Introduction
Early-onset epileptic encephalopathy (EOEE) is a
group of severe epilepsies characterized by refractory seizures
with progressive brain dysfunction in the early infantile period,
accompanied by complex etiologies (1,2).
In addition to perinatal brain injury, metabolic diseases and
structural brain malformations, genetic defects have also been
indicated to contribute to the pathogenesis of EOEE through
participating in processes such as the generation and pruning of
synapses and the differentiation and migration of neurons (3). With the development of
high-throughput sequencing technology, opportunities have been
provided to investigate the underlying molecular-genetic basis of
epilepsy. More than 100 genes have been identified to be related to
EOEE, such as Cdc42 guanine nucleotide exchange factor 9 [ARHGEF9;
Online Mendelian Inheritance in Man (OMIM) #300607],
cyclin-dependent kinase-like 5 (CDKL5; OMIM #300203), potassium
sodium-activated channel subfamily T member 1 (KCNT1; OMIM
#614959), sodium voltage-gated channel α subunit 8 (SCN8A; OMIM
#614558), solute carrier family 2 member 1 (SLC2A1; OMIM #614847)
syntaxin-binding protein 1 (STXBP1; OMIM #602926), polynucleotide
kinase 3′-phosphatase (PNKP; OMIM #605610) and potassium
voltage-gated channel subfamily Q member 2 (KCNQ2; OMIM #602235)
(4–14). Among these, KCNQ2 is the causative
gene for 7–10% of cases of EOEE (15–17).
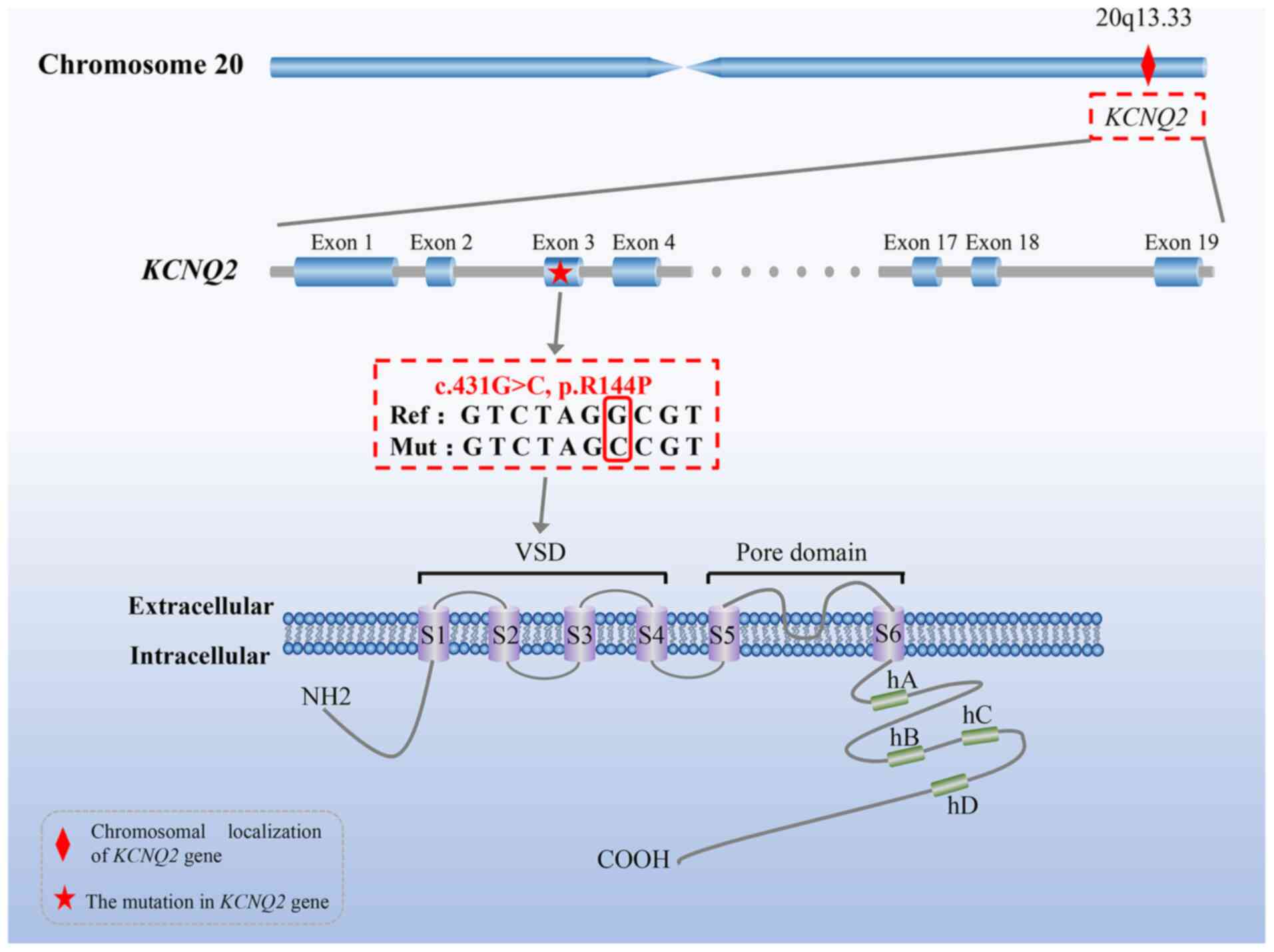
The KCNQ2 gene, a member of the KCNQ family, maps to
chromosome 20q13.33, consists of 19 exons and encodes a
voltage-gated potassium-channel subunit KV7.2 (also
known as KCNQ2 protein), which is widely distributed in the central
nervous system (18,19). The KCNQ2 protein is composed of
872 amino acids and contains an N-terminal cytoplasmic tail, a long
C-terminal region (including four α-helical regions A-D) and six
transmembrane segments (S1-S6) that form four voltage-sensing
domains (VSD) (S1-S4), as well as a pore domain (S5, S6 and the
loop between them) (Fig. 1). The
KCNQ2 protein and its homologous KCNQ3 protein constitute homo- and
hetero-tetrameric ion channels considered as the molecular basis of
neuronal M-channels, which induce an M-current (a slowly
deactivating, voltage-dependent and non-inactivating potassium
current) and have a major role in controlling neuronal excitability
through limiting repetitive firing of action potentials (20,21). A previous functional study by
Jentsch (22) suggested that a
25% down-regulation of M-currents may be sufficient to cause the
onset of epilepsy in infants. Echoing this finding, numerous
studies confirmed that mutations in the KCNQ2 gene usually resulted
in over-excitability of the neurons by affecting the generation of
M-currents, thus causing the occurrence of seizures (23,24). Consequently, haploinsufficiency
and a dominant-negative effect, resulting from the loss of KCNQ2
protein function caused by KCNQ2 mutations (such as c.740C>T,
c.853C>A, c.860C>T, etc.) (25,26), are currently recognized as the
primary drivers of KCNQ2-related EOEE (27,28). Furthermore, the severity of the
clinical phenotype is strongly related to the degree of protein
dysfunction or deficiency determined by various KCNQ2 mutations
(29). However, the
genotype-phenotype correlation in this disease has remained to be
fully elucidated.
