Introduction
Membranous nephropathy (MN) is a common glomerular
disease characterized by increased thickness of the diffuse
glomerular basement membrane and subepithelial immune deposits
(1). A total of one-third of MN
cases present primary glomerulonephritis and two-thirds present
secondary MN (2). In previous
years, the number of patients with MN has increased (3) and ~15% of them develop end-stage
renal disease (4). The prognosis
of patients with MN is poor, with 15–30% of patients relapsing
after remission (5).
The microflora, especially that in the intestine,
maintains human health and affects physiological functions
(6). A number of studies have
reported that patients with cancer, diabetes and chronic kidney
disease present an imbalance in the intestinal microbiota, and that
dysbiosis further stimulates disease development (7,8).
Furthermore, the composition of the intestinal microflora shows
different characteristics in different diseases. Yu et al
(9) found that the composition of
the gut microbiome in diabetic kidney disease and MN was markedly
different. Furthermore, the oral microbiota is a reflection of
health, and the dynamic changes in its diversity affect the balance
between disease and health (10).
It has also been reported that the oral microbiota is closely
related to chronic kidney disease. Duan et al (11) found that the diversity of the
saliva microbiota was increased, and its abundance and richness
were decreased in patients with end-stage renal disease. Our
previous study reported an association between the oral microbiota
and immunoglobulin A nephropathy (12). Nonetheless, studies on the
relationship between the oral microflora and kidney diseases are
rare, especially studies investigating the relationship between the
diversity and abundance of oral microbiota and MN. In the present
study, saliva samples from 22 patients with MN and 15 healthy
controls were collected for oral microbiota characterization and
the possibility of using the microbiota to diagnose patients with
MN was explored.
Materials and methods
Sample collection
Saliva was collected from 22 patients with MN (mean
age, 41.9 years) and 15 healthy controls (mean age, 42.1 years)
between March 1, 2019 and June 31, 2020, at Shenzhen Longhua
District Central Hospital (Shenzhen, China). The diagnosis of MN
was based on routine light and immunofluorescence microscopy
examination. The criteria proposed by Ehrenreich and Churg
(13) were used to determine
histological staging of MN. Subjects underwent comprehensive dental
and periodontal examinations by clinicians who performed clinical
status assessments. The subjects brushed their teeth in the morning
and 2 h after they were required to spit 4–5 ml of saliva directly
into sterile collection containers over 30 min. The collected
saliva was naturally produced, without any stimulation. After
collection, the samples were immediately mixed with RNA later (cat.
no. R0901; MilliporeSigma) and stored at −80°C until use. The
patients with MN did not receive hormones, antibiotics,
immunosuppressive therapy or alternate therapies when the saliva
samples were collected. The serum creatinine (CR), estimated
glomerular filtration rate (eGFR), systolic blood pressure (SBP),
diastolic blood pressure (DBP) and proteinuria in 24 h (Pro-24)
were measured before medication therapy and obtained from the
records of the patients, and these clinical characteristics are
listed in Table I. The present
study was approved by the ethics committee of Shenzhen Longhua
District Central Hospital (Shenzhen, China). All participants
enrolled voluntarily in the study and signed a written consent
form.
 | Table I.Clinical information of patients with
membranous nephropathy and healthy controls. |
Table I.
Clinical information of patients with
membranous nephropathy and healthy controls.
| A, Patients with
membranous nephropathy |
|---|
|
|---|
| ID | Sex | Age, years | CR, µmol/l | eGFR, ml/min/1.73
m2 | SBP, mmHg | DBP, mmHg | Pro-24, g/day | Pathological
results |
|---|
| 160801 | M | 51 | 304.4 | 20.1 | 137 | 69 | 5.55 | MN, stage III |
| 160901 | M | 30 | 68 | 126.2 | 122 | 83 | 4.36 | MN, stage II |
| 161004 | M | 46 | 90 | 83.8 | 112 | 67 | 7.26 | MN, stage II |
| 161005 | F | 43 | 48 | 130.2 | 126 | 72 | 1.92 | MN, stage II |
| 161007 | F | 44 | 61 | 98.2 | 123 | 83 | 3.66 | MN, stage II |
| 170102 | F | 39 | 67 | 90.3 | 122 | 75 | 4.36 | MN, stage II |
| 170103 | M | 53 | 82 | 90.6 | 139 | 82 | 8.90 | MN, stage II |
| 170201 | M | 42 | 93 | 82.2 | 117 | 77 | 2.20 | MN, stage II |
| 170302 | F | 45 | 83 | 68.5 | 139 | 79 | 1.87 | MN, stage I |
| 170402 | M | 50 | 90 | 82.4 | 161 | 100 | 3.63 | MN, stage II |
| 170403 | M | 30 | 95 | 85.8 | 149 | 73 | 7.50 | MN, stage II |
| 170407 | M | 34 | 64 | 132.0 | 113 | 81 | 5.45 | MN, stage II |
| 170502 | M | 44 | 80 | 96.8 | 162 | 106 | 9.33 | MN, stage II |
| 170903 | M | 50 | 220 | 29.4 | 130 | 74 | 6.33 | MN, stage III |
| 180102 | M | 37 | 79 | 101.8 | 119 | 61 | 0.78 | MN, stage II |
| 180304 | F | 43 | 71 | 82.8 | 139 | 81 | 1.80 | MN, stage III |
| 180305 | F | 24 | 64 | 105.1 | 105 | 70 | 2.94 | MN, stage II |
| 180602 | M | 46 | 77 | 100.3 | 137 | 92 | 5.96 | MN, stage II |
| 180702 | M | 44 | 78 | 99.7 | 105 | 56 | 3.26 | MN, stage II |
| 180801 | M | 35 | 88 | 90.9 | 121 | 81 | 2.93 | MN, stage II |
| 180902 | F | 47 | 54 | 111.6 | 135 | 85 | 3.93 | MN, stage I |
