Introduction
Hirschsprung-associated enterocolitis (HAEC) is the
most common complication of Hirschsprung disease (HSCR) (1,2),
which may occur during the preoperative or postoperative stages of
surgery, even after definitive pull-through surgery (3). Accumulating clinical evidence
suggests that abnormalities in the intestinal microbiome, impaired
intestinal mucosal barrier function, altered systemic immune system
function and bacterial translocation are all possible causes of
HAEC (4–6). The intestinal tract is the most
active immune organ in the human body and it is constantly
challenged by a large number of antigens. A previous study
indicated Clostridium difficile, E. coli and certain viruses
may be causative agents of enterocolitis development (7). However, the mechanisms by which gut
microbes influence the mucosal barrier and the development of
pathogenic bacteria-mediated HAEC remain to be fully elucidated; an
increasing number of studies have investigated the possible
implication of Clostridium difficile as a pathogen of HAEC
but remain inconclusive (8,9).
According to the results of Illumina-MiSeq high-throughput
sequencing for characterization of intestinal microbiomes of HAEC
(10), E. coli was the
most prominent genus detected in patients with HAEC and recurrence
of HAEC (11). However, the role
of Clostridium species as a cause of HAEC remains
controversial. Therefore, E. coli was selected as the
preferred pathogen in the present study (9–11).
Toll-like receptor 4 (TLR4) is a transmembrane
protein, which has important effects in initiating inflammatory
reactions (12). A recent study
suggested that defective murine TLR4 was responsible for
lipopolysaccharide (LPS) hyporesponsiveness in two mouse strains,
and furthermore, a study in TLR4-deficient mice revealed that TLR4
was essential for LPS-induced inflammatory signaling (13). A previous study reported that the
dysregulated expression of TLR4 signaling transduction led to
uncontrolled colitis, which was associated with the loss of mucosal
integrity, development of ulcerations and colonic bleeding
(14), consistent with what is
observed in patients with HAEC. Emerging evidence has indicated
that TLR4 expression is upregulated in several intestinal
inflammatory diseases, including inflammatory bowel colitis
(15) and necrotizing
enterocolitis (16). When TLR4 is
engaged by its ligands, the downstream signaling pathways,
including the NF-κB and MAPK p38 [phosphorylated (p-)p38] pathways,
are activated; this activation is essential for the initiation of
an inflammatory response by promoting and/or modulating the
transcription and translation of inflammation-related genes, such
as IL-6, TNF-α and IL-1β (17).
Inhibition of NF-κB has also been indicated to protect against
colonic epithelium damage and/or promote epithelial repair
(18,19).
In the present study, it was hypothesized that the
pathogenic organism E. coli JM83 promotes HAEC development
through TLR4/p-p38/NF-κB signaling, influencing intestinal mucosal
barrier integrity. Thus, whether TLR4/p-p38/NF-κB signaling
participates in the pathogenesis of HAEC during the invasive
infection of endothelin receptor B (Ednrb)−/− mice with
E. coli JM83 (used as a model of HAEC) was assessed in the
present study.
Materials and methods
Animals
Female wild-type (WT; age, 8 weeks; weight, 16–19 g;
n=20), female Ednrbflex3/flex3 (age, 8 weeks; weight,
15–19 g; n=8) mice and male Ednrbflex3/+ (age, 8 weeks;
weight, 15–19 g; n=8). Endothelin receptor B-null mice
(Ednrbflex3/flex3 and Ednrbflex3/+) were
established using C57/B6J mice. All mice were purchased from the
Institute of Model Animals of Wuhan University (Wuhan, China). In
brief, after mating Ednrbflex3/+ mice with
Ednrbflex3/flex3 mice, the homozygotes (herein referred
to as Ednrb−/−) were easily distinguished from the WT
and heterozygote littermates (herein referred to as
Ednrb+/+ and Ednrb+/−, respectively) by the
white fur color and gradually enlarging abdomens (due to the
absence of ganglion cells at the end of the rectum).
Ednrb+/+ and Ednrb+/− mice had a normal
phenotype and did not develop aganglionosis. Therefore, 20
Ednrb+/+ or Ednrb+/− animals were randomly
assigned to the WT group. A total of 20 Ednrb−/− animals
were obtained for the present study that displayed distal colonic
aganglionosis involving 5–10 mm of the colon. Naive mice were
defined as WT and Ednrb−/− mice without any
intervention. All experiments involving animals were performed in a
specific pathogen-free environment and were approved by the
Institutional Animal Research Committee of Zunyi Medical University
(Zunyi, China; approval no. IACUC-20191025028) and were in
accordance with the Zunyi Medical University Guidelines for Animal
Care. Animals were maintained under a 12-h light/dark cycle at a
temperature of 22±2°C in an air-conditioned room, and were given
access to food and water ad libitum. Male and female mice
were raised separately. A total of eight mice survived more than 5
weeks; however, two mice died of abdominal distension, diarrhea,
and severe dehydration before the date of sacrifice in the
Ednrb−/− group infected with E. coli. All mice
were sacrificed 5 weeks after modeling. Isoflurane inhalation
anesthesia was used (induction concentration, 3%; maintenance
concentration, 1%), followed by collection of the colon and blood.
Subsequently, mice were sacrificed by CO2 asphyxia (50%
CO2 volume displacement rate). When cessation of the
heartbeat and breathing of the mice was verified, and no reflexes
were detected, the death of mice was confirmed.
Bacterial strains
The E. coli JM83 (Jackson Laboratory) strain
was cultured on trypsin soybean agar plates (BD Biosciences)
supplemented with 5% sheep blood (BD Biosciences), which was in
turn supplemented with 0.2% yeast extract (Merck KGaA) at 37°C with
5% CO2 (durations indicated in individual
experiments).
Establishment of the HAEC model
WT and Ednrb−/− mice were infected with
0.1 ml Lb broth containing 1×109 colony-forming units of
E. coli JM83 (Jackson Laboratory) by oral gavage to
establish the WT+E. coli (n=5) and Ednrb−/−+E.
coli (n=5) groups. The colon samples from
Ednrb−/−+E. coli mice were stored at −80°C and
embedded in paraffin for subsequent H&E as well as
immunofluorescence staining analysis (described below) to verify
the establishment of the HAEC model. Inflammatory cell infiltration
of the crypts (cryptitis and crypt abscesses) was lighter in HAEC
mice than in human patients with HAEC. The WT and
Ednrb−/− mice were used as the control groups. The
severity of HAEC was evaluated according to the modified grading
system reported by Porokuokka et al (20) to reflect the epithelial pathology
in HAEC mice. Parts of the small bowel (jejunum and ileum) or large
bowel (cecum and colon) were excised by separation from the
mesentery to prepare a single intestinal cell suspension.
si-RNA transfection
In order to knockdown TLR4 expression in WT+E.
coli and Ednrb−/−+E. coli mice, small
interfering RNA (siRNA) targeting TLR4 (si-TLR4) and the
corresponding negative control (siRNA-NC) were used to generate
WT+siRNA-NC+E. coli (n=5), WT+si-TLR4+E.
coli (n=5) and Ednrb−/−+si-TLR4+E. coli (n=5)
mouse groups (21). In brief,
after anesthetization with isoflurane (induction concentration, 3%;
maintenance concentration, 1%), 20 nmol/kg siRNA was injected into
the caudal vein twice a week to target TLR4. The sequences of the
siRNAs were as follows: si-TLR4 sense, 5′-UUCGAGACUGGACAAGCC-3′ and
antisense, 5′-UGGCUUGUCCAGUCACGA-3′; siRNA-C sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-TTAAGAGGCUUGCACAGUGCA-3′ (100 nM; Guangzhou RiboBio Co., Ltd.).
Ednrb−/−+E. coli mice were used as a control
group. Knockdown of the TLR4 gene was confirmed at the protein
level by western blot analysis.
H&E staining and
immunohistochemistry (IHC)
The colon sections were removed following euthanasia
and cut into 3-mm sections, stained with H&E at room
temperature and imaged using light microscopy (Nikon Corporation;
magnification, ×200). The stained sections were assigned an
inflammatory score in a blinded manner, as previously described
(20). TLR4 protein expression
levels were detected by IHC. The IHC sections were incubated
overnight at 4°C in primary antibody solution containing anti-human
TLR4 antibody (cat. no. A0456; final dilution, 2 µg/l; 1:200;
OriGene Technologies, Inc.), and a biotin-streptavidin HRP
detection system for 12 h. The sections were incubated with
biotinylated goat anti-rabbit IgG (cat. no. 2019629; 1:200
dilution; OriGene Technologies, Inc.) at room temperature for 30
min, and followed by probing with an HRP-conjugated anti-rabbit IgG
secondary antibody (cat. no. 40295G; 1:200; BIOSS) at room
temperature for 30 min. Negative controls were treated with PBS
instead of primary antibodies. All sections were observed using a
light microscope (Olympus BH-2; Olympus Corporation; magnification,
×100).
