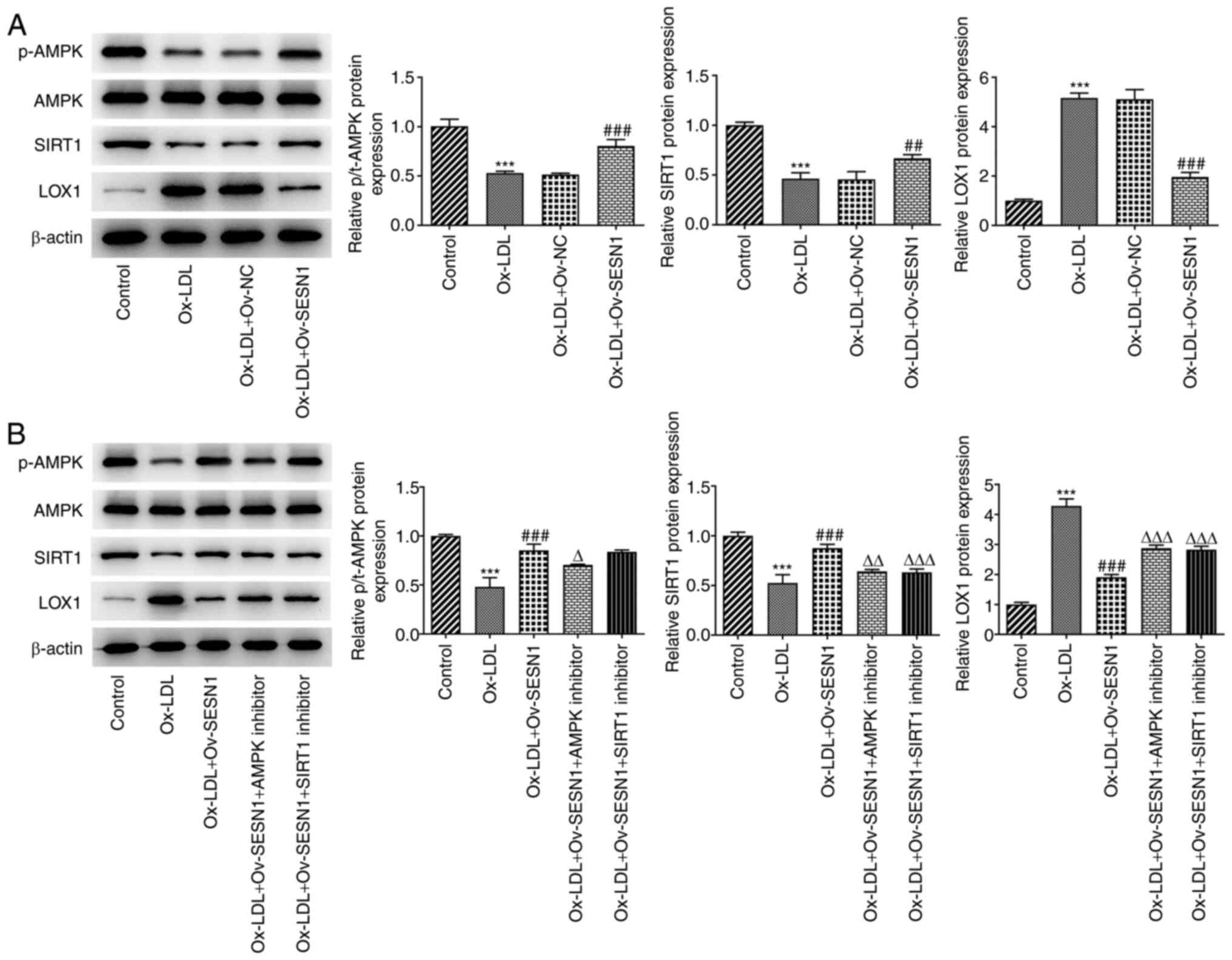 | Figure 5.SESN1 mediates AMPK/SIRT1/LOX1
signaling in Ox-LDL-stimulated HUVECs. (A) Western blot analysis
was used to examine p-AMPK, AMPK, SIRT1 and LOX1 protein levels in
HUVECs co-treated with Ox-LDL and Ov-SESN1. ***P<0.001 vs.
control; ##P<0.01, ###P<0.001 vs.