The present study reported on a female infant from
the Chinese Lisu minority suffering from EOEE caused by a novel
de novo KCNQ2 variant and the clinical and genetic
characteristics of this patient were determined.
Materials and methods
Subjects and clinical evaluation
A 3-month-old female patient and her parents were
recruited from the Department of Respiratory and Critical Care
Medicine, Kunming Children's Hospital (KCH; Kunming, China). This
patient was hospitalized in September 2021 due to recurrent
non-febrile convulsions. There was no consanguinity between the
parents. Written informed consent was obtained from the patient's
parents prior to performing clinical evaluations and collecting
blood samples from any subject (October 2021 and March 2022). The
present study was performed according to the Declaration of
Helsinki (2013 version) as well as relevant laws of China and
approved by the Ethics Committee of KCH (September 18, 2021).
Next-generation sequencing (NGS)
Sample collection and DNA extraction
Blood, urine and oral mucosa swab samples were
obtained from the proband. The genomic DNA was extracted from these
samples using a DNA Isolation Kit (BioTeke Corporation) according
to the manufacturer's protocol. Subsequently, the DNA concentration
was measured by the Qubit dsDNA HS Assay Kit on a Qubit fluorometer
(both from Invitrogen; Thermo Fisher Scientific, Inc.) and the DNA
quality was detected by electrophoresis on 1% agarose gels.
Construction of DNA library
The preparation of DNA libraries was performed by a
KAPA Library Preparation Kit (Kapa Biosystems; Roche Diagnostics)
following the manufacturer's instructions. Specifically, the
genomic DNA was randomly fragmented into ~200 bp pieces and these
fragments were then subjected to purification, end repair, poly-A
tailing reaction and adapter ligation. Finally, the prepared DNA
libraries were amplified through PCR (the compositions of the PCR
mixture included 10 µl Library DNA, 12.5 µl 2X KAPA HiFi HotStart
ReadyMix, 1 µl PCR Primer Premix and 1.5 µl water) with the
following thermocycling conditions: initial denaturation at 98°C
for 2 min, 8 cycles of denaturation at 98°C for 30 sec, annealing
at 65°C for 30 sec, extension at 72°C for 30 sec and final
extension at 72°C for 4 min.
Targeted gene capture
The capture of targeted genes was performed by
hybridization of the capture probes (cat. no. 5190-9494; Agilent
Technologies, Inc.) to the prepared DNA libraries and the removal
of non-hybridized library molecules. Dynabeads®
MyOne™ Streptavidin T1 (Invitrogen; Thermo Fisher
Scientific, Inc.) in binding buffer were added to the probe-library
hybridization mixture to absorb the probes with target fragments.
For purification and elution, the pooled magnetic beads were washed
with washing buffer 1 (25°C, 10 min), 3 (65°C, 15 min) and elution
buffer. Next, the captured DNA was amplified using PCR the
following PCR mixture: 21 µl 2X KAPA HiFi HotStart ReadyMix, 1 µl
of 5 µM primer and 20 µl captured library beads suspension. The PCR
amplification program was 98°C for 2 min; followed by 98°C for 30
sec, 65°C for 30 sec and 72°C for 30 sec, for 13 cycles; and
finally 72°C for 4 min. The products were subsequently purified by
Agencourt AMPure XP beads (Beckman Coulter, Inc.). The quality
inspection of the final product was performed with a Qubit dsDNA HS
Assay kit (Invitrogen; Thermo Fisher Scientific, Inc.) in
accordance with the manufacturer's instructions.
Sequencing
The resulting libraries were loaded onto the
Illumina HiSeq2500 platform (Illumina, Inc.) and then subjected to
NGS following the manufacturer's specifications to generate
paired-end 200-bp reads.