| 181003 | M | 44 | 124 | 58.4 | 108 | 73 | 10.36 | MN, stage II |
|
| B, Healthy
controls |
|
| ID | Sex | Age,
years | CR,
µmol/l | eGFR,
ml/min/1.73 m2 | SBP,
mmHg | DBP,
mmHg | Pro-24,
g/day | Pathological
results |
|
| HE1 | M | 40 | 69 | 117.1 | 121 | 56 | 0.04 | Healthy |
| HE2 | M | 55 | 80 | 92.5 | 98 | 73 | 0.03 | Healthy |
| HE3 | M | 34 | 68 | 123.0 | 105 | 79 | 0.10 | Healthy |
| HE4 | M | 33 | 78 | 105.7 | 123 | 55 | 0.034 | Healthy |
| HE5 | M | 42 | 70 | 114.0 | 130 | 85 | 0.06 | Healthy |
| HE6 | M | 34 | 73 | 113.4 | 116 | 79 | 0.05 | Healthy |
| HE7 | M | 40 | 75 | 106.4 | 108 | 88 | 0.08 | Healthy |
| HE8 | M | 45 | 67 | 118.3 | 123 | 72 | 0.12 | Healthy |
| HE9 | M | 50 | 82 | 91.7 | 113 | 59 | 0.09 | Healthy |
| HE10 | M | 43 | 54 | 119.9 | 125 | 86 | 0.08 | Healthy |
| HE11 | F | 58 | 58 | 98.5 | 108 | 84 | 0.10 | Healthy |
| HE12 | F | 43 | 54 | 113.6 | 107 | 76 | 0.08 | Healthy |
| HE13 | F | 38 | 67 | 90.8 | 98 | 82 | 0.12 | Healthy |
| HE14 | F | 34 | 50 | 130.2 | 106 | 75 | 0.03 | Healthy |
| HE15 | F | 42 | 57 | 107.3 | 113 | 83 | 0.04 | Healthy |
Exclusion criteria
Subjects who had diarrhea or other intestinal
diseases and had taken antibiotics, probiotics or laxatives in the
previous 4 weeks were excluded from the present study.
DNA extraction
A bacterial DNA kit (DP328; Tiangen Biotech Co.,
Ltd.) was used to extract the microbial DNA from saliva according
to the manufacturer's protocols. Agarose gel (1%) electrophoresis
was used to check the integrity of the extracted DNA, and the DNA
was stored at −20°C for future use.
Library construction for
next-generation sequencing based on 16S ribosomal RNA (16S
rRNA)
Illumina Bridge PCR-compatible primers, barcode
primers and a pair of primers were used to amplify the V4 region of
the 16S rRNA gene by DNA Taq polymerase (cat. no. P102-01, Vazyme
Biotechnology Co. Ltd.). The PCR conditions were as follows: 95°C
for 2 min; 30 cycles at 95°C for 15 sec, 56°C for 30 sec and 72°C
for 30 sec; and 72°C for 10 min. The amplicons were purified using
DNA binding beads (cat. no. DP705; Tiangen Biotech Co., Ltd.). The
sequences of the degenerate primers for amplification of the V4
region were designed by Primer 3 (version 2.4.0; http://primer3.ut.ee/) and they were: V4 forward,
5′-GTGCCAGCMGCCGCGGTAA-3′ and reverse, 5′-GGACTACHVGGGTWTCTAAT-3′.
An Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) was used
to assess the size of the amplicons, and Qubit 3.0 (Thermo Fisher
Scientific, Inc.) was used to measure the concentration. VAHTS
Universal DNA Library Prep Kit for Illumina V3 (cat. no. ND607,
Vazyme Biotechnology Co. Ltd.) was used to prepare sequencing
library. VAHTS Library Quantification kit for Illumina (cat. no.
NQ101, Vazyme Biotechnology Co. Ltd.) was used to quantify the
concentration and a loading concentration of 4 pM for sequencing.
The Illumina Hiseq X (Illumina, Inc., San Diego, CA, USA) was used
to sequence the libraries using read length of 250 bp from each end
(paired-end 250).
Quality control for raw data and data
assembly
The Trimmomatic (V0.33) software (14) was applied to pair-end raw data for
quality control, and all the parameters were set as default to
obtain clean reads. The mothur (V1.35.1) software (15) was used to categorize the clean
reads for each sample according to their barcodes. The FLASH
(V1.2.11) software (16) was used
to assemble the paired-end reads to obtain raw tags (original
splicing sequence). Clean tags (effective splicing fragments) were
obtained after quality control and filtering.
Bioinformatics analysis
All clean tags were clustered by USEARCH (v9.0.2132)
(17) and aligned to the
operational taxonomic units (OTUs) by UPARSE (V10.0.240) (18) to identify the taxonomy. The
parameters for identity were set as the default at 97%. Each
sequence was sorted out into OTUs by Quantitative Insights Into
Microbial Ecology (QIIME) (V1.9.1) (19) after singleton OTUs were removed by
USEARCH and chimeric sequences were removed by UCHIME (V4.1)
(20). QIIME was used to select
the best representative sequence from optimized QIIME-selected
sequences for the final OTU cluster to align with the Human Oral
Microbiome Database (21). The
species annotation information of OTUs was obtained and the OTUs
that were annotated as chloroplast or mitochondrion and those which
could not be annotated to any species were deleted. The relative
abundance of each OTU was each OTU read normalized to total OTUs
reads of each sample (in-house scripts). QIIME was also used to
perform α-diversity analysis of all OTU cluster data. The
nonparametric Mann-Whitney test (two-tailed; 95% CI) in GraphPad
Prism (version 6.0; GraphPad Software, Inc.) was performed to
analyze the abundance difference between patients with MN and
healthy controls at the genus level. P<0.05 was considered to
indicate a statistically significant difference. Analysis of
similarities (ANOSIM) was performed between the patients with MN
and healthy controls using the Vegan package (V2.4-3) of R language
(22). Chao1 and Shannon indices
between the patients with MN and healthy controls were analyzed
using QIIME. The Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway and Lefse analyses were predicted based on the abundance of
OTUs using the Phylogenetic Investigation of Communities by
Reconstruction of Unobserved States (PICRUST; V1.1.4) software
(23) and Lefse (V1.1.2)
(24).