Ox-LDL + Ov-NC. (B) Western blot analysis was used to examine
p-AMPK, AMPK, SIRT1 and LOX1 protein levels in the control, Ox-LDL,
Ox-LDL + Ov-SESN1, Ox-LDL + Ov-SESN1 + AMPK inhibitor and Ox-LDL +
Ov-SESN1 + SIRT1 inhibitor groups. ***P<0.001 vs. control;
###P<0.001 vs. Ox-LDL; ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. Ox-LDL +
Ov-SESN1. SESN1, sestrin 1; Ox-LDL, oxidized low-density
lipoprotein; Ov, overexpression; NC, negative control; LOX1,
low-density lipoprotein receptor-1; p-AMPK, phosphorylated
adenosine monophosphate-activated protein kinase catalytic subunit
α1; SIRT1, sirtuin 1; HUVECs, human umbilical vein endothelial
cells. |
SESN1 exerts protective effects on the
viability and inflammation of Ox-LDL-stimulated HUVECs via
activation of AMPK/SIRT1/LOX1 signaling
Subsequently, the results of a CCK-8 assay
demonstrated that the promotion of the viability of
Ox-LDL-stimulated HUVECs by SESN1 overexpression was attenuated by
the inhibitors of AMPK or SIRT1 (Fig.
6A). Similarly, the decreased expression of the inflammatory
factors, TNF-α, IL-6 and IL-1β [tested by RT-qPCR (Fig. 6B) and ELISA (Fig. 6C)], as well as iNOS and p-p65/p65
of NF-κB [detected by western blot analysis (Fig. 6D)] due to SESN1 overexpression was
abrogated by AMPK or SIRT1 inhibitor. These results suggested that
AMPK/SIRT1 signaling reversed the protective effects of SESN1
against the decrease in the viability and inflammation of HUVECs
mediated by Ox-LDL.
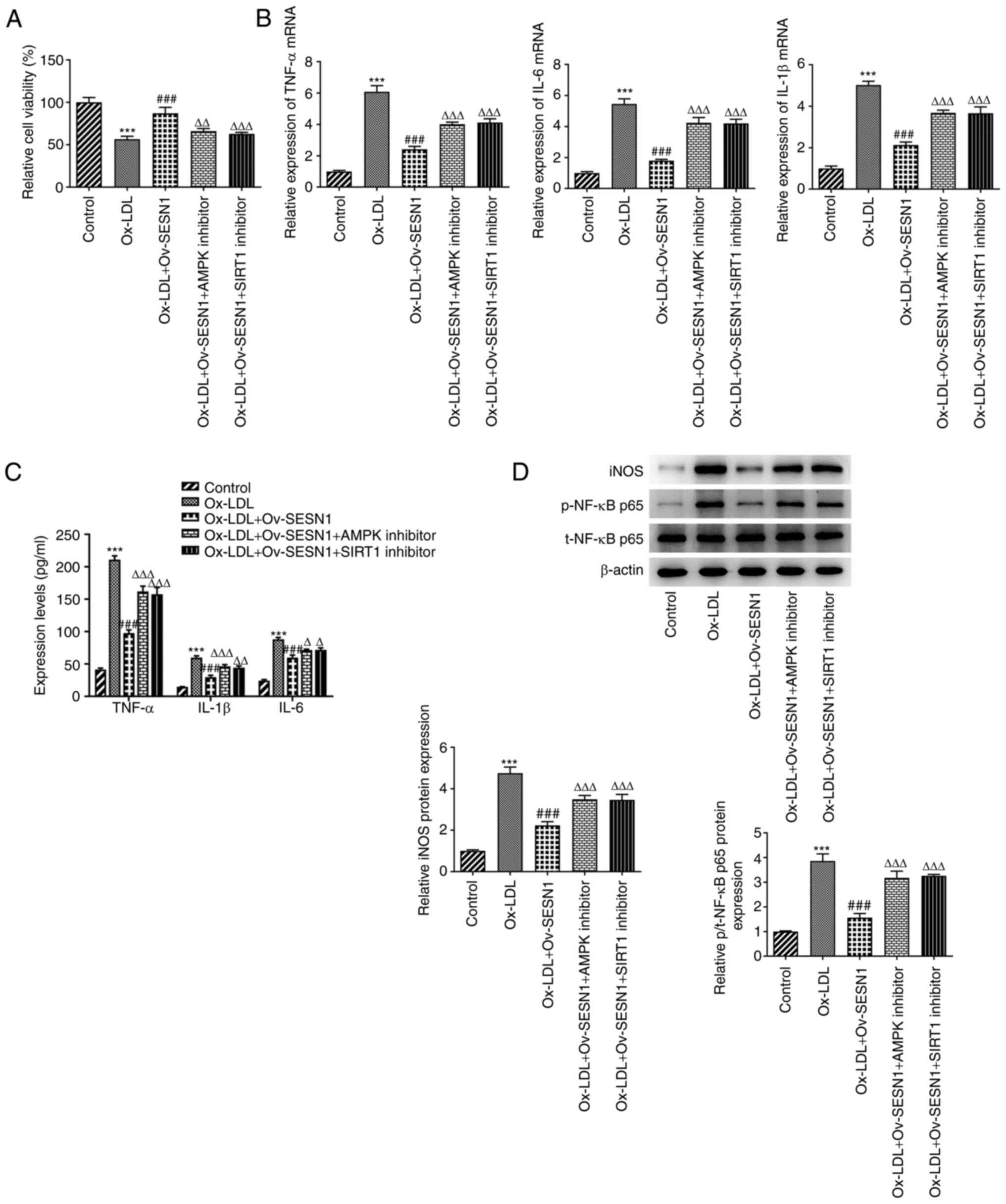 | Figure 6.SESN1 attenuates the decrease in cell
viability and inflammation in Ox-LDL-stimulated human umbilical
vein endothelial cells via activating AMPK/SIRT1/LOX1 signaling.