Bioinformatics analysis of NGS data
Raw image files from the Illumina HiSeq2500 platform
were preprocessed using Bcl2Fastq software (version 2.20;
http://support.illumina.com) for
BCL-to-FASTQ conversion and demultiplexing. The generated raw reads
were cleaned and filtered to remove low-quality data using Cutadapt
(version 1.2.1; http://cutadapt.readthedocs.io/en/stable/) and then
aligned to the human reference genome using the Short
Oligonucleotide Analysis Package (SOAP) aligner tool (version 2.21;
soap.genomics.org.cn/soapsnp.html). Genome Analysis Toolkit
(version 3.7; http://www.broadinstitute.org/gatk/) was utilized to
remove the redundant PCR duplicates and recalibrate the base
quality score. The obtained insertions and deletions as well as
single nucleotide polymorphisms were detected by SAMTOOLS (version
0.1.19; http://samtools.sourceforge.net/). Variant annotation
was performed with ANNOVAR (http://www.openbioinformatics.org/annovar/).
Pathogenicity prediction of the identified
variant
The potentially damaging effect of the identified
variant on protein function was predicted using Rare Exome Variant
Ensemble Learner (REVEL; http://sites.google.com/site/revelgenomics/downloads),
which is an ensemble pathogenicity analysis tool based on the
Random Forest algorithm.
PCR and Sanger sequencing
PCR and Sanger sequencing for the three subjects
were utilized to validate the candidate mutation verified by NGS.
In brief, genomic DNA from the parents of the patient was isolated
from peripheral blood using the genomic DNA extraction kit (Qiagen
China, Co., Ltd.) and the primers (forward,
5′-ACCACAGCCTCTGACTCCA-3′; and reverse, 5′-CAACCCTTCCTGCCCAGAG-3′)
for amplifying exon 3 of KCNQ2 were designed by online primer
design software PRIMER3 (http://frodo.wi.mit.edu/primer3). PCR amplification
(compositions of the PCR mixture: 10 µl target DNA, 12.5 µl 2X KAPA
HiFi HotStart ReadyMix, 1 µl PCR Primer Premix and 1.5 µl water;
amplification program: Initial denaturation at 95°C for 5 min,
followed by 32 cycles of denaturation at 95°C for 30 sec, annealing
at 60°C for 30 sec, extension at 72°C for 30 sec, and then a final
extension at 72°C for 7 min), purification of PCR products and
Sanger sequencing with an ABI 3730 Genetic Analyzer platform
(Applied Biosystems; Thermo Fisher Scientific, Inc.) were performed
sequentially. Sites of mutation were confirmed through comparing
the sequencing results with the human KCNQ2 reference sequence
(NM_172107.2) retrieved from GenBank (http://www.ncbi.nlm.nih.gov/Genbank/).
Molecular modeling of the mutant KCNQ2
protein
SWISS-Model, an automated web-based homology
modeling server (http://swissmodel.expasy.org/workspace/), was used to
calculate the three-dimensional (3D) structure models of the mutant
(Mut) and wild-type (Wt) KCNQ2 protein. The original and mutated
amino acid sequences of KCNQ2 were uploaded to the above
SWISS-Model workspace and the crystal structure 7CR1 fetched from
Protein Data Bank (http://www.rcsb.org/pdb/) was selected as the
best-matched template with a sequence identity of 62% and a
coverage of 73%. The modeled structures were visualized and
analyzed with PyMOL (https://pymol.org/2/).
Flow cytometry
The expression of KCNQ2 in all subjects was assessed
by flow cytometry. The isolation of peripheral blood mononuclear
cells (PBMCs) from peripheral blood of all subjects was performed
by Ficoll density gradient centrifugation (MilliporeSigma) using a
ratio of PBMC isolation reagents: Peripheral blood of 3:1. The
obtained PBMCs were washed three times with PBS and then treated
with fixation/permeabilization buffer (eBioscience; Thermo Fisher
Scientific, Inc.) for cell fixation and permeabilization. Rabbit
anti-KCNQ2 (cat. no. ab22897; Abcam,) primary antibody was applied
at 1:200 dilution overnight at 4°C, and subsequently, samples were
incubated with goat anti-rabbit IgG H&L (cat. no. ab150077;
Abcam) secondary antibody diluted at 1:400 for 30 min at room
temperature (protected from light). Measurement of fluorescence
intensity and analysis of flow cytometric data were performed using
a FACSCanto II flow cytometer (BD Bioscience; Thermo Fisher
Scientific, Inc.) and FlowJo v8.8 software (FlowJo LLC),
respectively.
Results
Clinical characteristics
The patient, born at full term via spontaneous
vaginal delivery without asphyxia, was a three-month-old female
infant from the Chinese Lisu minority, presenting with recurrent
non-febrile convulsions. The infant suffered 7 episodes of
unprovoked non-febrile seizures within 3 days prior to admission.
Each onset lasted ~3–5 min and was characterized by clenched fists,
upturned eyes, tonic spasm of limbs and unconsciousness. On
admission to KCH, the patient underwent a physical examination,
which indicated that the muscle strength, tension and reflexes of
the extremities were normal. There was no family history of
epilepsy for this patient.