Immunofluorescence
For immunofluorescence staining, colon tissues were
separated from mice. Colon tissues were fixed in PBS/4%
paraformaldehyde and 10% sucrose solution at 4°C for 1 h, followed
by cryoprotection in PBS/30% sucrose (Merck KGaA) for 3 days at
4°C. Tissue sections were cut into 20-mm sections using a Leica
Cryostat Microtome, blocked using a Streptavidin/Biotin Blocking
kit (Vector Laboratories, Inc.) according to the manufacturer's
protocol and stored at −80°C until required. Colon tissue samples
were stained with either mouse or rabbit anti-F-actin antibodies
(cat. no. 8927; 1:2,000 dilution; Merck KGaA) for 3 h at room
temperature and counterstained with DAPI (cat no. 6982; BioLegend,
Inc.) for 5 min at room temperature. Sections were observed using a
FV1000 laser-scanning confocal microscope (Olympus Corporation;
magnification, ×200).
Western blot analysis
Western blot analysis was performed following
protein extraction using RIPA lysis buffer (Beijing Solarbio
Science & Technology Co., Ltd.) according to the manufacturer's
protocol, and protein concentrations were measured using an
Enhanced Bicinchoninic Acid Protein Assay kit (Beyotime Institute
of Biotechnology). Tissue protein extracts (120 µl) were mixed with
SDS buffer (Beyotime Institute of Biotechnology) and heated at 90°C
for 5 min. Proteins (30 µg per lane) were resolved using 12%
SDS-PAGE and were subsequently transferred to PVDF membranes
(MilliporeSigma). The membranes were blocked with 5% fat-free milk
in Tris-buffered saline (TBS) for 45 min at room temperature and
washed with TBS containing Tween-20 (TBST), and then incubated with
the following primary rabbit anti-mouse antibodies: Anti-TLR4 (cat.
no. ab22048), anti-NF-κBp65 (cat. no. ab16502), anti-p-p38 (cat.
no. ab31828), anti-p38 (cat. no. ab2749) (all from Abcam; 1:200
dilution) and anti-GAPDH (cat. no. ab60004; Abcam; 1:200 dilution)
at 4°C for 16 h. The membranes were washed with TBST three times
and subsequently incubated with secondary antibodies [anti-rabbit
IgG (cat. no. sc-2357; 1:1,000 dilution) or anti-mouse IgG (cat.
no. sc-2942; 1:1,000 dilution), both from Beyotime Institute of
Biotechnology] for 2 h at room temperature. The protein bands were
visualized using an ECL Plus kit (cat. no. E1116; Beyotime
Institute of Biotechnology) and the Fusion FX7 Spectra
multifunction imaging system (Vilber Lourmat) was used to detect
bands. ImageJ version 1.8.0 (National Institutes of Health) was
used for densitometric analysis.
Reverse transcription-quantitative PCR
(RT-qPCR)
For RT-qPCR, total RNA was extracted from colon
tissues and macrophages using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. A NanoDrop 2000 spectrophotometer (Thermo
Fisher Scientific, Inc.) was used for RNA concentration analysis.
qPCR amplification was subsequently performed on an ABI 7900HT
Real-Time PCR Detection system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) to measure the mRNA expression levels. The
thermocycling conditions were as follows: Initial denaturation at
50°C for 2 min and 95°C for 10 min; followed by 40 cycles of 95°C
for 15 sec and 60°C for 60 sec. Expression levels were calculated
using the 2−ΔΔCq method (22). Relative expression levels were
normalized to GAPDH. The primer sequences for each gene were as
follows: TLR4 forward, 5′-CATGGATCAGAAACTCAGCAAAGTC-3′ and reverse,
5′-CATGCCATGCCTTGTCTTCA-3′; p38 forward, 5′-CGACTTGCTGGAGAAGATGC-3′
and reverse, 5′-TCCATCTCTTCTTGGTCAAGG-3′; NF-κB forward,
5′-AGACCTGGAGCAAGCCATTAG-3′ and reverse,
5′-CGGACCGCATTCAAGTCATAG-3′; and GAPDH forward,
5′-GACGGCCGCATCTTCTTGT-3′ and reverse,
5′-CACACCGACCTTCACCATTTT-3′.
ELISA
The levels of IL-10, TNF-α and TGF-β in colon
tissues were measured using commercially available ELISA kits
[IL-10 (cat. no. H009), TNF-α (cat. no. H052) and TGF-β (cat. no.
H034); all from Nanjing Jiancheng Bioengineering Institute]
according to the manufacturer's instructions.
Muscularis macrophage (MM) culture,
treatment and transfection
The colons of WT and Ednrb−/− mice were
carefully excised and separated from the mesentery, cleaned and
washed with Hank's Balanced Salt Solution (HBSS)
Mg2+Ca2+ (Gibco; Thermo Fisher Scientific,
Inc.). The colon tissues were opened in two, cut into 2-mm pieces
and digested with 400 U/ml collagenase D (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with HBSS
Mg2+Ca2+, 2% FBS (Gibco; Thermo Fisher
Scientific, Inc.) 1X NaPyr + 25 mM HEPES + 50 µg/ml DNase I + 2.5
U/ml dispase (Shanghai Maokang Biotechnology Co., Ltd.) at 4°C for
20 min. The digested tissue was homogenized and washed.
Subsequently, the dissociated tissue was filtered through a 70-µm
mesh cell strainer, washed with HBSS Mg2+Ca2+
and incubated with PBS containing 1% BSA, 10 mM EDTA, 0.02% sodium
azide containing Fc block and antibodies against CD16/CD32 (cat.
no. A0847; 1:200 dilution) (all from Gibco; Thermo Fisher
Scientific, Inc.) at 4°C for 30 min (23), washed and stained with
fluorophore-conjugated antibodies in PBS/2% FBS for 30 min at 4°C.
The obtained MMs were maintained in DMEM (Gibco; Thermo Fisher
Scientific, Inc.). siRNAs targeting TLR4 for knockdown TLR4
expression in Ednrb−/− MMs, and corresponding siRNA-NC
for WT MMs, synthesized by Guangzhou RiboBio Co., Ltd. were
transfected into the cells. The cell groups included: WT+ siRNA-NC
group, WT group, Ednrb−/− group and Ednrb−/−
+ si-TLR4 group. A total of 2 µg/ml siRNA was transfected into MMs
at 37°C for 10 h using Lipofectamine® 2000 transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. At 48 h post-transfection, the
WT+siRNA-NC MMs were supplemented with 10% PBS, while the WT,
Ednrb−/− and Ednrb−/−+si-TLR4 MMs were
supplemented with 100 ng/ml E. coli JM83. All cells (2,000
cells per well) were seeded in 96-well culture plates. WT+E.
coli and Ednrb−/−+E. coli were control
groups. Cell Counting Kit-8 reagent was used according to the
manufacturer's instructions (Beyotime Institute of Biotechnology)
after 0, 24 or 48 h to assess cell proliferation. The expression of
TLR4/p-p38/NF-κB signaling pathway members was assessed by western
blot analysis and RT-qPCR, and the collected media were analyzed
for IL-10, TNF-α and TGF-β protein levels by ELISA.