Statistical analysis
The differences of clinical indicators between
patients with MN and healthy controls were analysed by one way
ANOVA using Tukey's post hoc. Metastats analysis allows a
comparison of metagenomic samples (represented as counts of
individual features such as organisms, genes and functional groups)
from two treatment populations (for example, healthy vs. disease)
and identifies those features that statistically distinguish the
two populations. Spearman was used to analyse the correlation
between OTUs and clinical indicators using Stats package of R
language (22).
Results
Characterization of the sequencing
results
The salivary microbiota of patients with MN and
healthy controls were analyzed by sequencing of the 16S rRNA gene
using Illumina platform. After quality control, the total number of
salivary microbial sequences was 1,534,154 reads at an average of
69,734 reads per sample for the 22 patients with MN, and 1,414,400
at an average of 88,400 reads per sample for the 15 healthy
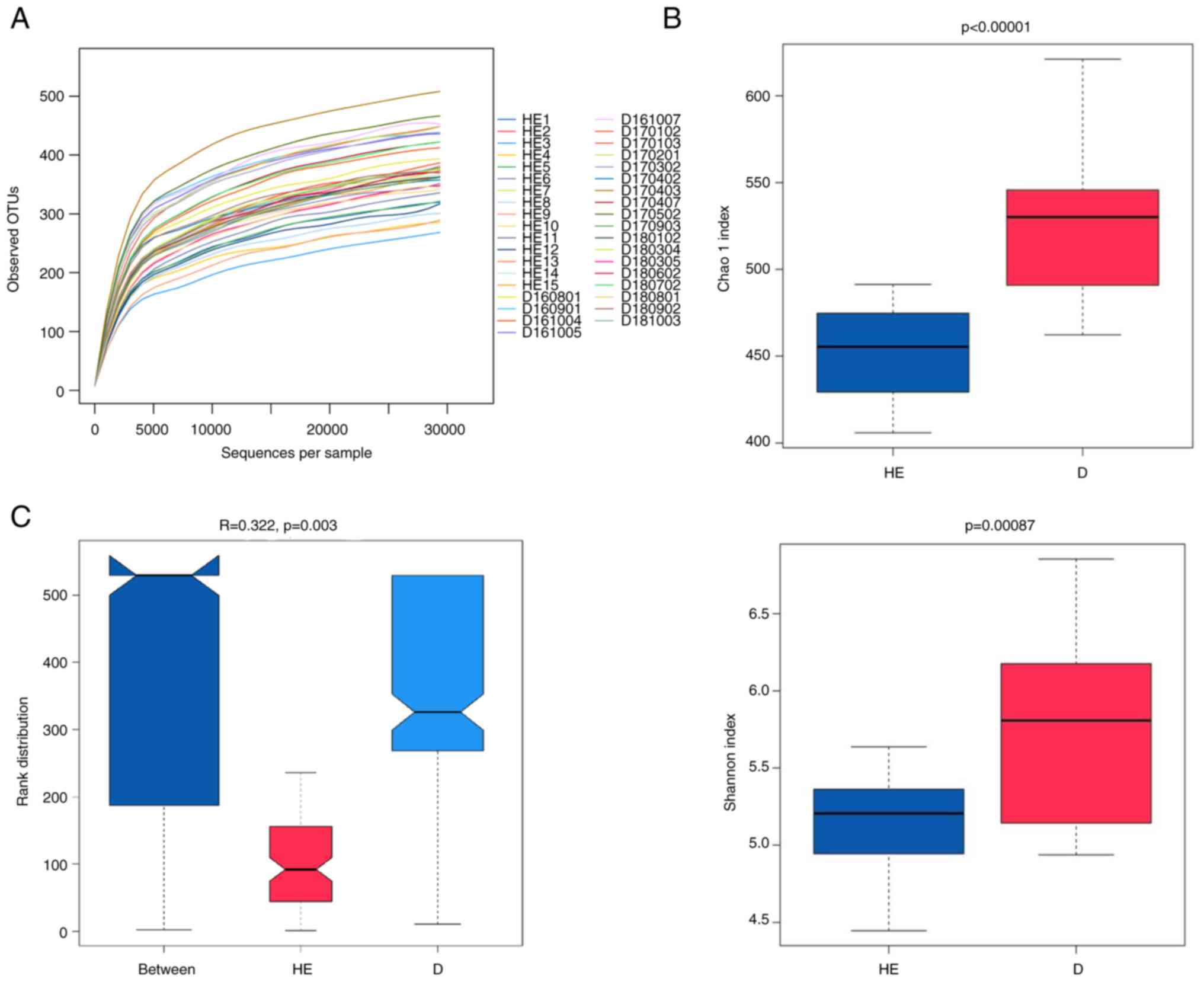
controls. The rarefaction curve plateaued, indicating that the
sequencing depth could be used to analyze the composition of the
salivary microbiota (Fig.
1A).
Taxonomic analysis of the
microbiota
Through the OTU clustering of sequencing reads, a
total of 1,362 OTUs were found in the 22 samples of the MN group.
The number of OTUs in each sample ranged between 391 and 559, with
an average value of 462. The healthy group had fewer OTUs than the
group of patients with MN. The number of OTUs in each sample was
344–453, with an average of 401. Bacterial species richness (Chao1
index) and diversity (Shannon index) of the patients with MN were
significantly higher than those of the healthy controls (Fig. 1B). The Chao1 index (a value of
521) of patients with MN was higher than that of the healthy
controls (which presented a Chao1 index of 452). The P-values for
the Chao1 index and Shannon index were <0.00001 and 0.00087,
respectively. ANOSIM is a non-parametric statistical test widely
used to assess the similarities between groups. According to ANOSIM
analysis, the microbial composition in the MN group was
statistically different from that in the healthy group (P=0.003;
Fig. 1C).