(A) Cell viability was detected using a Cell Counting Kit-8 assay.
(B) Reverse transcription-quantitative PCR was to analyze the
expression of inflammatory factors. (C) ELISA kits were used to
determine the levels of TNF-α, IL-6 and IL-1β in cell culture
supernatant. (D) Western blot analysis was used to examine the
iNOS, p-NF-κB p65 and t-NF-κB p65 protein levels. ***P<0.001 vs.
control; ###P<0.001 vs. Ox-LDL;
∆P<0.05,∆∆P<0.01,
∆∆∆P<0.001 vs. Ox-LDL + Ov-SESN1. SESN1, sestrin 1;
Ox-LDL, oxidized low-density lipoprotein; Ov, overexpression; LOX1,
low-density lipoprotein receptor-1; SIRT1, sirtuin 1; AMPK,
adenosine monophosphate-activated protein kinase catalytic subunit
α1; iNOS, inducible nitric oxide synthase; p-, phosphorylated
protein; t-, total protein. |
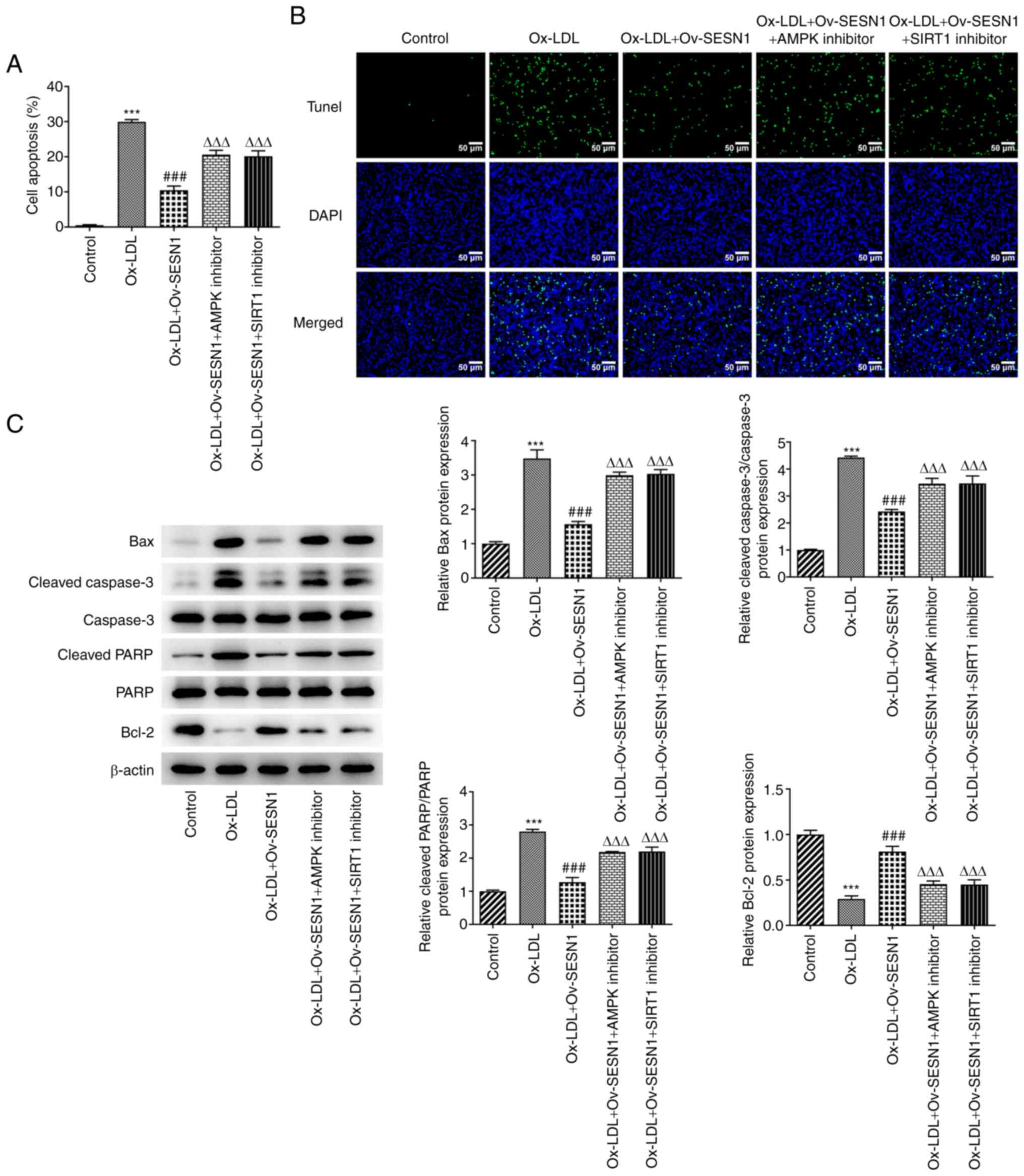
Overexpression of SESN1 prevents
Ox-LDL-mediated HUVEC apoptosis by regulating AMPK/SIRT1/LOX1
signaling
Analysis of cell apoptosis using the TUNEL assay
revealed that the suppression of apoptosis of Ox-LDL-stimulated
HUVECs associated with SESN1 overexpression was abrogated by AMPK
or SIRT1 inhibitor (Fig. 7A and
B). Western blot analysis indicated that the decrease in the
protein levels of Bax, cleaved caspase-3/caspase-3 and cleaved
PARP/PARP, and the increase in the protein level of Bcl-2 in
Ox-LDL-stimulated HUVECs induced by SESN1 overexpression were all
reversed by treatment with an AMPK or SIRT1 inhibitor; these
observations were consistent with the results of TUNEL assay
(Fig. 7C). Taken together, it was
indicated that SESN1 impeded Ox-LDL-induced HUVEC apoptosis via the
activation of AMPK/SIRT1/LOX1 signaling.
Inhibition of AMPK/SIRT1/LOX1
signaling attenuates the protective effects of SESN1 against the
Ox-LDL-induced EndMT of HUVECs
Immunofluorescence staining confirmed that SESN1
enhanced CD31 expression and decreased α-SMA expression upon