Statistical analysis
SPSS version 19.0 (IBM Corp.) and GraphPad Prism
version 8.0 (GraphPad software Inc.) were used for statistical
analysis. Normally distributed data are presented as the mean ±
standard deviation and were statistically analyzed using one-way
ANOVA, followed by Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
The TLR4/p-p38/NF-κB signaling pathway
participates in the pathogenesis of HAEC in Ednrb−/−
mice
The Ednrb-null mouse (Fig. S1A and B) was used to resemble the
pathological features of HSCR, including aganglionosis in the
rectum and distal colon, as well as enterocolitis (21). First, WT and Ednrb−/−
mice were infected with E. coli JM83 and inflammation scores
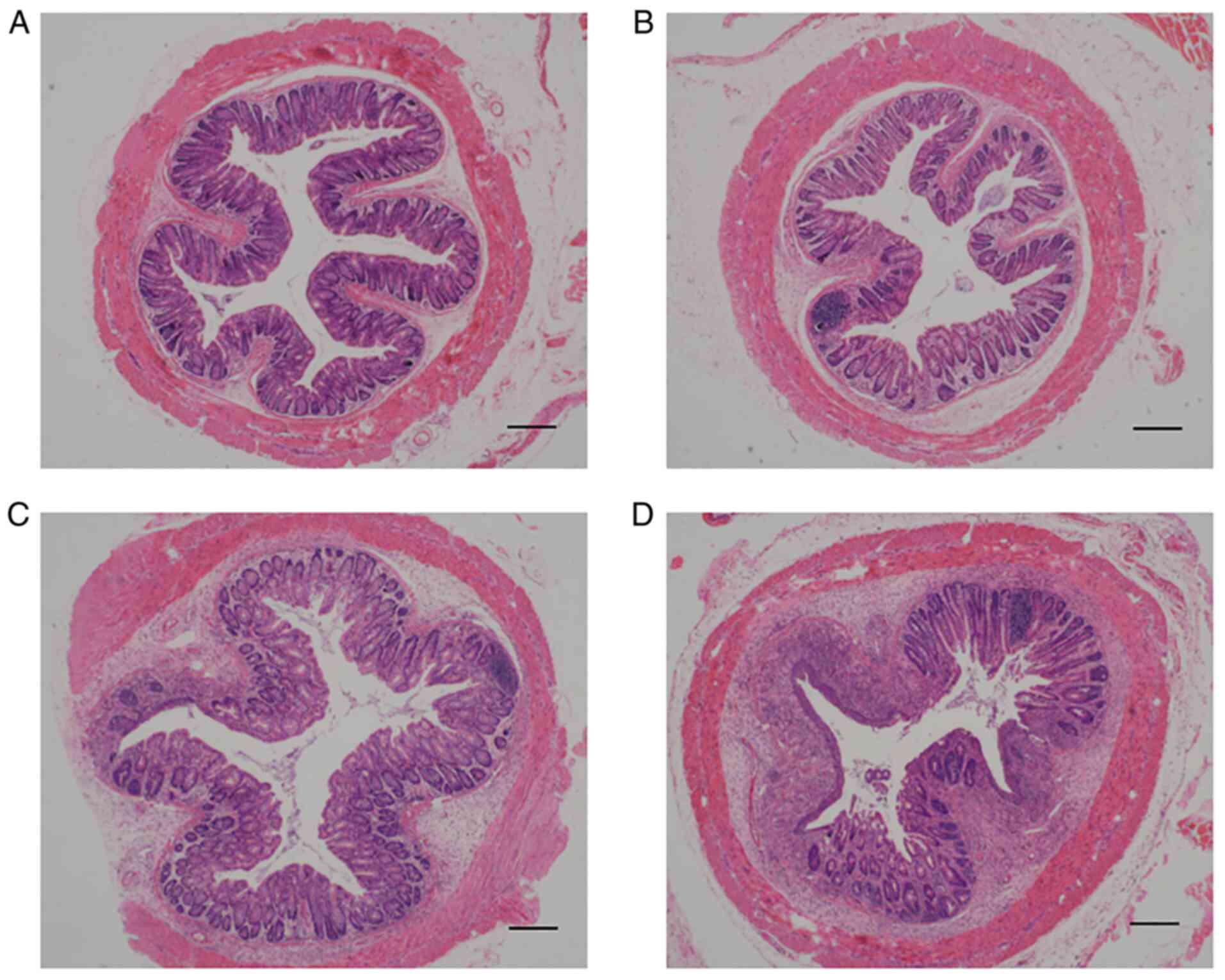
in the colon were histologically assessed. As presented in Fig. 1, naive Ednrb−/− mice
developed enterocolitis spontaneously after 5 weeks (Fig. 1C), whereas Ednrb−/−
mice infected with E. coli devloped enterocolitis after 3
weeks. The degree of epithelial damage and leukocyte infiltration
in E. coli-infected Ednrb−/− mice were more than
naive Ednrb−/− mice when they were sacrificed at 5 weeks
(Fig. 1D). By contrast, WT mice
infected with E. coli (Fig.
1B) only developed mild inflammation in the colon compared with
WT mice (Fig. 1A).
Ednrb increases TLR4/p-p38/NF-κB
signaling to promote enterocolitis
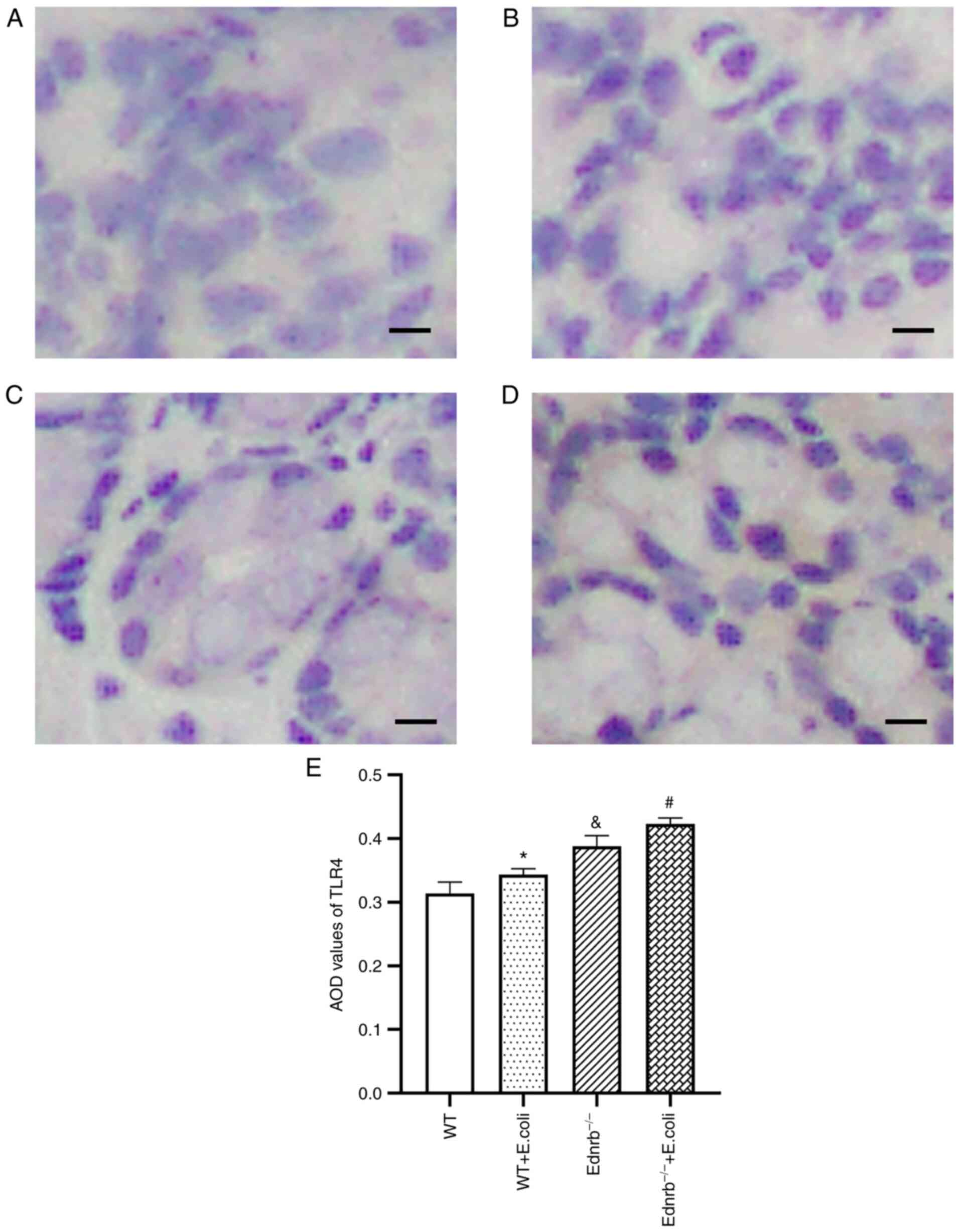
As presented in Fig.
2, IHC analysis revealed intense brown staining, resembling
TLR4 expression in the colon of Ednrb−/− (Fig. 2C) and E. coli-infected
Ednrb−/− mice (Fig.
2D), compared with WT (Fig.