Heatmap analysis
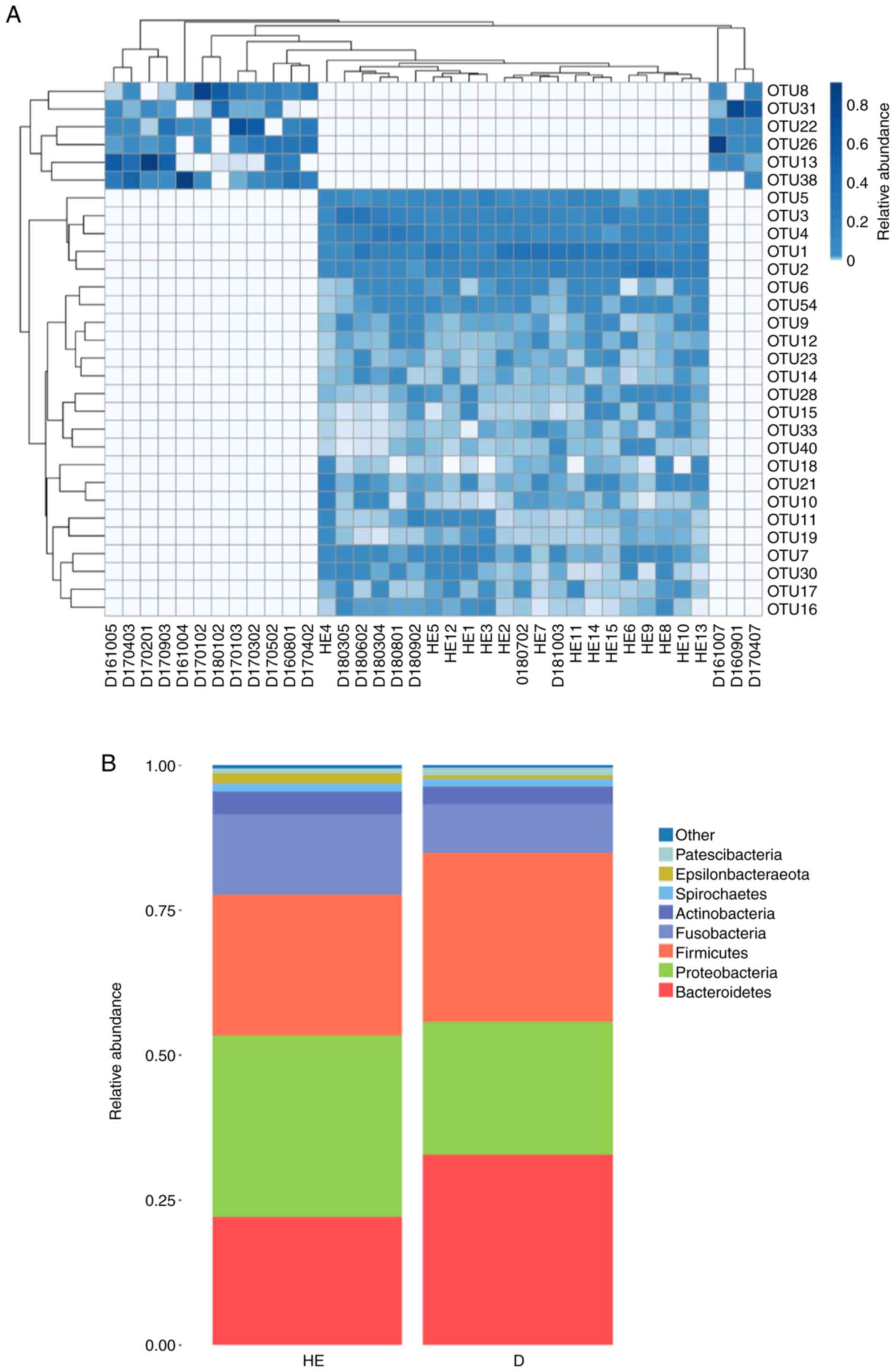
The OTU heatmap showed that the MN group and healthy
control group were mostly clustered separately (Fig. 2A; Table SI). In the microbial
classification, eight phyla (Bacteroidetes, Firmicutes,
Proteobacteria, Fusobacteria, Actinobacteria, Spirochaetes,
Patescibacteria and Epsilonbacteraeota) accounted for
>99% of the microbiota in the MN group and >99% in the
healthy control group (Fig. 2B).
Metastat analysis was conducted at the genus level and revealed
that there were 47 different genera between the MN group and the
healthy control group (P<0.05; Table SII).
Correlation analysis of biochemical
indicators and OTUs
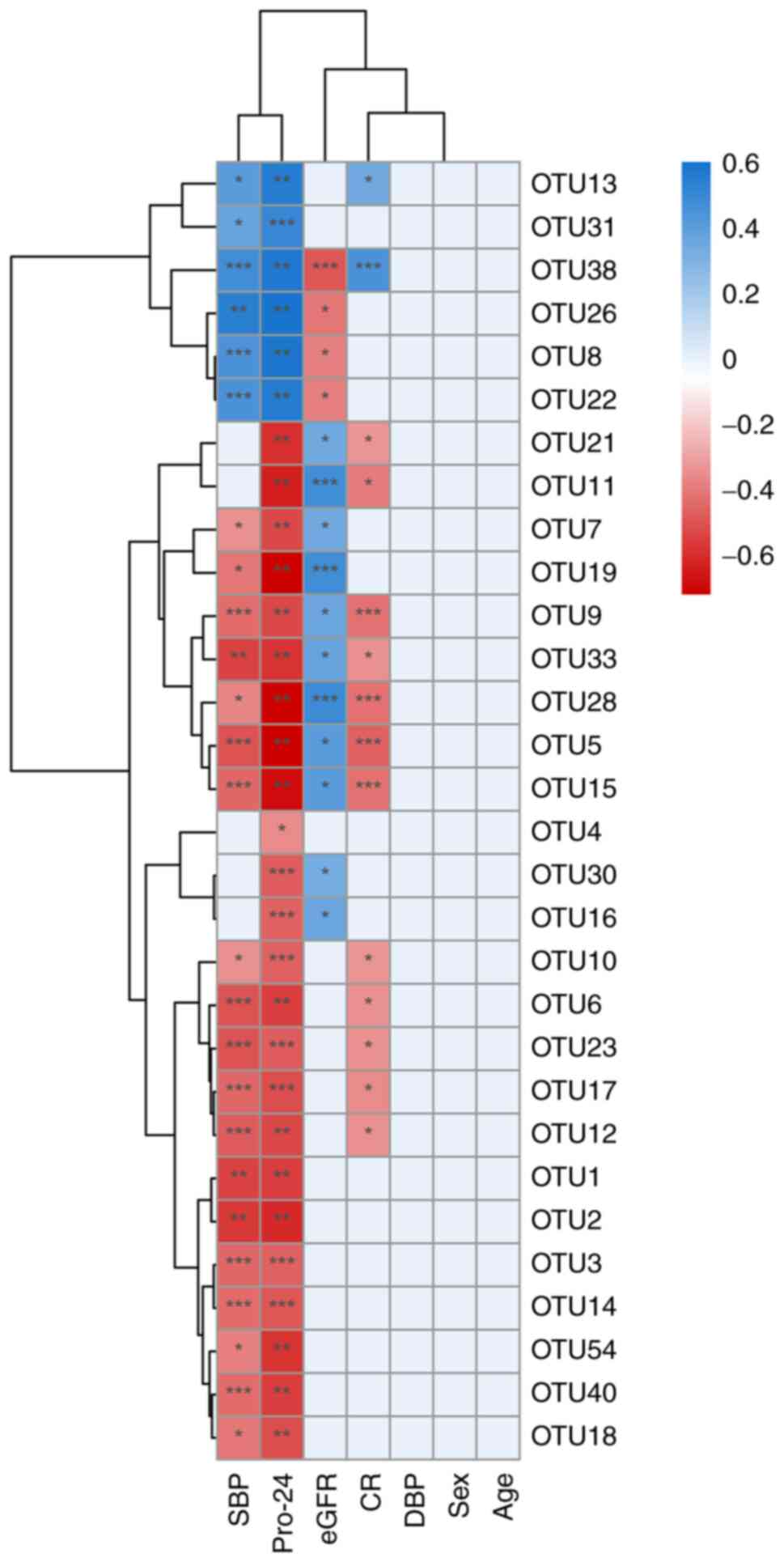
OTUs of each sample were used to analyze the
correlation with the biochemical indicators CR, Pro-24, eGFR, SBP