The laboratory parameters revealed no significant
abnormalities in 25-hydroxyvitamin D, thyroid function, blood
glucose and bilirubin, thereby excluding tetany of vitamin D
deficiency and convulsions caused by hypoglycemic or bilirubin
encephalopathy. Neuroimaging tests and echocardiography were also
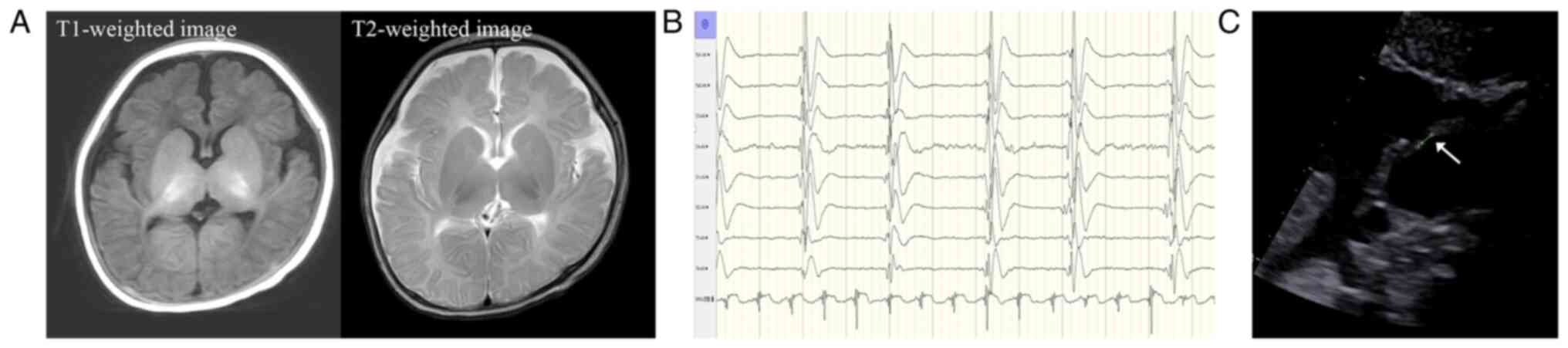
completed after admission. Specifically, brain MRI of the patient
indicated no pathological signs (Fig.
2A), while the electroencephalogram (EEG) revealed abnormal
background activity at the time of admission (September 2021) and a
burst suppression pattern at the most recent follow-up (March 2022)
(Fig. 2B). In addition, an atrial
septal defect (ASD) (diameter of 2.5 mm) was observed by
echocardiography (Fig. 2C).
Identification of a novel de novo
KCNQ2 variant
The proband and the proband's parents underwent
genetic testing to investigate the potentially causative variant.
According to the comparison of the sequencing result with the human
reference genome, a novel heterozygous missense variant
(c.431G>C) in exon 3 of the KCNQ2 gene (Fig. 3A) was identified in the proband
(II-1). This single base substitution from G to C at nucleotide 431
led to an arginine (R)-to-proline (P) amino acid change at position
144 (p.R144P), thus possibly resulting in dysfunction of the
encoded protein due to the fact that R144 in KCNQ2 is highly
conserved among multiple species (Fig. 3B). In order to exclude the
potential possibility of chimera, urine and oral samples from the
proband were also used for DNA extraction and high-throughput
sequencing. The KCNQ2 c.431G>C variant was also detected in the
above two samples (Fig. 3A) and
this finding corresponded with the sequencing result from the blood
sample of the proband. To our knowledge, there have not been any
previous reports about the c.431G>C variant in KCNQ2 up to now
and this variant with an unknown frequency in the general
population was predicted to be deleterious by REVEL. Neither
previously reported pathogenic mutations for EOEE in KCNQ2 nor
mutations in other EOEE-related genes (ARHGEF9, CDKL5, KCNT1,
SCN8A, SCN2A, SLC2A1, STXBP1, etc.) were found in this patient.
This novel missense variant was speculated to be the molecular
pathological cause for the clinical phenotype of the proband.
Neither of the proband's unaffected parents (the father, I-1; the
mother, I-2) were carriers of this novel variant (Fig. 3C). Furthermore, the confirmation
of the identity of the proband's biological parents was performed
through paternity analysis (data not shown), thereby verifying that
the KCNQ2 c.431G>C variant occurred de novo in the
proband.
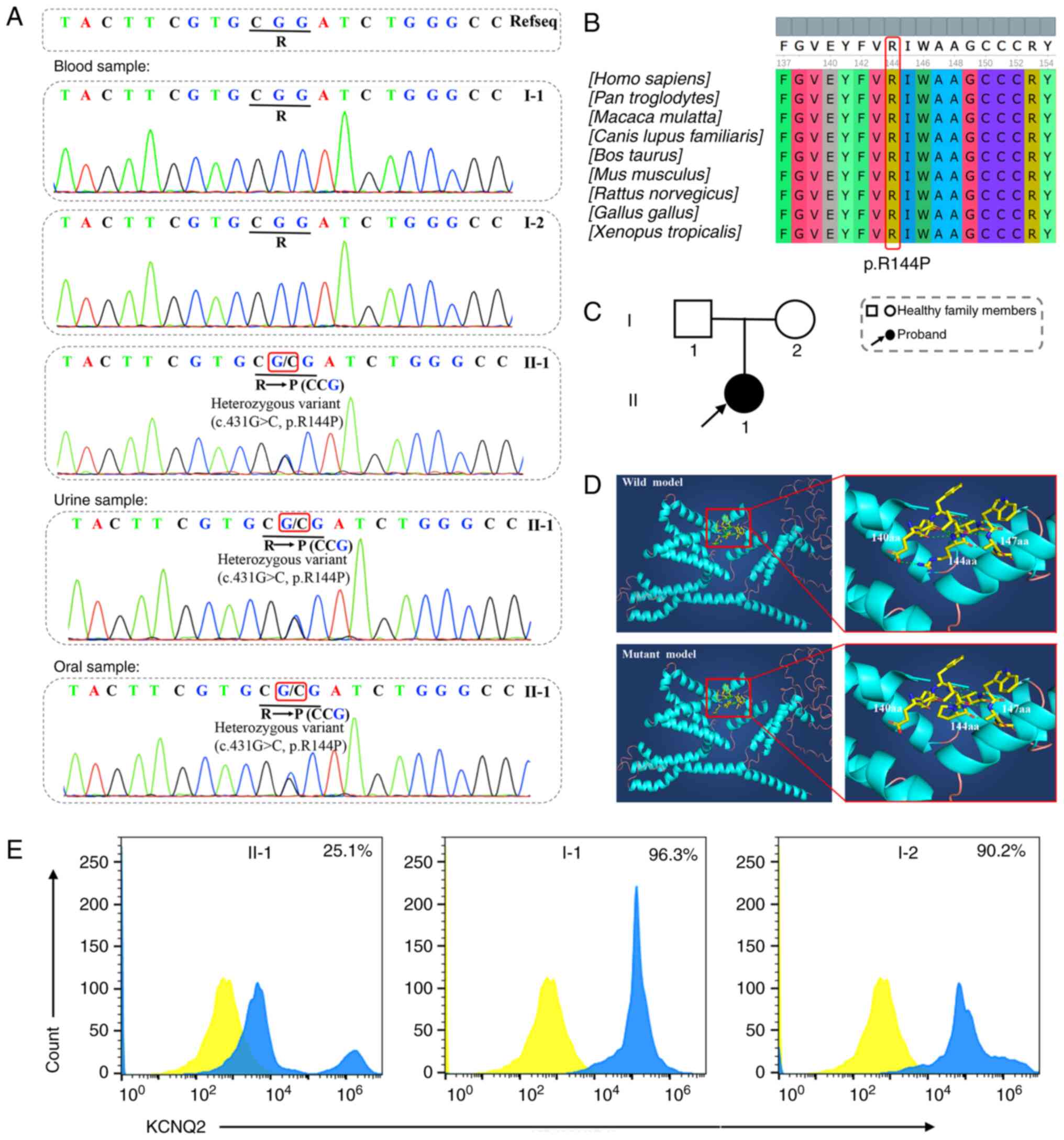 | Figure 3.Mutational analysis of all
participants and analysis of mutation pathogenicity. (A) Results of
genetic testing. The proband had a novel de novo c.431G>C
mutation in exon 3 of the KCNQ2 gene, while there was no mutation
detected in the proband's parents. This novel de novo mutation was
also detected in urine and oral samples from the proband through
high-throughput sequencing. The identified mutation was marked with
red boxes. (B) Evolutionary conservation analysis of amino acid 144
in KCNQ2. Conservation of the altered amino acid was revealed
through sequence alignment. (C) The pedigree of the proband's