2A) and E. coli-infected WT mice (Fig. 2B). The average optical density
values of TLR4 in E. coli-infected Ednrb−/− mice
were higher than those in the Ednrb−/− and WT mice
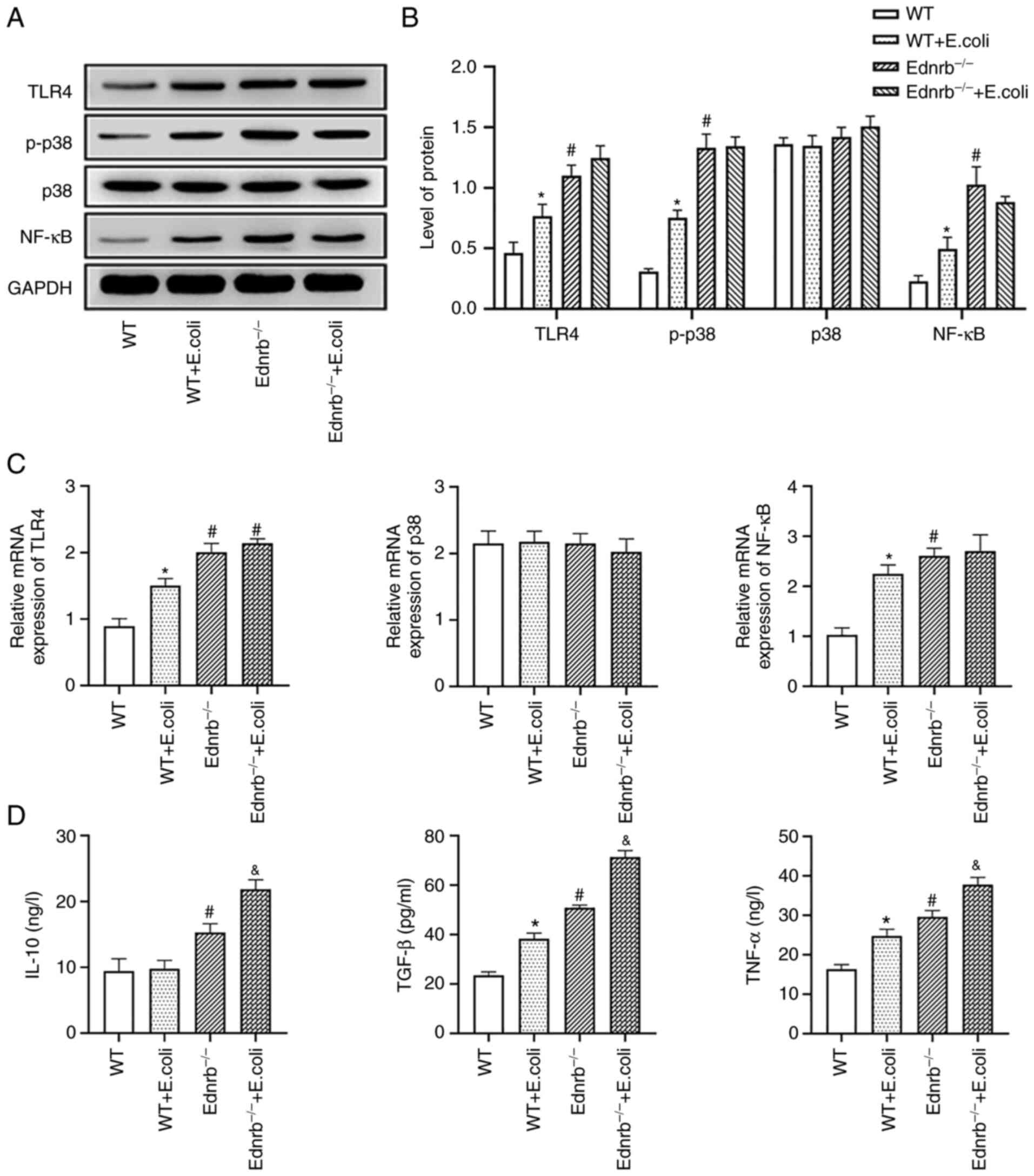
(Fig. 2E). Western blot analysis
and RT-qPCR further confirmed that the protein and mRNA levels of
TLR4, NF-κB and p-p38 were significantly increased in
Ednrb−/− and E. coli-infected Ednrb−/−
mice (P<0.05 vs. WT and E. coli-infected WT mice), with
no significant difference identified between the
Ednrb−/− and E. coli-infected Ednrb−/−
mice (P>0.05; Fig. 3A-C). In
addition, the expression levels of TNF-α, TGF-β and IL-10 were
increased in Ednrb−/− and E. coli-infected
Ednrb−/− mice, compared with those in WT and E.
coli-infected WT mice (P<0.05; Fig. 3D). Of note, E.
coli-infected Ednrb−/− mice exhibited markedly
higher TNF-α, TGF-β and IL-10 levels than Ednrb−/− mice
(P<0.05; Fig. 3D).
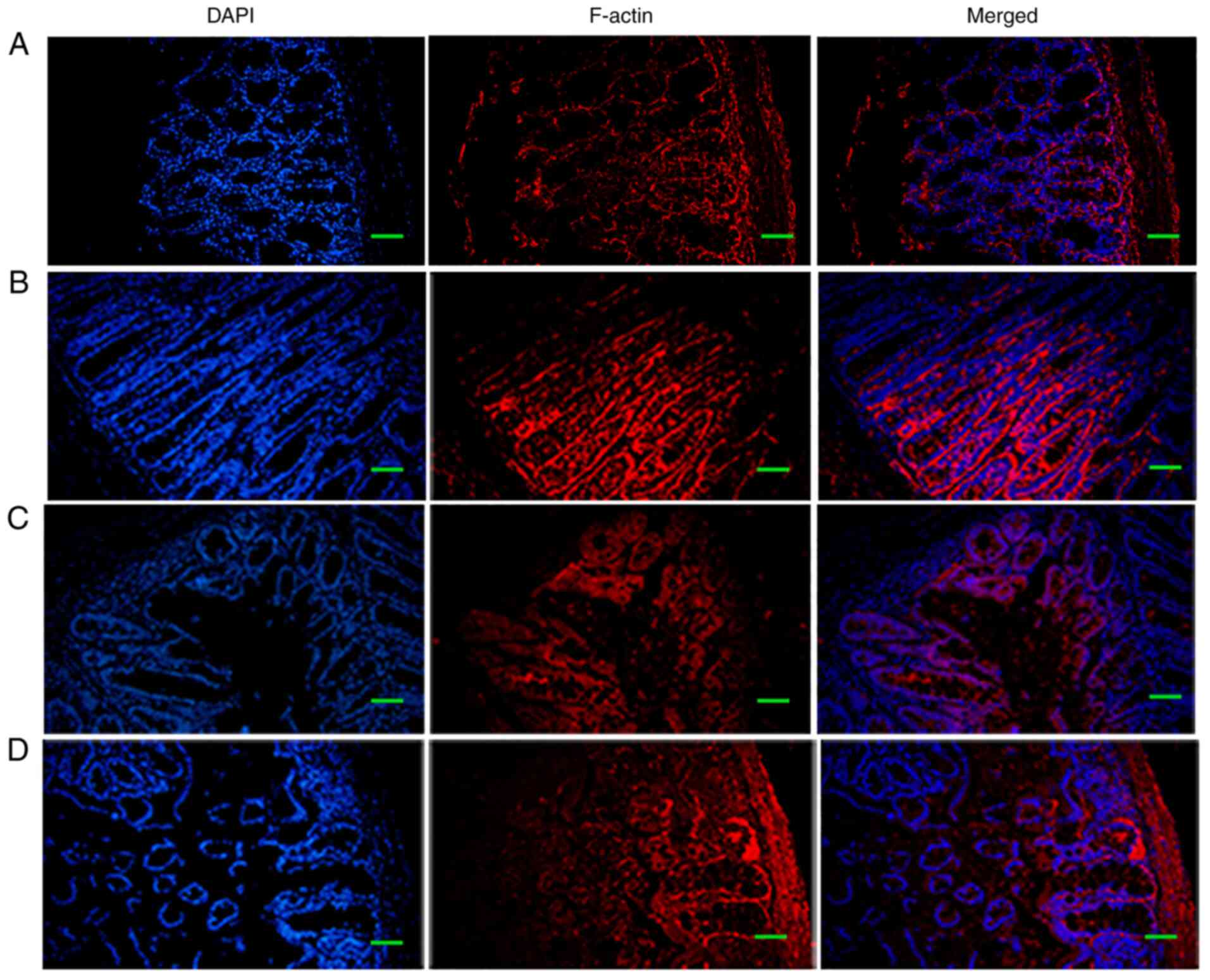
TLR4 knockdown reverses intestinal
inflammation
To assess the extent of damage to the intestinal
mucosal barrier and the role of TLR4/p-p38/NF-κB signaling in E.
coli JM83 infection-induced HAEC, the severity of enterocolitis
and cytoskeletal F-actin expression was assessed following TLR4
knockdown in WT and Ednrb−/− mice. It has been reported
that changes in F-actin in the intestinal epithelial cytoskeleton
may occur as a mechanism of damage to intestinal barrier function
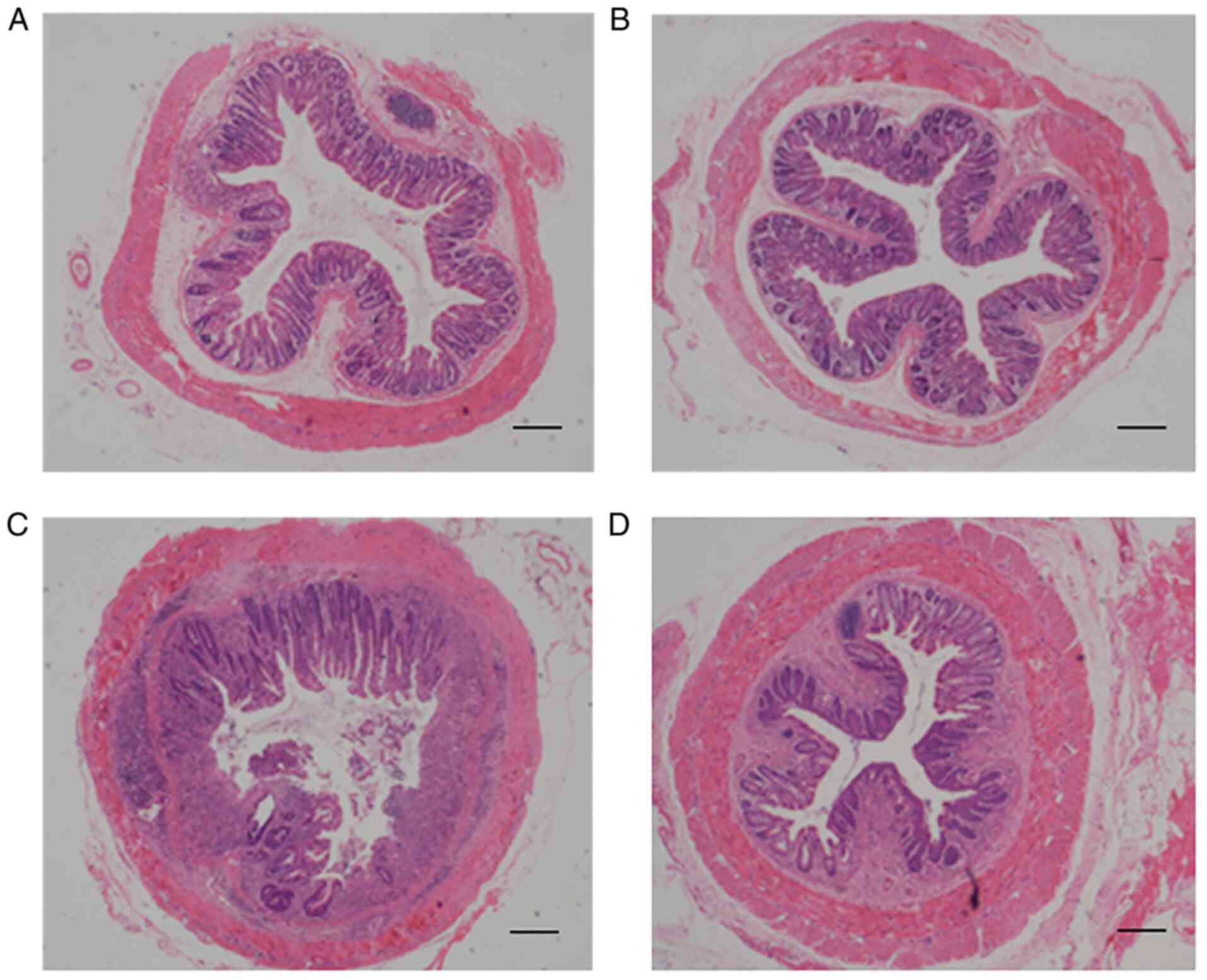
(24). As indicated in Fig. 4, a marked interruption in mucosal
structures was present, and large numbers of inflammatory cells and
abscesses were observed to have infiltrated the mucosa and
sub-mucosa upon E. coli infection in Ednrb−/−
mice (Fig. 4C), whereas the
severity of E. coli infection-induced enterocolitis was
markedly alleviated in Ednrb−/− mice following TLR4
knockdown (Fig. 4D). In addition,
the mild degree of inflammation in the colon observed in E.