and DBP, which are used clinically (Table SIII). Pro-24, SBP and eGFR were
found to be significantly different between the patients with MN
and healthy controls (P<0.05; Table SIV). The abundance of OTUs was
considered to be related to clinical indicators. Serum CR was
negatively correlated with 12 OTUs, of which OTU12, OTU17, OTU23,
OTU6, OTU10, OTU33, OTU21 and OTU11 had a P-value <0.05, and
OTU5, OTU9, OTU15 and OTU28 had a P-value <0.001, and it was
positively correlated with OTU13 (P<0.05) and OTU38
(P<0.001). Pro-24 was negatively correlated with 24 OTUs (OTU4
with P<0.05; OTU1, OTU2, OTU5, OTU6, OTU7, OTU9, OTU11, OTU12,
OTU15, OTU18, OTU19, OTU21, OTU28, OTU33, OTU40 and OTU54 with
P<0.01; OTU3, OTU10, OTU14, OTU16, OTU17, OTU23 and OTU30 with
P<0.001) and positively correlated with 6 OTUs (OTU8, OTU13,
OTU22, OTU26 and OTU38 with P<0.05; and OTU31 with P<0.001).
eGFR was negatively correlated with 4 OTUs (OTU8, OTU22 and OTU26
with P<0.05; OTU38 with P<0.001) and positively correlated
with 11 OTUs (OTU5, OTU7, OTU9, OTU15, OTU16, OTU21, OTU30 and
OTU33 with P<0.05; OTU11, OTU19 and OTU28 with P<0.001). The
SBP was negatively correlated with 19 OTUs and positively
correlated with 6 OTUs. No OTUs were associated with DBP, age or
sex (Fig. 3). Among all OTUs,
OTU5, OTU9, OTU15, OTU28 and OTU33 were significantly correlated
with SBP, Pro-24, eGFR and CR.
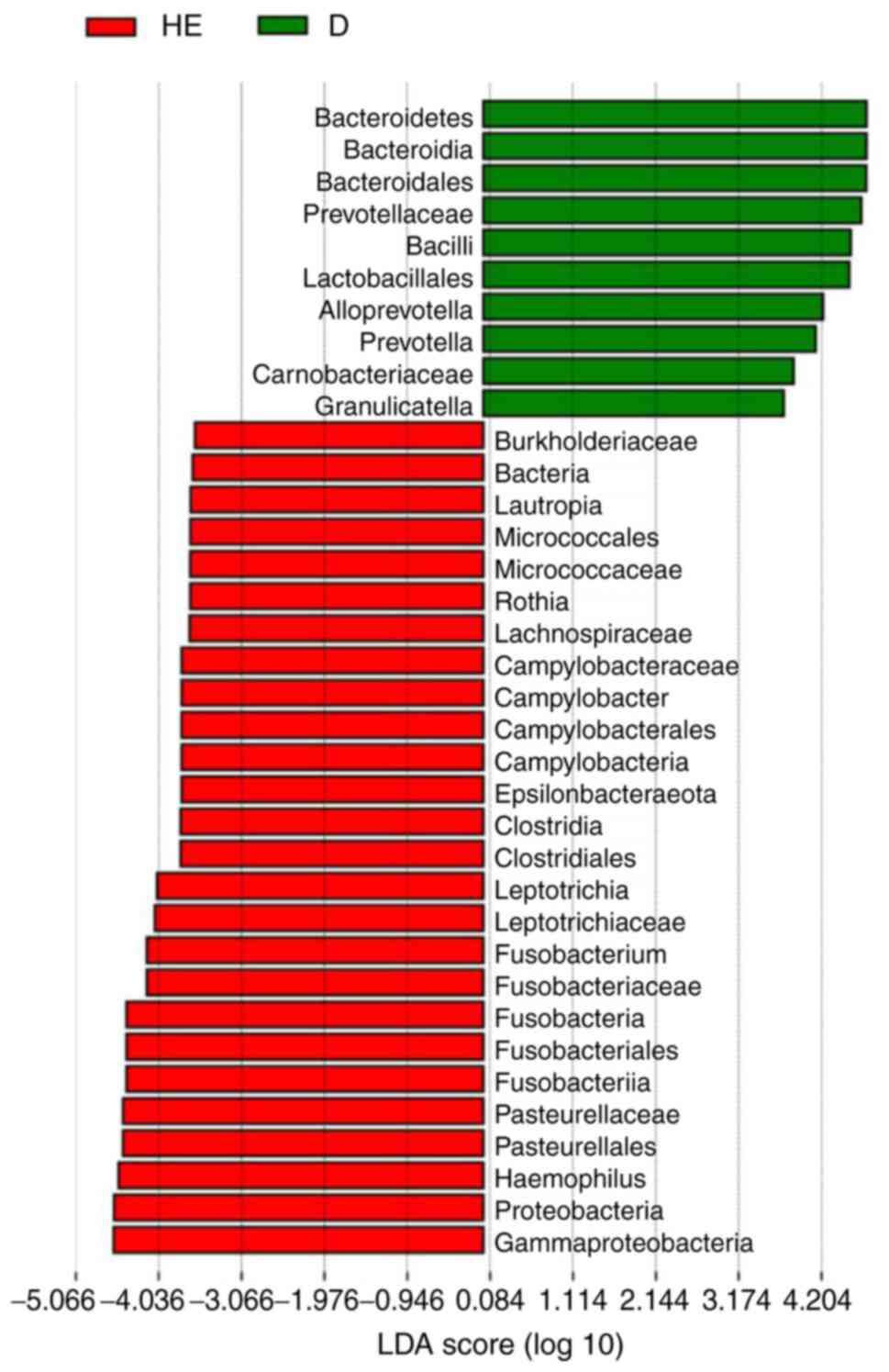
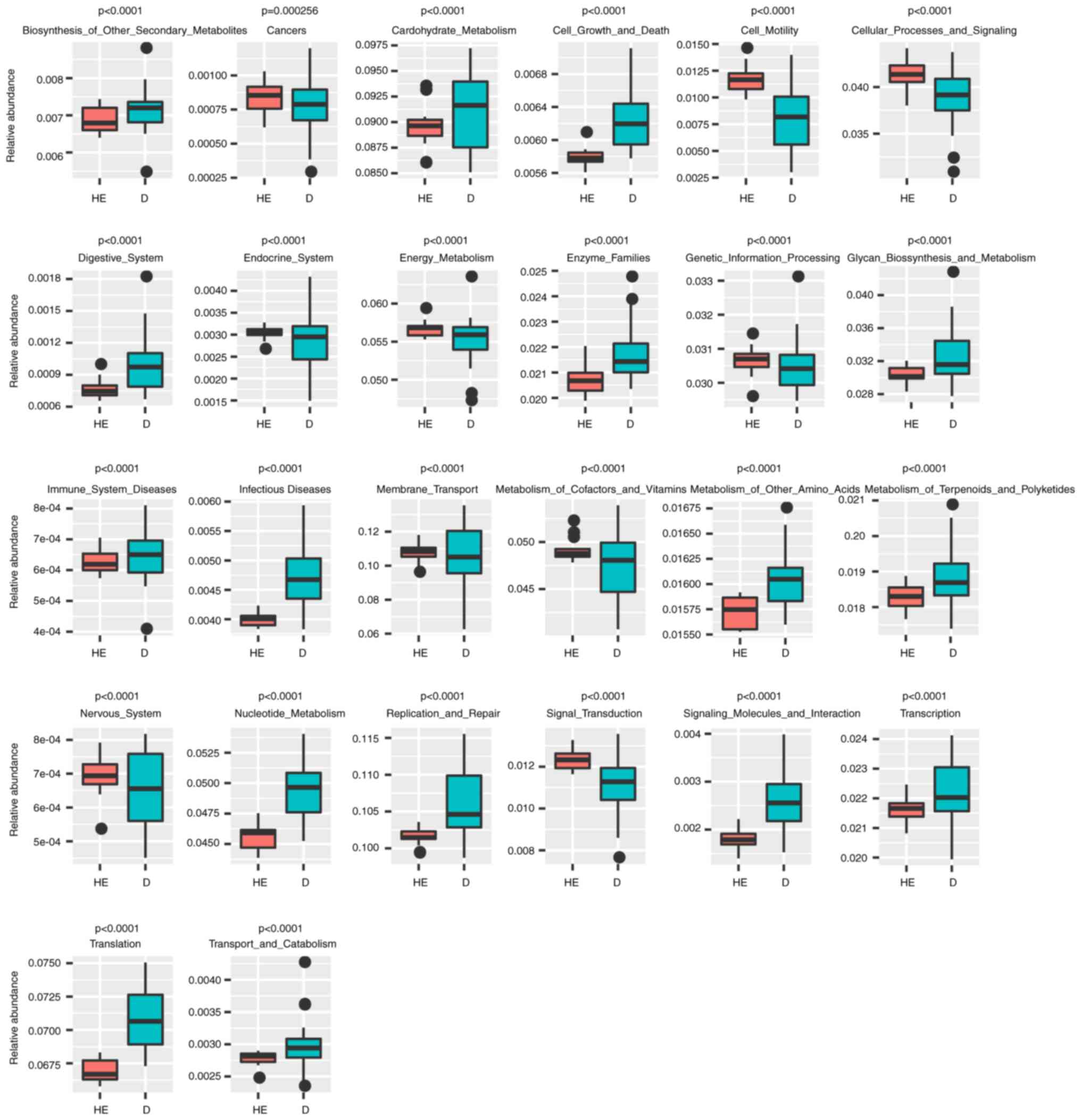
Lefse and PICRUST analyses
Lefse analysis was conducted to explore possible
biomarkers and PICRUST analysis was conducted to explore pathways.
Lefse analysis showed that 10 bacteria could be possibly used as
biomarkers for patients with MN (Fig.