exposure to Ox-LDL; this effect was reversed by AMPK or SIRT1
inhibitor (Fig. 8A and B).
Furthermore, AMPK or SIRT1 inhibitor reversed the promoting effects
of SESN1 on the expression of CD31 and vWF, and abrogated the
suppressive effects of SESN1 on α-SMA and vimentin expression in
Ox-LDL-stimulated HUVECs (Fig. 8C and
D). Collectively, SESN1 impeded the Ox-LDL-mediated EndMT of
HUVECs via the activation of AMPK/SIRT1/LOX1 signaling.
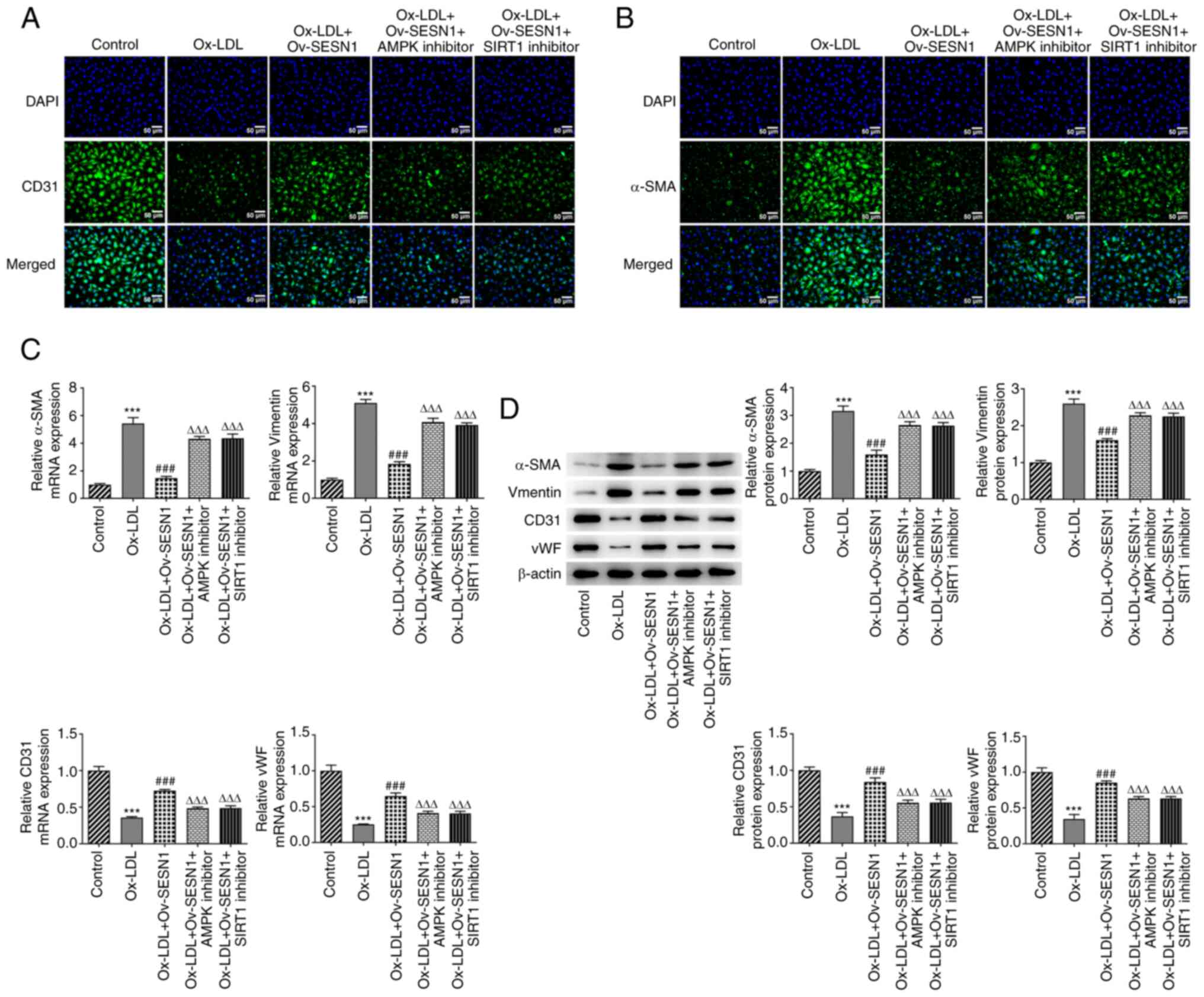 | Figure 8.Inhibition of AMPK/SIRT1/LOX1
signaling attenuates the protective effects of SESN1 against the
Ox-LDL-induced endothelial-mesenchymal transition of human
umbilical vein endothelial cells. (A) CD31 and (B) α-SMA expression
was assessed using immunofluorescence staining (scale bars, 50 µm).
(C) Reverse transcription-quantitative PCR and (D) western blot
analysis were used to examine the expression of CD31, vWF, α-SMA
and vimentin. ***P<0.001 vs. control; ###P<0.001
vs. Ox-LDL; ∆∆∆P<0.001 vs. Ox-LDL + Ov-SESN1. SESN1,
sestrin 1; Ox-LDL, oxidized low-density lipoprotein; Ov,
overexpression; LOX1, low-density lipoprotein receptor-1; SIRT1,
sirtuin 1; SMA, smooth muscle actin; vWF, von Willebrand factor;
AMPK, adenosine monophosphate-activated protein kinase catalytic
subunit α1. |
Discussion
Endothelial cells are considered active metabolic
components of biological tissue that have crucial physiological
functions (32). It is widely
acknowledged that endothelial cell injury is highly associated with
various human diseases, including chronic cardiovascular, renal and
metabolic diseases (33). With
regard to cardiovascular diseases, endothelial cell injury is
considered to be an early and enduring feature, preceding the
development of cardiovascular disease (34). As a risk factor for
atherosclerosis, Ox-LDL contributes to atherosclerotic plaque