coli-infected WT+siRNA-NC mice (Fig. 4A) was almost completely reversed
by TLR4 knockdown (Fig. 4A).
F-actin expression was increased in the cytoplasm of intestinal
epithelial cells, alongside increased tight junction integrity in
the intestinal mucosal barrier in both WT+siRNA-NC and
si-TLR4-transfected WT mice 5 weeks after E. coli infection
(Fig. 5A and B). By contrast,
Ednrb−/− mice exhibited a substantially decreased
density of F-actin protein and severely disordered tight junction
structures in response to E. coli infection (Fig. 5C). Of note, TLR4 knockdown in
Ednrb−/− mice gradually increased the density of F-actin
and partly reversed tight junction integrity (Fig. 5D).
TLR4 knockdown reduces inflammatory
response in Ednrb−/− mice and suppresses downstream
TLR4/p-p38/NF-κB signaling
To ascertain whether TLR4/p-p38/NF-κB signaling was
critical to the progression of HAEC, TLR4-mediated downstream
pathways and inflammatory cytokines were examined following TLR4
knockdown in WT and Ednrb−/− mice after E. coli
infection. The activation of downstream TLR4/p-p38/NF-κB signaling
pathways, including NF-κB and p-p38/p38, was further examined. As
presented in Fig. 6A,
transfection of both WT and Ednrb−/− mice with si-TLR4
efficiently knocked down TLR4 protein expression. The TLR4 protein
was significantly upregulated in E. coli-infected
Ednrb−/− mice, which were subjected to a rescue
experiment (P<0.05 vs. WT+siRNA-NC and E. coli-infected
Ednrb−/−+si-TLR4 mice). Of note, TLR4 knockdown in E.
coli-infected Ednrb−/− mice markedly suppressed
NF-κB and p-p38 expression at the protein level (P<0.05 vs.
E. coli-infected Ednrb−/− mice; Fig. 6B and C). Furthermore, TLR4
knockdown significantly attenuated TNF-α, TGF-β and IL-10
expression in E. coli-infected Ednrb−/− mice
(P<0.05 vs. E. coli-infected Ednrb−/− mice;
Fig. 6D). However, the expression
levels of p-p38, TNF-α, TGF-β and IL-10 were increased in
si-TLR4-transfected E. coli-infected Ednrb−/−
mice, compared with those in si-TLR4-transfected E.
coli-infected WT mice (P<0.05; Fig. 6C and D).
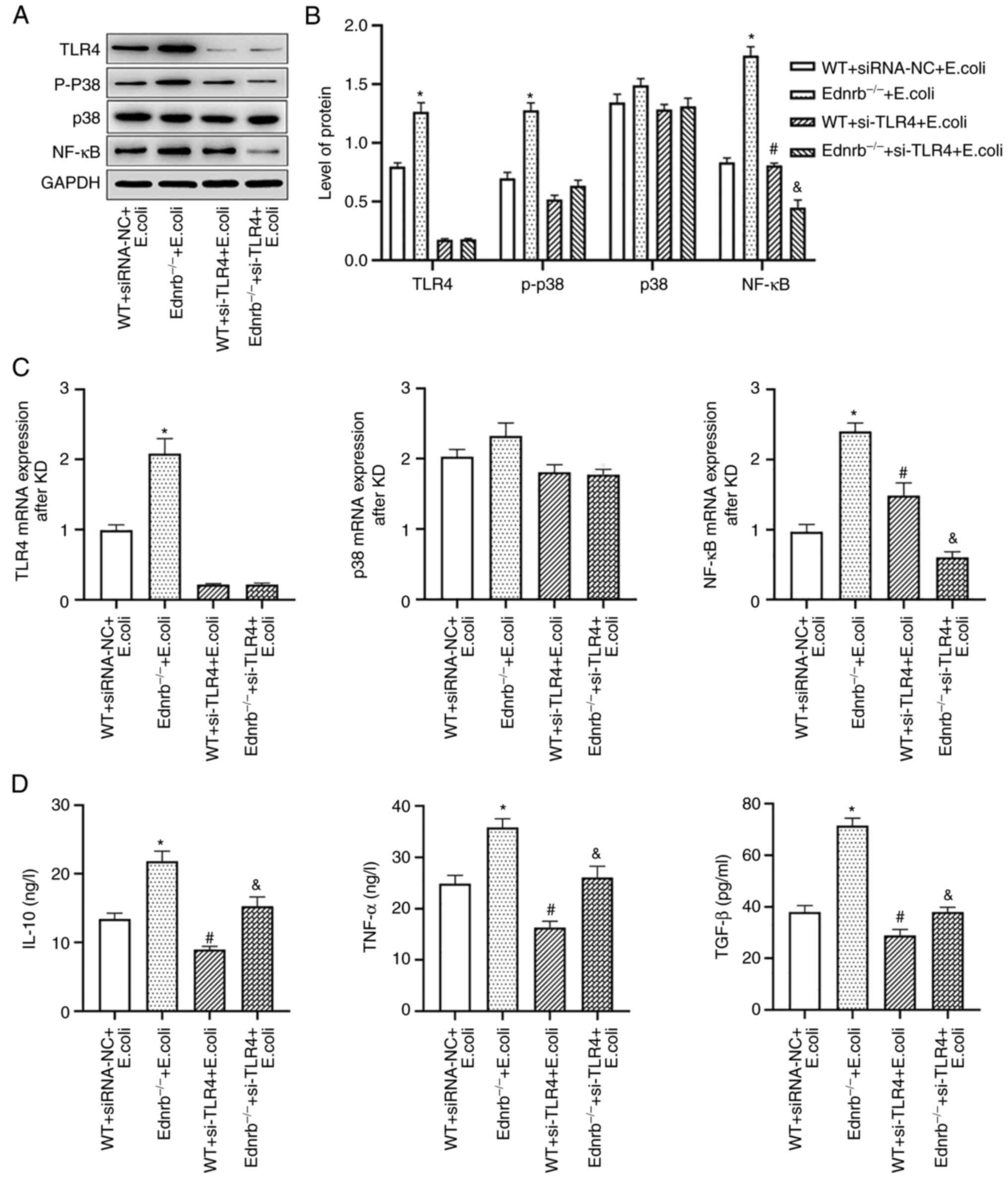 | Figure 6.TLR4 knockdown inhibits the secretion
of intestinal inflammatory cytokines. (A and B) Western blot
analysis of TLR4, p-p38, p38 and NF-κB protein expression levels in
colonic tissues. (A) Representative western blot image and (B)
quantified expression levels. (C) mRNA expression levels of TLR4,
p38 and NF-κB in colonic tissues. (D) Secreted TNF-α, TGF-β and
IL-10 expression in the colonic tissues. *P<0.05 vs.