4), including Bacteroidales (order,
P=6.59×10−5), Prevotellaceae (family,
P=2.62×10−4), Lactobacillales (order, P=0.0412),
Alloprevotella (genus, P=7.81×10−3),
Prevotella (family, P=0.0145), Carnobacteriaceae
(family, P=0.0172) and Granulicatella (genus, P=0.0328). A
total of 26 KEGG pathways were obtained from significant OTUs using
PICRUST (Fig. 5; Table SV), including
‘Immune_System_Diseases’ (P=1.81×10−6),
‘Glycan_Biosynthesis_and_Metabolism’ (P=1.91×10−6),
‘Energy_Metabolism’ (P=7.76×10−7), ‘Endocrine_System’
(P=4.05×10−7) and ‘Digestive_System’
(P=1.31×10−4).
Discussion
MN is a common glomerular disease; however, there
are few studies on the oral microbiota of patients with MN. Through
high-throughput sequencing of the 16S rRNA gene of oral microbiota
and the subsequent bioinformatics analysis, the composition and
characterization of oral microbiota in patients with MN were
thoroughly investigated and the associations between clinically
used biochemical indicators and OTUs were evaluated. ANOSIM
analysis revealed that there was a significant difference in the
composition of microbial species between the patients with MN and
healthy controls (P=0.003). α-diversity analysis showed that The
Chao1 index of patients with MN was higher than that of the healthy
controls, indicating that the total number of oral microbes in the
patients with MN was higher than that in the healthy controls.
Although α-diversity is normally lower in patients than in
controls, some reports have indicated that α-diversity is higher in
patients than in the control group and might be associated with
clinical benefits (25–27). The exact meaning of the
observation that α-diversity in patients with MN was higher than in
the controls needs further exploration. Additionally, the Shannon
index of the patients with MN was higher than that of the healthy
controls (5.72 vs. 5.13), indicating that the microbial diversity
of the patients with MN was also higher than that of the healthy
controls.
According to Lefse analysis, there were 10
significantly different bacteria in patients with MN. At the phylum
and class level, Bacteroidetes, Bacteroidia and
Bacilli may be candidate biomarkers. Lactobacillales
and Bacteroidales at the order level, Prevotellaceae,
Carnobacteriaceae and Prevotella at the family level,
and Alloprevotella and Granulicatella at the genus
level may serve as potential biomarkers for patients with MN.
Streptococcus and Granulicatella produce
antimicrobial compounds that inhibit bacterial growth, and thus,
are beneficial to the oral cavity (28). The abundance of
Streptococcus and Granulicatella was significantly
higher in patients with MN than in the control group, which was
also observed in the case of immunoglobulin A nephropathy (12). However, the present findings were
different from those reported by Piccolo et al (29), and this may be due to the
difference in ethnicities. Meanwhile, the phylum
Bacteroidetes normally implies poor health in the host
(9).
The potential KEGG pathway function of
microorganisms was predicted using PICRUST (30). The abundance of the pathway
‘Immune_System_Diseases’ in the MN group was significantly higher
than that in the healthy individuals (Fig. 5), indicating that the microbial
environment of patients with MN is likely to change according to
the immune system condition.
Regarding clinical indicators, CR is a product of
muscle metabolism in the human body and is mainly excreted from the
body by glomerular filtration. When acute or chronic
glomerulonephritis causes the glomerular filtration function to
decrease, serum CR levels can increase. eGFR is an indicator of
kidney function. It mainly refers to the excretion capacity of the
kidneys per unit of time and is used to assess kidney function.
Pro-24 is the 24 h urine protein content. Increased urine protein
is often present in various glomerular diseases, such as acute
nephritis, chronic nephritis, nephrotic syndrome and lupus
nephritis (31). Patients with
nephritis also often present high blood pressure (32,33). The correlations between the OTU
abundance of the 22 patients with MN and these clinical indicators
were analyzed to explore the relationship between the microbes and
the clinical indicators and the possibility of using them to
diagnose and assess the status of the patients with MN. Six OTUs
(OTU5, OTU28, OTU9, OTU15, OTU33 and OTU38) were found to be
significantly correlated with CR, Pro-24, eGFR and SBP, implying
that they could be used as biomarkers for the diagnosis of patients
with MN.
The present study has certain limitations. First,
the sample size was relatively small. This was a preliminary
proof-of-concept study that indicated that the salivary microbiota
was associated with MN. The sample size will be increased in
subsequent research. Second, it was a retrospective study. For the
biomarkers to be used in clinical settings, a prospective study is
necessary. Third, the present study enrolled patients from southern
China. Their diet is different from patients from northern China
(34). Whether and how the
different diets affect our conclusions requires further
investigation.
In conclusion, to the best of our knowledge, there
are no previous reports on the relationship between oral microbiota
and MN. The present study was the first to investigate the oral
microbiota in patients with MN. The present results revealed that
there were significant differences between the microbiota of the
patients with MN and that of healthy controls and that certain
microbial strains and OTUs (OTU5, OTU28, OTU9, OTU15, OTU33 and
OTU38) can be used as biomarkers to facilitate the diagnosis of
patients with MN in clinical settings.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The work was supported by grants from the National Natural
Science Foundation of China (grant no. 82001336), Guangdong Basic
and Applied Basic Research Foundation (grant nos. 2019A1515011009,
2021A1515010683, 2020A1515010225 and 2021A1515010955), Shenzhen
Foundation of Science and Technology (grant nos.
JCYJ20180306172449376, JCYJ20180306172459580 and
JCYJ20180306172502097), Nanjing Municipal Health Science and
Technology Development Special Fund Project (grant no. YKK19161)
and Shenzhen Longhua District Foundation of Science and Technology
(grant no. SZLHQJCYJ202002).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the National Center for
Biotechnology Information repository (no. PRJNA698736; http://www.ncbi.nlm.nih.gov/bioproject/PRJNA698736).
Authors' contributions
BH and HG conceived and supervised the study. SL,
SZ, LP and WH wrote the draft of the manuscript and designed the
experiments. HC and XWe analyzed the data. RL, CL, PZ, XWa and WL
collected the samples and clinical information and analyzed the
data. SL, ZX and YZ designed the experiments, revised the
manuscript and provided helpful advice. SL and HG confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by The Ethics
Committee of Shenzhen Longhua District Central Hospital (Shenzhen,
China). Informed written consent was obtained from each patient and
healthy subject enrolled in the present study.