formation and progression via several mechanisms, including
endothelial cell dysfunction (25). Under pathological conditions,
Ox-LDL is known to induce endothelial cell injury during
atherosclerosis (35). SESN1 has
been reported to be involved in cell metabolism and cardiovascular
and age-related diseases (36).
Furthermore, SESN1 has been demonstrated to inhibit NOD-like
receptor family pyrin domain containing 3 inflammasome activation
in lipopolysaccharide-primed macrophages induced by Ox-LDL via the
inactivation of NF-κB signaling (20). The present study confirmed the low
expression of SESN1 in Ox-LDL-stimulated HUVECs and Ox-LDL
stimulation decreased cell viability in a concentration-dependent
manner.
Endothelial cell apoptosis has a key role in the
process of atherosclerosis, which is hallmarked by the inflammatory
response (37). For instance,
iNOS, a pro-inflammatory cytokine, is considered to be a major
contributor to atherosclerosis (38). NF-κB transcription factors formed
by the dimerization of Rel proteins [RelA (p65), c-Rel, RelB, p50,
p52] have been confirmed to have crucial roles in inflammation,
immunity, cell proliferation and apoptosis (39,40). In addition, as has been
demonstrated, EndMT is the process through which endothelial cells
undergo a series of molecular events, which leads to the adoption
of a mesenchymal-like phenotype (41). Furthermore, endothelial cells
undergoing EndMT lose expression of endothelial cell-specific
proteins, including CD31 and vWF, and this initiates the expression
of mesenchymal cell-specific genes and the production of their
encoded proteins, including α-SMA and vimentin (42). For instance, α-SMA, an
EndMT-related marker, has been reported to be involved in
cardiogenesis and cardiovascular diseases (43). As an EndMT-related marker, CD31
has also been confirmed as a potential therapeutic target in
atherosclerosis (44).