WT+siRNA-NC+E. coli, Ednrb−/−+E. coli and
si-TLR4-transfected Ednrb−/−+E. coli groups;
#P<0.05 vs. si-TLR4-transfected
Ednrb−/−+E. coli group; &P<0.05
vs. WT+siRNA-NC+E. coli, si-TLR4-transfected WT+E.
coli and Ednrb−/−+E. coli groups (n=5 per
group). Ednrb, endothelin receptor B; WT, wild-type; p-,
phosphorylated; siRNA, small interfering RNA; NC, negative control;
si-TLR4, siRNA targeting TLR4; TLR, Toll-like receptor. |
Ednrb suppresses MM proliferation via
activation of TLR4/p-p38/NF-κB signaling to promote
inflammation
As presented in Fig.
7A-D, a large number of pseudopods were observed in both WT and
si-TLR4-transfected Ednrb−/− MMs in response to E.
coli infection compared with the WT+siRNA-NC group. By
contrast, a small number of pseudopods was present in
Ednrb−/− MMs upon E. coli infection. The
proliferative ability of MMs in the Ednrb−/− group was
significantly decreased compared with that in the
Ednrb−/−+si-TLR4 group (P<0.05; Fig. 7E). WT MMs exhibited upregulated
expression of NF-κB and p-p38 with significantly increased TGF-β
and TNF-α levels, but diminished IL-10 levels in response to E.
coli infection compared with WT+siRNA-NC MMs (P<0.05;
Fig. 8A-C). Of note, in the
presence of E. coli infection, Ednrb−/− MMs
exhibited downregulated expression of NF-κB and p-p38, as well as
lower TNF-α and TGF-β levels and increased IL-10 levels (P<0.05
vs. WT and Ednrb−/−+si-TLR4; Fig. 8B and C). Knockdown of TLR4 in
Ednrb−/− MMs resulted in substantially increased levels
of TNF-α and TGF-β, and decreased levels of IL-10 upon E.
coli infection (P<0.05 vs. Ednrb−/−; Fig. 8B and C).
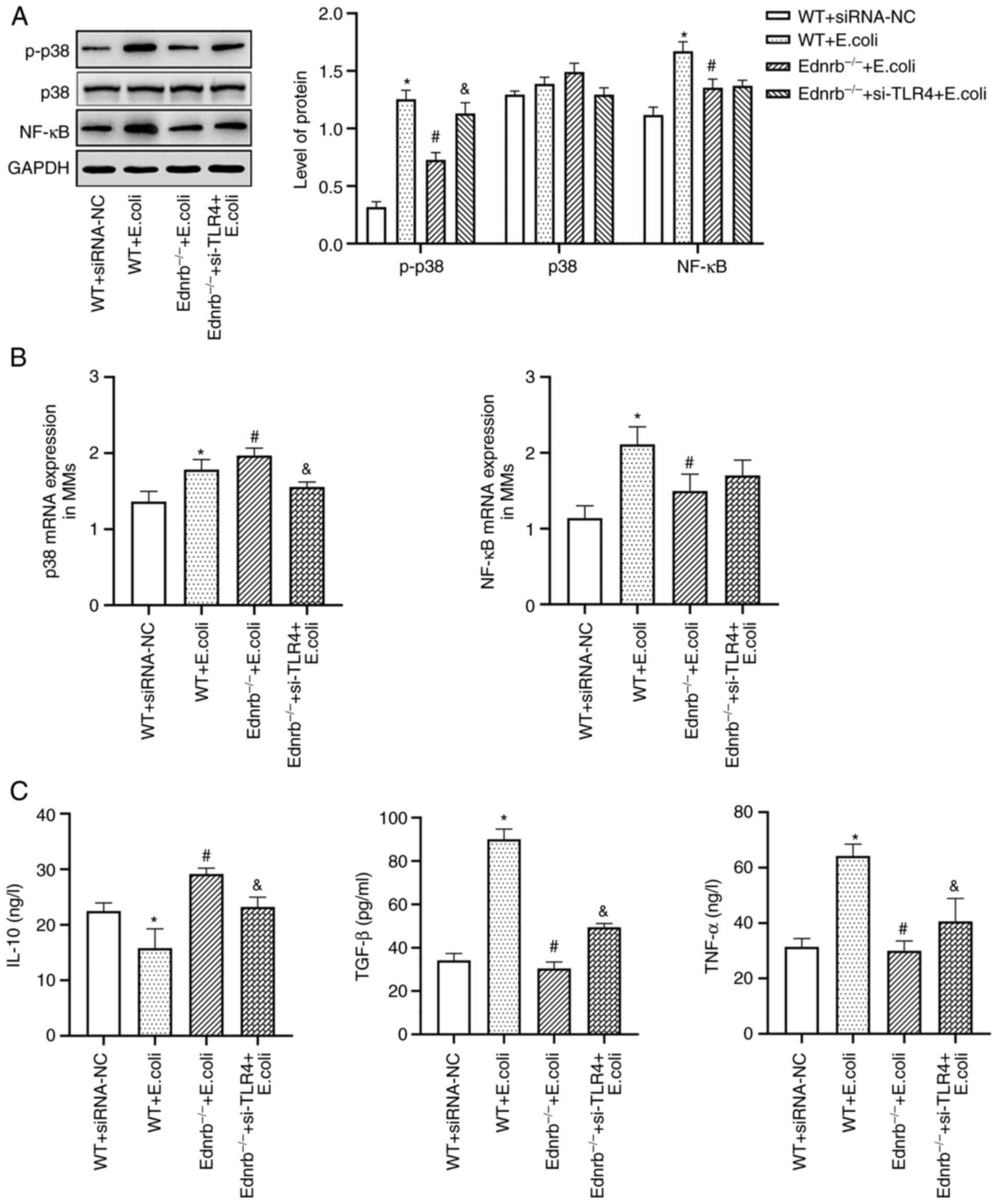 | Figure 8.TLR4/p-p38/NF-κB signaling is
primarily responsible for induction of the anti-inflammatory
activity in MMs. (A) Protein expression levels of NF-κB, p38 and
p-p38. (B) mRNA levels of p38 and NF-κB. (C) Secretion levels of
TNF-α, TGF-β and IL-10 in the culture supernatants. *P<0.05 vs.
WT+siRNA-NC+E. coli, Ednrb−/−+E. coli and
si-TLR4-transfected Ednrb−/−+E. coli groups;
#P<0.05 vs. si-TLR4-transfected
Ednrb−/−+E. coli group; &P<0.05
vs. WT+siRNA-NC, WT+E. coli and Ednrb−/−+E.
coli groups (n=5 per group). Ednrb, endothelin receptor B; TLR,
Toll-like receptor; NC, negative control; si-TLR4, siRNA targeting
TLR4; WT, wild-type; p-, phosphorylated; siRNA, small interfering
RNA; MM, Muscularis macrophage. |
Discussion
HAEC may occur at any time prior to, during or even
after endorectal pull-through surgery, which is the definitive
procedure for HSCR (25). There
exists a wide variation in the reported incidence of HAEC, which
occurs in 2–33% of patients with common-type and 50% of patients
with long-type HSCR (4).
Clinically, HAEC is characterized by discomfort, a loss of
appetite, abdominal distention, loose foul-smelling stools, fever
and sepsis (26). Previous
studies suggested that postoperative HAEC was related to surgical
factors, such as anastomotic stricture or leak, and bowel
obstructions (1,5,11,27–29). More recent studies have indicated
that the pathogenesis of HAEC is related to the mucosal barrier,
intestinal microbiota and immune function (10,30,31). At least partly due to the wide use
of Ednrb−/− animal models in HAEC research, the order in
which these HAEC-related etiological features change is gradually
becoming increasingly understood, which has promoted improvements
in the treatment and prevention of HAEC (32–34). A previous multicenter study
determined that HAEC patients exhibited a reduced abundance of the
phyla Firmicutes and an increased abundance of the phyla
Bacteroidetes and Proteobacteria, by comparing the
bacterial microbiome composition of pediatric patients with HSCR to
those who had a history of HAEC (9). These results strongly indicate a
dysequilibrium in the gut microbial ecosystem of patients with
HAEC, such that the dominance of bacteria (E. coli)
predisposes a patient to the development of HAEC.
In the present study, E. coli JM83 was used
as the pathogenic bacterium to infect the intestines of
Ednrb−/− mice to establish a mouse model of HAEC. A
previous study reported that Ednrb−/− mice developed
HAEC on post-natal days 24–26 and 100% mortality was recorded by
day 28 after birth (32).
Clinical histopathological features of patients with HAEC mainly
include colon crypt dilatation, mucin retention, enterocyte
adherence of bacteria, epithelial damage, leukocyte infiltration
and ulceration, and in the terminal stages, transmural necrosis and
perforation (35,36). In the present study, the
histopathological results indicated that a certain amount of
inflammatory cells infiltrated the mucosa and submucosa of the
intestinal wall, and even abscesses were observed in E. coli
JM83-infected Ednrb−/− mice, in accordance with the
manifestation observed in humans. Ten Ednrb−/− mice
developed HAEC 3 weeks after E. coli JM83 infection; eight
mice survive for more than 5 weeks, with two mice dying of
abdominal distention, diarrhea and dehydration; the 80% survival
rate was higher than that reported in a previous study (20).