family. II-1, the proband, is suffering from early-onset epileptic
encephalopathy, while I-1 (proband's father) and I-2 (proband's
mother) are healthy. (D) Structural model of the wild- and
mutant-type (p.R144P) KCNQ2 protein. The novel mutation disrupted
the hydrogen bonds between the 140 and 144th amino acids and
between the 147 and 144th amino acids. (E) Flow cytometry results.
A significant reduction but not complete loss of native KCNQ2
protein expression was identified in peripheral blood mononuclear
cells from the proband (II-1, 25.1%). By contrast, no apparent
downregulation of KCNQ2 expression was detected in the proband's
father (I-1, 96.3%) and mother (I-2, 90.2%). In the image for II-1,
two blue peaks were observed, which may be explained by the fact
that a few PBMCs of the proband could express KCNQ2 normally. The
peaks of the blank control were labeled in yellow and the peaks of
the subjects were labeled in blue. |
Structural analysis of KCNQ2 protein
affected by the novel c.431G>C variant
To explore the possible role of the KCNQ2
c.431G>C variant in the protein structure and function, 3D
structures of the Mut- and Wt-KCNQ2 protein were modeled (Fig. 3D). The comparative structural
analysis of Mut- and Wt-KCNQ2 suggested that the identified variant
(c.431G>C, p.R144P) disrupted the hydrogen bonds between the 140
and 144th amino acids and between the 147 and 144th amino acids.
The breakage of hydrogen bonds may alter the local secondary
structure and function of the protein.
Analysis of KCNQ2 protein
expression
The expression levels of native KCNQ2 protein in all
subjects were assessed by flow cytometry to validate the
pathogenicity of the novel KCNQ2 c.431G>C mutation. The
acquired data were processed by FlowJo v8.8 software and converted
into histograms. As speculated, the flow cytometry results
(Fig. 3E) indicated a significant
reduction but not complete loss of native KCNQ2 protein expression
in PBMCs from the proband (II-1, 25.1%). By contrast, the values of
the KCNQ2 protein expression in the proband's father (I-1) and
mother (I-2) were 96.3 and 90.2%, respectively, and no prominent
abnormalities were found.
Discussion
KCNQ2 variants, responsible for cell excitability
control and maintenance of ion balance, were first discovered by
Biervert et al (19) and
Singh et al (21) in 1998
and were considered a major genetic cause of EOEE. The precise
diagnosis and classification of EOEE rely on the combination of
clinical data analysis and molecular genetic testing. Recently,
through searching the Human Gene Mutation Database (HGMD;
http://www.hgmd.org/), a total of 325 reported
variant sites in the KCNQ2 gene were retrieved. Among these
variants, 42 were verified in Chinese patients (16,30–41) and 83% (35/42) of them were
missense variants, which were also primarily responsible for severe
EOEE (Table I). In the present
study, a novel de novo missense variant of the KCNQ2 gene
(c.431G>C, p.R144P) was identified in a Chinese Lisu infant with
KCNQ2-related EOEE. It was noted that this patient with the KCNQ2
c.431G>C variant had a milder phenotype without significant
psychomotor retardations in comparison to previously reported
typical cases of KCNQ2-related EOEE (25,26).
 | Table I.List of reported KCNQ2 variant sites
in Chinese patients. |
Table I.