Furthermore, apoptosis, inflammation and EndMT induced by Ox-LDL
are vital for the development of atherosclerosis (45). Previous studies have indicated
that SESN1 has a crucial role in cell apoptosis and the
inflammatory response. For instance, SESN1 is targeted by
microRNA-16-5p and influences myoblast proliferation and apoptosis
(16). SESN1 also inhibits
macrophage-mediated inflammation of the aorta (20). SESN1 has a crucial role in aerobic
exercise and suppresses the activation of inflammatory signaling
(46). In the present study,
functional experiments demonstrated that stimulation with 100 µg/ml
Ox-LDL promoted apoptosis of HUVECs. Furthermore, Ox-LDL
stimulation elevated the TNF-α, IL-6 and IL-1β mRNA levels, as well
as the iNOS, p/t-NF-κB p65 protein levels. In addition, Ox-LDL
increased α-SMA and vimentin expression, whereas it decreased CD31
and vWF expression. All these aforementioned effects were reversed
by SESN1 overexpression.
AMPK is a central regulator of endothelial cell
metabolism and has a crucial role in diabetes, cancers and vascular
diseases (47,48). SIRT1 protein, the product of the
longevity gene, is involved in a wide variety of cellular processes
(49). It has been reported that
SIRT1 mediates endothelial functions (50). Furthermore, SIRT1 is an important
activator of AMPK (30). Of note,
SIRT1 suppresses the expression of LOX1, which is a 50-kDa
transmembrane glycoprotein that serves as a receptor for Ox-LDL
(30,51). As has been previously reported,
SESNs are the primary regulators of AMPK (52). SESN1 has been indicated to
directly interact with AMPK, thereby participating in several human
diseases. For instance, Li et al (53) demonstrated that SESN1 protected
cardiomyocytes from doxorubicin-induced damage by upregulating AMPK
expression. The SESN1-2/AMPK/mTOR axis is regulated by AMPK
phosphorylation, which has a crucial role in pancreatic cancer cell
autophagy (54). In the present
study, it was observed that the overexpression of SESN1 enhanced
the decreased protein levels of p-AMPK/AMPK and SIRT1, and
decreased the elevated protein levels of LOX1 following exposure to
Ox-LDL. This indicated that SESN1 led to the activation of
AMPK/SIRT1 signaling to inhibit LOX1 expression in
Ox-LDL-stimulated HUVECs. At the same time, it was noted that AMPK
or SIRT1 inhibitor attenuated the protective effects of SESN1
against the Ox-LDL-induced apoptosis, inflammation and EndMT of
HUVECs.
In conclusion, the present study demonstrated that
SESN1 suppressed Ox-LDL-induced endothelial EndMT, inflammation and
apoptosis by regulating the AMPK/SIRT1/LOX1 signaling pathway.
SESN1 may thus prove to be an effective biomarker for protection
from endothelial cell injury, thus highlighting a novel target for
the treatment of atherosclerosis. The lack of elucidation of
upstream mechanisms of SESN1 and an in vivo animal study are
limitations of the present study and comprehensive analysis is
required in the future.
Acknowledgements
Not applicable.
Funding
Funding: Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FG and YZ conceived and designed the study. BZ and
CX performed the experiments. ZS, YG and XD analyzed and
interpreted the experimental data. FG and XD wrote and revised the
manuscript. YZ and BZ confirmed the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Getz GS and Reardon CA: Animal models of
atherosclerosis. Arterioscler Thromb Vasc Biol. 32:1104–1115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frostegård J: Immunity, atherosclerosis
and cardiovascular disease. BMC medicine. 11:1172013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jensen HA and Mehta JL: Endothelial cell
dysfunction as a novel therapeutic target in atherosclerosis.
Expert Rev Cardiovasc Ther. 14:1021–1033. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Falk E: Pathogenesis of atherosclerosis. J
Am Coll Cardiol. 47 (8 Suppl):C7–C12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bai X, Wang X and Xu Q: Endothelial damage
and stem cell repair in atherosclerosis. Vascul Pharmacol.
52:224–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lao KH, Zeng L and Xu Q: Endothelial and
smooth muscle cell transformation in atherosclerosis. Curr Opin
Lipidol. 26:449–456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin B, Yang H and Xiao B: Role of
microRNAs in endothelial inflammation and senescence. Mol Biol Rep.
39:4509–4518. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Winn RK and Harlan JM: The role of
endothelial cell apoptosis in inflammatory and immune diseases. J
Thromb Haemost. 3:1815–1824. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Souilhol C, Harmsen MC, Evans PC and
Krenning G: Endothelial-mesenchymal transition in atherosclerosis.