A previous study indicated passive transport of
E. coli through the mucosal barrier in Ednrb−/−
mice and E. coli transport was significantly reduced in the
proximal colon compared with the distal colon (37). Previous studies also suggested
that the dysfunction of the intestinal epithelium contributed to
the reduction in expression and changes in distribution of F-actin,
influencing barrier function and increasing permeability (24,38). The results of the present study
indicated that the expression of F-actin gradually decreased with
the activation of the TLR4/p-p38/NF-κB signaling pathway and thus
eventually led to intestinal mucosal damage, and the levels of
IL-10 gradually increased, consistent with the degree of intestinal
barrier damage. These results are supported by those of previous
studies (39,40), suggesting that IL-10 is a
pleiotropic cytokine, the activity of which attempts to limit
inflammatory responses. TNF-α and TGF-β expression were also
significantly increased in HAEC mice in the present study, which
supported the finding of a previous study that the secretion of
pro-inflammatory cytokines, such as TNF-α, IFN-γ, TGF-β and IL-6,
and other inflammatory mediators and the resultant cascade
reactions aggravate inflammation and destroy intestinal barrier
function in patients with HAEC (41). Certain studies have indicated that
IL-10 decreased the secretion of TNF-α. However, the high levels of
TNF-α reported in inflammatory bowel diseases are produced by
recruitment of immune cells rather than by resident colitogenic
cells (41,42). Studies have reported that TLR4
activated by bacteria may be a major mediator of activating
intestinal mucosal immunity, progression of intestinal inflammation
and promotion of the immune response (16,43). Therefore, the results of the
present study correspond with the fact that HAEC may occur after
postoperative pull-through surgery.
The TLR4/p-p38/NF-κB signaling pathway is
transmitted through adaptor proteins and signaling through MyD88
may be necessary to drive phagocytosis (15,44,45). Studies have indicated that the
primary function of TLR4 signaling in macrophages is to induce
inflammatory responses and protect the host from pathogenic
bacteria (19,46). The in vivo experiments of
the present study revealed that TLR4 protein expression was
upregulated in the colon for 3 weeks following stimulation with
E. coli JM83. TLR4 stimulated NF-κB through MyD88 in a mouse
model and the levels of NF-κB and p-p38/p38 were increased in the
colon wall following stimulation with E. coli JM83.
Likewise, NF-κB induced an increase in TNF-α and TGF-β expression
with the degree of enterocolitis in Ednrb−/− mice, and
therefore eventually led to intestinal mucosal damage. The above
results indicate that the TLR4/p-p38/NF-κB signaling pathway has a
central role in the initiation of innate cellular immune responses
and in the development of subsequent adaptive immune responses to
invading bacterial infection, and eventually promote intestinal
mucosal tissue damage in HAEC. This process is consistent with the
pathogenesis of inflammatory bowel disease in previous adult
studies (47,48). By contrast, mucosal barrier
integrity was maintained without the development of enterocolitis
following TLR4 knockdown. A previous study indicated that
upregulated TLR4 expression is related to mortality in a model of
sepsis (46).
However, siRNA-mediated knockdown of TLR4 to
inhibit TLR4/p-p38/NF-κB signaling reversed the inflammatory
effects caused by E. coli infection, indicating that
TLR4/p-p38/NF-κB signaling has a central role in maintaining the
balance of gut homeostasis during the pathogenesis of HAEC. Under
certain conditions, this downregulation of TLR4 signaling may
ameliorate the degree of immune-mediated enterocolitis, providing a
novel idea for the treatment and prevention of HAEC.
The limitation of the present study was that it did
not analyze the side effects of inhibition of the TLR4/p-p38/NF-κB
signaling pathway. Since the TLR4 receptor is expressed in a number
of cells, we aim to focus on intestine-specific inhibitors of the
TLR4/p-p38/NF-κB pathway and assess their protective effect on HAEC
in future studies.
In conclusion, the present study highlighted the
response of the intestinal mucosal barrier to HAEC induced by
pathogenic E. coli. In addition, the activation of
TLR4/p-p38/NF-κB signaling in Ednrb−/− mice by E.
coli JM83 led to the development of inflammation and the
underlying mechanism was indicated to be this signaling pathway.
Furthermore, inhibition of TLR4/p-p38/NF-κB signaling may be of
potential benefit for the treatment and prevention of HAEC,
highlighting a novel means for improving intestinal mucosal
integrity.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Joint Fund of the Department of
Guizhou Science and Technology of China (Guizhou, China; grant nos.
20177100, 20204Y005 and ZK2021361).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
ZZ performed the majority of experiments, analyzed
the data and wrote, reviewed and edited the manuscript. MG, CT, LH
and YG curated and analyzed the data. JW and YL conceived and
designed the experiments and contributed to the analytical tools.
All of the authors have read and approved the final manuscript. JW
and YL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experimental protocols complied with the
Guide for the Care and Use of Laboratory Animals published by the
National Institutes of Health. The present study was approved by
the Institutional Animal Research Committee of Zunyi Medical
University (Guizhou, China; approval no. IACUC-20191025028).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HAEC
|
Hirschsprung-associated
enterocolitis
|
|
HSCR
|
Hirschsprung disease
|
|
TLR4
|
Toll-like receptor 4
|
|
WT
|
wild-type
|
|
Ednrb
|
endothelin receptor B
|
References
|
1
|
Le-Nguyen A, Righini-Grunder F, Piché N,
Faure C and Aspirot A: Factors influencing the incidence of
Hirschsprung associated enterocolitis (HAEC). J Pediatr Surg.
54:959–963. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakamura H, Lim T and Puri P: Probiotics
for the prevention of Hirschsprung-associated enterocolitis: A
systematic review and meta-analysis. Pediatr Surg Int. 34:189–193.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fang YF, Bai JX, Zhang B, Wu DM, Lin Y and
Liu MK: Laparoscopic Soave procedure for long-segment
Hirschsprung's disease-single-center experience. Videosurgery
Miniinv. 15:234–238. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Austin KM: The pathogenesis of
Hirschsprung's disease-associated enterocolitis. Semin Pediatr
Surg. 21:319–327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng S, Wang J, Pan W, Yan W, Shi J, Guan
W, Wang Y and Cai W: Pathologically assessed grade of
Hirschsprung-associated enterocolitis in resected colon in children
with Hirschsprung's disease predicts postoperative bowel function.
J Pediatr Surg. 52:1776–1781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frykman PK, Nordenskjöld A, Kawaguchi A,
Hui TT, Granström AL, Cheng Z, Tang J, Underhill DM, Iliev I,
Funari VA, et al: Characterization of bacterial and fungal
microbiome in children with Hirschsprung disease with and without a
history of enterocolitis: A multicenter study. PLoS One.
10:e01241722015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Poroyko V, Yan Z, Pan L, Feng Y,
Zhao P, Xie Z and Hong L: Characterization of intestinal
microbiomes of Hirschsprung's disease patients with or without
enterocolitis using illumina-MiSeq high-throughput sequencing. PLoS
One. 11:e01620792016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neuvonen MI, Korpela K, Kyrklund K,
Salonen A, de Vos W, Rintala RJ and Pakarinen MP: Intestinal
microbiota in Hirschsprung disease. J Pediatr Gastroenterol Nutr.
67:594–600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prato AP, Bartow-McKenney C, Hudspeth K,
Mosconi M, Rossi V, Avanzini S, Faticato MG, Ceccherini I, Lantieri
F, Mattioli G, et al: A metagenomics study on Hirschsprung's
disease associated enterocolitis: Biodiversity and gut microbial
homeostasis depend on resection length and patient's clinical
history. Front Pediatr. 7:3262019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singer G, Kashofer K, Castellani C and
Till H: Hirschsprung's associated enterocolitis (HAEC) personalized
treatment with probiotics based on gene sequencing analysis of the
fecal microbiome. Case Rep Pediatr. 2018:32923092018.PubMed/NCBI
|
|
11
|
Tang W, Su Y, Yuan C, Zhang Y, Zhou L,
Peng L, Wang P, Chen G, Li Y, Li H, et al: Prospective study
reveals a microbiome signature that predicts the occurrence of
post-operative enterocolitis in Hirschsprung disease (HSCR)
patients. Gut Microbes. 11:842–854. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bigorgne AE, John B, Ebrahimkhani MR,
Shimizu-Albergine M, Campbell JS and Crispe IN: TLR4-dependent
secretion by hepatic stellate cells of the
neutrophil-chemoattractant CXCL1 mediates liver response to gut
microbiota. PLoS One. 11:e01510632016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cheng Y, Zhu Y, Huang X, Zhang W, Han Z
and Liu S: Association between TLR2 and TLR4 gene polymorphisms and
the susceptibility to inflammatory bowel disease: A meta-analysis.
PLoS One. 10:e01268032015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Płóciennikowska A, Hromada-Judycka A,
Borzęcka K and Kwiatkowska K: Co-operation of TLR4 and raft
proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life
Sci. 72:557–581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rosadini CV, Zanoni I, Odendall C, Green
ER, Paczosa MK, Philip NH, Brodsky IE, Mecsas J and Kagan JC: A
single bacterial immune evasion strategy dismantles both MyD88 and
TRIF signaling pathways downstream of TLR4. Cell Host Microbe.