Patient consent for publication
Consent for publication was obtained from all
patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Francis JM, Beck LH Jr and Salant DJ:
Membranous nephropathy: A journey from bench to bedside. Am J
Kidney Dis. 68:138–147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai Q and Hendricks AR: Membranous
nephropathy: A ten-year journey of discoveries. Semin Diagn Pathol.
37:116–120. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang L, Yao J, Kong X, Sun Q, Wang Z,
Zhang Y, Wang P, Liu Y, Li W, Cui M, et al: Increasing prevalence
of membranous nephropathy in patients with primary glomerular
diseases: A cross-sectional study in China. Nephrology (Carlton).
22:168–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cattran DC: Idiopathic membranous
glomerulonephritis. Kidney Int. 59:1983–1994. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cattran D: Management of membranous
nephropathy: When and what for treatment. J Am Soc Nephrol.
16:1188–1194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sekirov I, Russell SL, Antunes LC and
Finlay BB: Gut microbiota in health and disease. Physiol Rev.
90:859–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma S and Tripathi P: Gut microbiome
and type 2 diabetes: Where we are and where to go? J Nutr Biochem.
63:101–108. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin C, Cai X, Zhang J, Wang W, Sheng Q,
Hua H and Zhou X: Role of gut microbiota in the development and
treatment of colorectal cancer. Digestion. 100:72–78. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu W, Shang J, Guo R, Zhang F, Zhang W,
Zhang Y, Wu F, Ren H, Liu C, Xiao J and Zhao Z: The gut microbiome
in differential diagnosis of diabetic kidney disease and membranous
nephropathy. Ren Fail. 42:1100–1110. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pflughoeft KJ and Versalovic J: Human
microbiome in health and disease. Annu Rev Pathol. 7:99–122. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duan X, Chen X, Gupta M, Seriwatanachai D,
Xue H, Xiong Q, Xu T, Li D, Mo A, Tang X, et al: Salivary
microbiome in patients undergoing hemodialysis and its associations
with the duration of the dialysis. BMC Nephrol. 21:4142020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luan S, Zhang S, Zhong H, Zhang Y, Wei X,
Lin R, Li C, Zeng P, Wang X, Li W and Gao H: Salivary microbial
analysis of Chinese patients with immunoglobulin A nephropathy. Mol
Med Rep. 20:2219–2226. 2019.PubMed/NCBI
|
|
13
|
Ehrenreich T and Churg J: Pathology of
membranous nephropathy. Pathol Ann. 145–186. 1968.
|
|
14
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schloss PD, Westcott SL, Ryabin T, Hall
JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH,
Robinson CJ, et al: Introducing mothur: Open-source,
platform-independent, community-supported software for describing
and comparing microbial communities. Appl Environ Microbiol.
75:7537–7541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Magoc T and Salzberg SL: FLASH: Fast
length adjustment of short reads to improve genome assemblies.
Bioinformatics. 27:2957–2963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Edgar R: Taxonomy annotation and guide
tree errors in 16S rRNA databases. PeerJ. 6:e50302018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Edgar RC: UPARSE: Highly accurate OTU
sequences from microbial amplicon reads. Nat Methods. 10:996–998.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Edgar RC, Haas BJ, Clemente JC, Quince C
and Knight R: UCHIME improves sensitivity and speed of chimera
detection. Bioinformatics. 27:2194–2200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Escapa IF, Chen T, Huang Y, Gajare P,
Dewhirst FE and Lemon KP: New insights into human nostril
microbiome from the expanded human oral microbiome database
(eHOMD): A resource for the microbiome of the human aerodigestive
tract. mSystems. 3:e00187–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
R Core Team: R, . A language and
environment for statistical computing (V 4.1.1). R Foundation for
Statistical Computing; Vienna: 2021, http://www.R-project.org/
|
|
23
|
Langille MG, Zaneveld J, Caporaso JG,
McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega
Thurber RL, Knight R, et al: Predictive functional profiling of
microbial communities using 16S rRNA marker gene sequences. Nat
Biotechnol. 31:814–821. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12:R602011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Gu X, Yang J, Wei Y and Zhao Y:
Gut microbiota dysbiosis and increased plasma LPS and TMAO levels
in patients with preeclampsia. Front Cell Infect Microbiol.
9:4092019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu M, Zhang X, Liang Y, Lin S, Qian W and
Fan S: Alterations in vaginal microbiota and associated metabolome
in women with recurrent implantation failure. mBio. 11:e03242–19.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riquelme E, Zhang Y, Zhang L, Montiel M,
Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et
al: Tumor microbiome diversity and composition influence pancreatic
cancer outcomes. Cell. 178:795–806.e12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar PS, Griffen AL, Moeschberger ML and
Leys EJ: Identification of candidate periodontal pathogens and
beneficial species by quantitative 16S clonal analysis. J Clin
Microbiol. 43:3944–3955. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piccolo M, De Angelis M, Lauriero G,
Montemurno E, Di Cagno R, Gesualdo L and Gobbetti M: Salivary
microbiota associated with immunoglobulin a nephropathy. Microb
Ecol. 70:557–565. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanehisa M, Furumichi M, Sato Y,
Ishiguro-Watanabe M and Tanabe M: KEGG: Integrating viruses and
cellular organisms. Nucleic Acids Res. 49:D545–D551. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Levey AS, Becker C and Inker LA:
Glomerular filtration rate and albuminuria for detection and
staging of acute and chronic kidney disease in adults: A systematic
review. JAMA. 313:837–846. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Azevedo FVA, Maia DG, de Carvalho JF
and Rodrigues CEM: Renal involvement in antiphospholipid syndrome.
Rheumatol Int. 38:1777–1789. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Turrent-Carriles A, Herrera-Felix JP and
Amigo MC: Renal involvement in antiphospholipid syndrome. Front
Immunol. 9:10082018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang L, Xing Y, Yu X, Ming J, Liu X, Li X,
Fu J, Zhou J, Gao B, Hu D, et al: Greater macrovascular and
microvascular morbidity from type 2 diabetes in northern compared
with southern China: A cross-sectional study. J Diabetes Investig.
11:1285–1294. 2020. View Article : Google Scholar : PubMed/NCBI
|