List of reported KCNQ2 variant sites
in Chinese patients.
| Nucleotide
change | Amino acid
change | Variant type | Variant class | (Refs.) |
|---|
| c.850T>C | p.Y284H | Missense | DM | (16) |
| c.871A>G | p.R291G | Missense | DM | (16) |
| c.710A>T | p.Y237F | Missense | DM | (16) |
| c.868G>A | p.G290S | Missense | DM | (16) |
| c.838T>C | p.Y280H | Missense | DM | (16) |
| c.1452G>C | p.E484D | Missense | DM | (16) |
| c.1284delG | p.Q429Rfs*5 | Frameshift | DM | (16) |
| c.913T>C | p.F305L | Missense | DM | (16) |
| c.736G>C | p.A246P | Missense | DM | (16) |
| c.793G>A | p.A265T | Missense | DM | (16) |
| c.748G>T | p.V250L | Missense | DM | (16) |
| c.821C>T | p.T274M | Missense | DM | (16) |
| c.637C>T | p.R213W | Missense | DM | (16) |
| c.781T>A | p.F261I | Missense | DM | (30) |
| c.1742G>A | p.A518G | Missense | DM | (30) |
| c.1048A>C | p.N350H | Missense | DM | (31) |
| c.242T>C | p.L81P | Missense | DM | (31) |
| c.2506G>T | p.E836* | Nonsense | DM | (31) |
| c.958G>A | p.V320I | Missense | DM | (31) |
| c.998G>A | p.R333Q | Missense | DM | (31) |
| c.775G>A | p.D259N | Missense | DM | (31) |
| c.237T>G | p.N79K | Missense | DM | (31) |
| c.185C>T | p.A62V | Missense |
DM? | (32) |
| c.839A>G | p.Y280C | Missense |
DM? | (32) |
| c.2331delC | del 1 bp codon
777 | Frameshift | DM | (32) |
| c.1948dupG | p.E650fs | Frameshift | DM | (33) |
| c.641G>A | p.R214Q | Missense | DM | (33) |
| c.916G>C | p.A306P | Missense | DM | (33) |
| c.1678C>T | p.R560W | Missense | DM | (33) |
| c.1019T>C | p.I340T | Missense | DM | (33) |
| c.766G>A | p.G256R | Missense | DM | (33) |
| c.365C>T | p.S122L | Missense | DM | (34) |
| c.956A>C | p.K319T | Missense | DM | (34) |
| c.830C>T | p.T277I | Missense | DM | (34) |
| c.1655A>C | p.K552T | Missense | DM | (34) |
| c.743dupT | ins 1 bp codon
248 | Frameshift | DM | (35) |
| c.944G>A | p.G315E | Missense | DM | (36) |
| c.878T>C | p.L293P | Missense | DM | (37) |
| c.1057C>T | p.R353C | Missense | DM | (38) |
| c.1286G>A | p.C429Y | Missense |
DM? | (39) |
| c.2015delG | del 1 bp codon
672 | Frameshift | DM | (40) |
|
c.2513_2514delAG | del 2 bp codon
838 | Frameshift | DM | (41) |
The causative missense KCNQ2 variants associated
with EOEE were mainly distributed in four hotspots, including the
VSD (particularly in S4), the pore domain and two calmodulin
(CaM)-binding α-helical regions (helix A and B) in the cytoplasmic
C-terminal domain (42), and the
function of the KCNQ2 protein was regulated by the above regions
through different mechanisms (25). Specifically, the intracellular
localization of KCNQ2 and the assembly as well as transport
activity of the potassium channel were mediated by the interaction
between the helix regions (A and B) with CaM. The P-loop (between
S5 and S6) in the pore domain was found to have high potassium ion
selectivity and permeability, which are critical for neuronal
excitability. Of note, the voltage sensitivity of the
KV7.2 channel was conferred by the VSD. KCNQ2 mutations
occurring in the VSD would reduce the voltage sensitivity by
destabilizing the active VSD configuration (43). The voltage-sensing function of the
VSD was endowed by charged residues, particularly the first four
residues in S4, which was therefore regarded as the most crucial
functional region in the VSD (44). The variants at the distal region
of S4 disrupted the stability of the active VSD configuration but
were not directly involved in voltage sensing. Conversely, the
variants near S4 were able to squarely mediate the activation of
gating pore currents. As Miceli et al (45) reported earlier, the effect of the
KCNQ2 variants in the S1-S2 linker and S2 transmembrane segment on
the voltage sensitivity and voltage-dependent activation of the
KV7.2 channel was smaller than that of KCNQ2 variants in
S4 (43). Based on this, it may
be speculated that the variants closer to S4 may result in more
severe clinical phenotypes and this speculation may in part explain
the phenomenon that the patient of the present study, who carried
the c.431G>C variant located between S2 and S3, had a milder
phenotype. Resonating with the above finding, the comparative
structural analysis of Mut- and Wt-KCNQ2 protein in the present
study suggested that the novel mutant (c.431G>C, p.R144P) led to
the breaking of hydrogen bonds between several amino acids as
compared to Wt-KCNQ2. Although this alteration would probably
impact the local secondary protein structure of KCNQ2, no dramatic
changes in the overall conformation of this protein were observed.