18:682–693. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leaphart CL, Cavallo J, Gribar SC, Cetin
S, Li J, Branca MF, Dubowski TD, Sodhi CP and Hackam DJ: A critical
role for TLR4 in the pathogenesis of necrotizing enterocolitis by
modulating intestinal injury and repair. J Immunol. 179:4808–4820.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu S, Gallo DJ, Green AM, Williams DL,
Gong X, Shapiro RA, Gambotto AA, Humphris EL, Vodovotz Y and
Billiar TR: Role of toll-like receptors in changes in gene
expression and NF-kappa B activation in mouse hepatocytes
stimulated with lipopolysaccharide. Infect Immun. 70:3433–3442.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gibson DL, Ma C, Rosenberger CM, Bergstrom
KS, Valdez Y, Huang JT, Khan MA and Vallance BA: Toll-like receptor
2 plays a critical role in maintaining mucosal integrity during
citrobacter rodentium-induced colitis. Cell Microbiol. 10:388–403.
2008.PubMed/NCBI
|
|
19
|
Zhang J, Zheng Y, Luo Y, Du Y, Zhang X and
Fu J: Curcumin inhibits LPS-induced neuroinflammation by promoting
microglial M2 polarization via TREM2/TLR4/NF-κB pathways in BV2
cells. Mol Immunol. 116:29–37. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Porokuokka LL, Virtanen HT, Lindén J,
Sidorova Y, Danilova T, Lindahl M, Saarma M and Andressoo JO: Gfra1
underexpression causes Hirschsprung's disease and associated
enterocolitis in mice. Cell Mol Gastroenterol Hepatol. 7:655–678.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frykman PK, Cheng Z, Wang X and Dhall D:
Enterocolitis causes profound lymphoid depletion in endothelin
receptor B- and endothelin 3-null mouse models of
Hirschsprung-associated enterocolitis. Eur J Immunol. 45:807–817.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TJ: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Muller PA, Koscsó B, Rajani GM, Stevanovic
K, Berres ML, Hashimoto D, Mortha A, Leboeuf M, Li XM, Mucida D, et
al: Crosstalk between muscularis macrophages and enteric neurons
regulates gastrointestinal motility. Cell. 158:300–313. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ye X and Sun M: AGR2 ameliorates tumor
necrosis factor-α-induced epithelial barrier dysfunction via
suppression of NF-κB p65-mediated MLCK/p-MLC pathway activation.
Int J Mol Med. 39:1206–1214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mao YZ, Tang ST and Li S: Duhamel
operation vs transanal endorectal pull-through procedure for
Hirschsprung disease: A systematic review and meta-analysis. J
Pediatr Surg. 53:1710–1715. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dore M, Vilanova Sanchez A, Triana Junco
P, Barrena S, De Ceano-Vivas M, Jimenez Gomez J, Andres Moreno AM,
Lopez Santamaria M and Martinez L: Reliability of the
Hirschsprung-associated enterocolitis score in clinical practice.
Eur J Pediatr Surg. 29:132–137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang X, Li L, Li SL, Li SX, Wang XY and
Tang ST: Primary laparoscopic endorectal pull-through procedure
with or without a postoperative rectal tube for Hirschsprung
disease: A multicenter perspective study. J Pediatr Surg.
55:381–386. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pruitt LCC, Skarda DE, Rollins MD and
Bucher BT: Hirschsprung-associated enterocolitis in children
treated at US children's hospitals. J Pediatr Surg. 55:535–540.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taylor MA, Bucher BT, Reeder RW, Avansino
JR, Durham M, Calkins CM, Wood RJ, Levitt MA, Drake K and Rollins
MD: Comparison of Hirschsprung disease characteristics between
those with a history of postoperative enterocolitis and those
without: Results from the pediatric colorectal and pelvic learning
consortium. Eur J Pediatr Surg. 31:207–213. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng Z, Zhao L, Dhall D, Ruegger PM,
Borneman J and Frykman PK: Bacterial microbiome dynamics in post
pull-through Hirschsprung-associated enterocolitis (HAEC): An
experimental study employing the endothelin receptor B-null mouse
model. Front Surg. 5:302018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Halleran DR, Ahmad H, Maloof E, Paradiso
M, Lehmkuhl H, Minneci PC, Levitt MA and Wood RJ: Does
Hirschsprung-associated enterocolitis differ in children with and
without down syndrome? J Surg Res. 245:564–568. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng Z, Wang X, Dhall D, Zhao L, Bresee
C, Doherty TM and Frykman PK: Splenic lymphopenia in the endothelin
receptor B-null mouse: Implications for Hirschsprung associated
enterocolitis. Pediatr Surg Int. 27:145–150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fattahi F, Steinbeck JA, Kriks S, Tchieu
J, Zimmer B, Kishinevsky S, Zeltner N, Mica Y, El-Nachef W, Zhao H,
et al: Deriving human ENS lineages for cell therapy and drug
discovery in Hirschsprung disease. Nature. 531:105–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soret R, Schneider S, Bernas G,
Christophers B, Souchkova O, Charrier B, Righini-Grunder F, Aspirot
A, Landry M, Kembel SW, et al: Glial cell-derived neurotrophic
factor induces enteric neurogenesis and improves colon structure
and function in mouse models of Hirschsprung disease.
Gastroenterology. 159:1824–1838.e17. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gosain A, Frykman PK, Cowles RA, Horton J,
Levitt M, Rothstein DH, Langer JC and Goldstein AM; American
Pediatric Surgical Association Hirschsprung Disease Interest Group,
: Guidelines for the diagnosis and management of
Hirschsprung-associated enterocolitis. Pediatr Surg Int.
33:517–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mattar AF, Coran AG and Teitelbaum DH:
MUC-2 mucin production in Hirschsprung's disease: Possible
association with enterocolitis development. J Pediatr Surg.
38:417–421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yildiz HM, Carlson TL, Goldstein AM and
Carrier RL: Mucus Barriers to microparticles and microbes are
altered in Hirschsprung's disease. Macromol Biosci. 15:712–718.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song H, Zhang J, He W, Wang P and Wang F:
Activation of cofilin increases intestinal permeability via
depolymerization of F-actin during hypoxia in vitro. Front Physiol.
10:14552019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu D, Yin X, Olyha SJ, Nascimento MSL,
Chen P, White T, Gowthaman U, Zhang T, Gertie JA, Zhang B, et al:
IL-10-dependent crosstalk between murine marginal zone B cells,
macrophages, and CD8α+ dendritic cells promotes listeria
monocytogenes infection. Immunity. 51:64–76.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zigmond E, Bernshtein B, Friedlander G,
Walker CR, Yona S, Kim KW, Brenner O, Krauthgamer R, Varol C,
Müller W and Jung S: Macrophage-restricted interleukin-10 receptor
deficiency, but not IL-10 deficiency, causes severe spontaneous
colitis. Immunity. 40:720–733. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schreurs RRCE, Baumdick ME, Sagebiel AF,
Kaufmann M, Mokry M, Klarenbeek PL, Schaltenberg N, Steinert FL,
van Rijn JM, Drewniak A, et al: Human fetal
TNF-α-cytokine-producing CD4+ effector memory T cells
promote intestinal development and mediate inflammation early in
life. Immunity. 50:462–476.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ahn J, Son S, Oliveira SC and Barber GN:
STING-dependent signaling underlies IL-10 controlled inflammatory
colitis. Cell Rep. 21:3873–3884. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tan Y, Zanoni I, Cullen TW, Goodman AL and
Kagan JC: Mechanisms of toll-like receptor 4 endocytosis reveal a
common immune-evasion strategy used by pathogenic and commensal
bacteria. Immunity. 43:909–922. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang W, Weng J, Yu L, Huang Q, Jiang Y and
Guo X: Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced
pulmonary epithelial hyperpermeability. BMC Pulm Med. 18:1782018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang D, Li S, Duan X, Ren J, Liang S,
Yakoumatos L, Kang Y, Uriarte SM, Shang J, Li W and Wang H: TLR4
induced Wnt3a-Dvl3 restrains the intensity of inflammation and
protects against endotoxin-driven organ failure through
GSK3β/β-catenin signaling. Mol Immunol. 118:153–164. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen
J, Jiao S, Gao Y, Liu C, Duan Z, et al: Activation of vascular
endothelial growth factor receptor-3 in macrophages restrains
TLR4-NF-κB signaling and protects against endotoxin shock.
Immunity. 40:501–514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Meroni E, Stakenborg N, Viola MF and
Boeckxstaens GE: Intestinal macrophages and their interaction with
the enteric nervous system in health and inflammatory bowel
disease. Acta Physiol (Oxf). 225:e131632019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shouval DS, Biswas A, Goettel JA, McCann
K, Conaway E, Redhu NS, Mascanfroni ID, Al Adham Z, Lavoie S,
Ibourk M, et al: Interleukin-10 receptor signaling in innate immune
cells regulates mucosal immune tolerance and anti-inflammatory
macrophage function. Immunity. 40:706–719. 2014. View Article : Google Scholar : PubMed/NCBI
|