Cardiovasc Res. 114:565–577. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu L, Gong X, Gong J, Xuan Y, Fu T, Ni S,
Xu L and Ji N: Notoginsenoside R1 upregulates miR-221-3p expression
to alleviate ox-LDL-induced apoptosis, inflammation, and oxidative
stress by inhibiting the TLR4/NF-κB pathway in HUVECs. Braz J Med
Biol Res. 53:e93462020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu K, Liu X, Ren G, Yin D, Guo S and Zhao
Y: Depletion of CPEB1 protects against oxidized LDL-induced
endothelial apoptosis and inflammation though SIRT1/LOX-1
signalling pathway. Life Sci. 239:1168742019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim M, Kowalsky AH and Lee JH: Sestrins in
physiological stress responses. Annu Rev Physiol. 83:381–403. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang M, Xu Y, Liu J, Ye J, Yuan W, Jiang
H, Wang Z, Jiang H and Wan J: Recent insights into the biological
functions of sestrins in health and disease. Cell Physiol Biochem.
43:1731–1741. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sanchis-Gomar F: Sestrins: Novel
antioxidant and AMPK-modulating functions regulated by exercise? J
Cell Physiol. 228:1647–1650. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Narita N, Ito Y, Takabayashi T, Okamoto M,
Imoto Y, Ogi K, Tokunaga T, Matsumoto H and Fujieda S: Suppression
of SESN1 reduces cisplatin and hyperthermia resistance through
increasing reactive oxygen species (ROS) in human maxillary cancer
cells. Int J Hyperthermia. 35:269–278. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai B, Ma M, Chen B, Li Z, Abdalla BA, Nie
Q and Zhang X: MiR-16-5p targets SESN1 to regulate the p53
signaling pathway, affecting myoblast proliferation and apoptosis,
and is involved in myoblast differentiation. Cell Death Dis.
9:3672018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong XC: The potential of sestrins as
therapeutic targets for diabetes. Expert Opin Ther Targets.
19:1011–1015. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JH, Cho US and Karin M: Sestrin
regulation of TORC1: Is Sestrin a leucine sensor? Sci Signal.
9:re52016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Budanov AV: SESTRINs regulate mTORC1 via
RRAGs: The riddle of GATOR. Mol Cell Oncol. 2:e9971132015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Keping Y, Yunfeng S, Pengzhuo X, Liang L,
Chenhong X and Jinghua M: Sestrin1 inhibits oxidized low-density
lipoprotein-induced activation of NLRP3 inflammasome in macrophages
in a murine atherosclerosis model. Eur J Immunol. 50:1154–1166.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang J, Wang Z, Zhang J, Zuo G, Li B, Mao
W and Chen S: Rapamycin attenuates endothelial apoptosis induced by
low shear stress via mTOR and sestrin1 related redox regulation.
Mediators Inflamm. 2014:7696082014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xue R, Zeng J, Chen Y, Chen C, Tan W, Zhao
J, Dong B, Sun Y, Dong Y and Liu C: Sestrin 1 ameliorates cardiac
hypertrophy via autophagy activation. J Cell Mol Med. 21:1193–1205.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fulco M and Sartorelli V: Comparing and
contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell
Cycle. 7:3669–3679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang H, Yang G, Zhang Q, Liang X, Liu Y,
Gao M, Guo Y and Chen L: Apremilast ameliorates ox-LDL-induced
endothelial dysfunction mediated by KLF6. Aging (Albany NY).
12:19012–19021. 2020.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm.
2013:1527862013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ou HC, Chou WC, Hung CH, Chu PM, Hsieh PL,
Chan SH and Tsai KL: Galectin-3 aggravates ox-LDL-induced
endothelial dysfunction through LOX-1 mediated signaling pathway.
Environ Toxicol. 34:825–835. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hwang ES and Song SB: Nicotinamide is an
inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell
Mol Life Sci. 74:3347–3362. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng S, Li W, Xu M, Bai X, Zhou Z, Han J,
Shyy JY and Wang X: Calcitonin gene-related peptide promotes
angiogenesis via AMP-activated protein kinase. Am J Physiol Cell
Physiol. 299:C1485–C1492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu J, Mitra S, Wang X, Khaidakov M and
Mehta JL: Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in
atherogenesis and tumorigenesis. Antioxid Redox Signal.