This suggested that there may be a certain degree of KCNQ2 function
damage, but this did not appear to be severe. Besides molecular
modeling, flow cytometry was also performed to measure the
expression of native KCNQ2 protein in the proband and the proband's
parents. The result indicated that the novel missense mutation
changed the normal structure of KCNQ2, resulting in the failure of
protein encoded by the mutant KCNQ2 to bind to the normal
anti-KCNQ2 antibody, which was reflected as the defect in
expression of native KCNQ2. This evidence partially supported the
pathogenicity of the novel mutation. However, the expression of
native KCNQ2 protein in the proband (25.1%) was not completely
absent, and this perhaps explained why the patient of the present
study had a relatively mild phenotype compared to other reported
KCNQ2-related EOEE cases. Another possible factor influencing the
severity of the clinical phenotype is the type of mutation. In a
previous study, the homozygous KCNQ2 variant was lethal for mice;
by contrast, no fatalities were observed in heterozygous mice
despite the exhibition of neuronal hyperexcitation caused by
reduced expression of KCNQ2 protein (46). Consistent with the previous study,
the patient of the present study, who carried a heterozygous KCNQ2
variant only, presented with a mild EOEE phenotype without severe
psychomotor retardations. According to the assessment of the
patient's condition, the antiepileptic drug topiramate and certain
conventional symptomatic (midazolam for sedation, etc.) and
supportive therapies (levocarnitine for the facilitation of lipid
metabolism, etc.) were eventually implemented for the patient.
Certainly, poor neurocognitive development in EOEE is also a
concern in spite of this abnormality not currently being observed
in the patient. Follow-up surveillance for neurodevelopment in this
patient will be maintained.
Apart from KCNQ2-related EOEE, ASD with a diameter
of 2.5 mm was detected in this patient via echocardiography. ASD is
one of the most common congenital heart diseases with an incidence
of 1.6 per 1,000 live births and is anatomically characterized by
absent tissue in the interatrial septum (47,48). Persistent left-to-right shunt via
the defect may cause secondary pulmonary arterial hypertension with
resultant fatal right heart failure and Eisenmenger syndrome
(49,50). However, no related symptoms have
been detected in the patient of the present study, and the ASD
(only 2.5 mm in diameter) was considered to have a strong
likelihood of healing spontaneously during childhood. Thus, no
interventions for ASD were performed. However, high altitude may be
a potential factor to take into account during the assessment for
heart disease development. A higher risk of pulmonary hypertension
and right heart failure was observed in the individuals living at
high altitudes (51,52). Yunnan, where the patient resided,
is a highland area located in the southwest of China with an
average altitude of 2,000 m (53). Based on the aforementioned
reasons, echocardiography will be performed regularly to monitor
the patient's cardiac health.
In addition, the patient of the present study is
from the Chinese Lisu minority in Yunnan, where >50% of Chinese
ethnic minorities are settled (People's Government of Yunnan;
http://www.yn.gov.cn). The rate of consanguineous
marriage among the Lisu population is highest in China despite no
consanguinity between the parents of the patient of the present
study. The high incidence of genetic defects in ethnic minority
groups remains a significant concern. Their unique cultural customs
(particularly consanguineous marriage) and relatively isolated
residential areas may predispose them to genetic disorders, which
may perhaps also be a result of natural selection. Genome-wide
association studies will be a valuable tool for the investigation
of genetic variants in various populations.
Of note, there are three limitations to the present
study. The first is that this novel mutation was not detected in
any cohort of patients with EOEE. Furthermore, the study lacked an
in vitro expression model of Mut- and Wt-KCNQ2. Finally, the
study did not investigate the detailed relationship between the
disruption of the hydrogen bonds (between the 140 and 144th amino
acids) and damage to KCNQ2 function.
In conclusion, genetic analysis is crucial for
diagnosing EOEE with high genetic and clinical heterogeneity. In
the present study, a novel de novo KCNQ2 variant, which
resulted in impaired function of the KCNQ2 protein, was identified
in a Chinese Lisu minority infant through NGS and Sanger
sequencing. This variant was confirmed to underlie the EOEE in the
patient of the present study. Overall, investigating and reporting
novel causal KCNQ2 variants will expand the mutation spectrum of
KCNQ2 and further contribute to genetic counseling and prenatal
diagnosis. Future research should be dedicated to uncovering the
exquisite molecular mechanisms of EOEE and clarifying the
relationship between genotype and phenotype.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The sequencing data that support the findings of the
present study are available from the public DNA Data Bank of Japan
(accession no. LC706559; http://getentry.ddbj.nig.ac.jp/getentry/na/LC706559/?format=flatfile&filetype=html&trace=true&show_suppressed=false&limit=10).
The datasets used and/or analyzed during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
HFL and HMF conceived and designed the study; TYY,
JWY, FL and FW collected the clinical and genetic data; TYY, FL and
FW performed flow cytometry; HFL, TYY and JWY analyzed the data;
and HFL and HMF prepared the manuscript. All authors read and
approved the final manuscript. HMF and HFL confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Ethical approval for the present study was obtained
from the Ethics Committee of KCH (Kunming, China).
Patient consent for publication
Informed consent was obtained from the parents of
the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARHGEF9
|
Cdc42 guanine nucleotide exchange
factor 9
|
|
ASD
|
atrial septal defect
|
|
CDKL5
|
cyclin-dependent kinase-like 5
|
|
EEG
|
electroencephalogram
|
|
EOEE
|
early-onset epileptic
encephalopathy
|
|
KCH
|
Kunming Children's Hospital
|
|
KCNT1
|
potassium sodium-activated channel
subfamily T member 1
|
|
NGS
|
next-generation sequencing
|
|
PBMCs
|
peripheral blood mononuclear cells
|
|
PNKP
|
polynucleotide kinase
3′-phosphatase
|
|
REVEL
|
Rare Exome Variant Ensemble
Learner
|
|
SCN8A
|
sodium voltage-gated channel α subunit
8
|
|
SLC2A1
|
solute carrier family 2 member 1
|
|
SOAP
|
Short Oligonucleotide Analysis
Package
|
|
STXBP1
|
syntaxin-binding protein 1
|
|
VSD
|
voltage-sensing domain
|
References
|
1
|
Nieh SE and Sherr EH: Epileptic
encephalopathies: New genes and new pathways. Neurotherapeutics.