15:2301–2333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cetrullo S, D'Adamo S, Tantini B, Borzi RM
and Flamigni F: mTOR, AMPK, and Sirt1: Key players in metabolic
stress management. Crit Rev Eukaryot Gene Expr. 25:59–75. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lawal AO, Davids LM and Marnewick JL:
Diesel exhaust particles and endothelial cells dysfunction: An
update. Toxicol In Vitro. 32:92–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang X, Sun D, Song JW, Zullo J,
Lipphardt M, Coneh-Gould L and Goligorsky MS: Endothelial cell
dysfunction and glycocalyx-A vicious circle. Matrix Biol.
71-72:421–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Monteiro JP, Bennett M, Rodor J,
Caudrillier A, Ulitsky I and Baker AH: Endothelial function and
dysfunction in the cardiovascular system: The long non-coding road.
Cardiovasc Res. 115:1692–1704. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su Q, Sun Y, Ye Z, Yang H, Kong B and Li
L: Pinocembrin protects endothelial cells from oxidized LDL-induced
injury. Cytokine. 111:475–480. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun W, Wang Y, Zheng Y and Quan N: The
emerging role of sestrin2 in cell metabolism, and cardiovascular
and age-related diseases. Aging Dis. 11:154–163. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang T, Tian F, Wang J, Jing J, Zhou SS
and Chen YD: Atherosclerosis-associated endothelial cell apoptosis
by MiR-429-mediated down regulation of Bcl-2. Cell Physiol Biochem.
37:1421–1430. 2015. View Article : Google Scholar : View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lind M, Hayes A, Caprnda M, Petrovic D,
Rodrigo L, Kruzliak P and Zulli A: Inducible nitric oxide synthase:
Good or bad? Biomed Pharmacother. 93:370–375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lawrence T and Fong C: The resolution of
inflammation: Anti-inflammatory roles for NF-kappaB. Int J Biochem
Cell Biol. 42:519–523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hulshoff MS, Del Monte-Nieto G, Kovacic J
and Krenning G: Non-coding RNA in endothelial-to-mesenchymal
transition. Cardiovasc Res. 115:1716–1731. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Piera-Velazquez S and Jimenez SA:
Endothelial to mesenchymal transition: Role in physiology and in
the pathogenesis of human diseases. Physiol Rev. 99:1281–1324.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anbara T, Sharifi M and Aboutaleb N:
Endothelial to mesenchymal transition in the cardiogenesis and
cardiovascular diseases. Curr Cardiol Rev. 16:306–314. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Caligiuri G: CD31 as a therapeutic target
in atherosclerosis. Circ Res. 126:1178–1189. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao H, Liu M, Liu H, Suo R and Lu C:
Naringin protects endothelial cells from apoptosis and inflammation
by regulating the Hippo-YAP pathway. Biosci Rep.
40:BSR201934312020. View Article : Google Scholar : View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun Y, Wu Y, Jiang Y and Liu H: Aerobic
exercise inhibits inflammatory response in atherosclerosis via
sestrin1 protein. Exp Gerontol. 155:1115812021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qi D and Young LH: AMPK: Energy sensor and
survival mechanism in the ischemic heart. Trends Endocrinol Metab.
26:422–429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mount PF, Lane N, Venkatesan S, Steinberg
GR, Fraser SA, Kemp BE and Power DA: Bradykinin stimulates
endothelial cell fatty acid oxidation by CaMKK-dependent activation
of AMPK. Atherosclerosis. 200:28–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Quiñones M, Al-Massadi O, Fernø J and
Nogueiras R: Cross-talk between SIRT1 and endocrine factors:
Effects on energy homeostasis. Mol Cell Endocrinol. 397:42–50.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hwang JW, Yao H, Caito S, Sundar IK and
Rahman I: Redox regulation of SIRT1 in inflammation and cellular
senescence. Free Radic Biol Med. 61:95–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hung CH, Chan SH, Chu PM, Lin HC and Tsai
KL: Metformin regulates oxLDL-facilitated endothelial dysfunction
by modulation of SIRT1 through repressing LOX-1-modulated oxidative
signaling. Oncotarget. 7:10773–10787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shen T, Alvarez-Garcia O, Li Y, Olmer M
and Lotz MK: Suppression of Sestrins in aging and osteoarthritic
cartilage: Dysfunction of an important stress defense mechanism.
Osteoarthritis Cartilage. 25:287–296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li EZ, Sun Y, Lv GX, Li Y, Zhang Z, Hu Z
and Cao W: Sinoporphyrin sodium based sonodynamic therapy induces
anti-tumor effects in hepatocellular carcinoma and activates
p53/caspase 3 axis. Int J Biochem Cell Biol. 113:104–114. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fiorini C, Menegazzi M, Padroni C, Dando
I, Dalla Pozza E, Gregorelli A, Costanzo C, Palmieri M and
Donadelli M: Autophagy induced by p53-reactivating molecules
protects pancreatic cancer cells from apoptosis. Apoptosis.
18:337–346. 2013. View Article : Google Scholar : PubMed/NCBI
|