11:796–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alam S and Lux AL: Epilepsies in infancy.
Arch Dis Child. 97:985–992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galanopoulou AS and Moshé SL: The
epileptic hypothesis: Developmentally related arguments based on
animal models. Epilepsia. 50 (Suppl 7):S37–S42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang JY, Zhou P, Wang J, Tang B, Su T, Liu
XR, Li BM, Meng H, Shi YW, Yi YH, et al: ARHGEF9 mutations in
epileptic encephalopathy/intellectual disability: Toward
understanding the mechanism underlying phenotypic variation.
Neurogenetics. 19:9–16. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jakimiec M, Paprocka J and Śmigiel R:
CDKL5 deficiency disorder-A complex epileptic encephalopathy. Brain
Sci. 10:1072020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gertler T, Bearden D, Bhattacharjee A and
Carvill G: KCNT1-related epilepsy. Adam MP, Mirzaa GM, Pagon RA,
Wallace SE, Bean LJH, Gripp KW and Amemiya A:
GeneReviews® Seattle (WA): University of Washington;
Seattle: 1993–2022. 2018
|
|
7
|
Bonardi CM, Heyne HO, Fiannacca M,
Fitzgerald MP, Gardella E, Gunning B, Olofsson K, Lesca G, Verbeek
N, Stamberger H, et al: KCNT1-related epilepsies and epileptic
encephalopathies: Phenotypic and mutational spectrum. Brain.
144:3635–3650. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Talwar D and Hammer MF: SCN8A epilepsy,
developmental encephalopathy, and related disorders. Pediatr
Neurol. 122:76–83. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hammer MF, Wagnon JL, Mefford HC and
Meisler MH: SCN8A-related epilepsy with encephalopathy.
GeneReviews®. Adam MP, Mirzaa GM, Pagon RA, Wallace SE,
Bean LJH, Gripp KW and Amemiya A: University of Washington;
Seattle, Seattle, WA: 1993-2022. 2016
|
|
10
|
Reynolds C, King MD and Gorman KM: The
phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr
Neurol. 24:117–122. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hedrich UBS, Lauxmann S and Lerche H:
SCN2A channelopathies: Mechanisms and models. Epilepsia. 60 (Suppl
3):S68–S76. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Agostinelli S, Traverso M, Accorsi P,
Beccaria F, Belcastro V, Capovilla G, Cappanera S, Coppola A, Dalla
Bernardina B, Darra F, et al: Early-onset absence epilepsy: SLC2A1
gene analysis and treatment evolution. Eur J Neurol. 20:856–859.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wild B and Nelson S: STXBP1-Related
developmental and epileptic encephalopathy. Pediatr Neurol Briefs.
33:62019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stamberger H, Nikanorova M, Willemsen MH,
Accorsi P, Angriman M, Baier H, Benkel-Herrenbrueck I, Benoit V,
Budetta M, Caliebe A, et al: STXBP1 encephalopathy: A
neurodevelopmental disorder including epilepsy. Neurology.
86:954–962. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Lian Y and Xie N: Early onset
epileptic encephalopathy with a novel GABRB3 mutation treated
effectively with clonazepam: A case report. Medicine (Baltimore).
96:e92732017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Q, Li J, Zhao Y, Bao X, Wei L and
Wang J: Gene mutation analysis of 175 Chinese patients with
early-onset epileptic encephalopathy. Clin Genet. 91:717–724. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mastrangelo M and Leuzzi V: Genes of
early-onset epileptic encephalopathies: From genotype to phenotype.
Pediatr Neurol. 46:24–31. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leppert M, Anderson VE, Quattlebaum T,
Stauffer D, O'Connell P, Nakamura Y, Lalouel JM and White R: Benign
familial neonatal convulsions linked to genetic markers on
chromosome 20. Nature. 337:647–648. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biervert C, Schroeder BC, Kubisch C,
Berkovic SF, Propping P, Jentsch TJ and Steinlein OK: A potassium
channel mutation in neonatal human epilepsy. Science. 279:403–406.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Pan Z, Shi W, Brown BS, Wymore RS,
Cohen IS, Dixon JE and MacKinnon D: KCNQ2 and KCNQ3 potassium
channel subunits: Molecular correlates of the M-channel. Science.
282:1890–1893. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh NA, Charlier C, Stauffer D, DuPont
BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy
JV, et al: A novel potassium channel gene, KCNQ2, is mutated in an
inherited epilepsy of newborns. Nat Genet. 18:25–29. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jentsch TJ: Neuronal KCNQ potassium
channels: Physiology and role in disease. Nat Rev Neurosci.
1:21–30. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cooper EC and Jan LY: M-channels:
Neurological diseases, neuromodulation, and drug development. Arch
Neurol. 60:4962003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goto A, Ishii A, Shibata M, Ihara Y,
Cooper EC and Hirose S: Characteristics of KCNQ2 variants causing
either benign neonatal epilepsy or developmental and epileptic
encephalopathy. Epilepsia. 60:1870–1880. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee IC, Yang JJ, Wong SH, Liou YM and Li
SY: Heteromeric Kv7.2 current changes caused by loss-of-function of
KCNQ2 mutations are correlated with long-term neurodevelopmental
outcomes. Sci Rep. 10:133752020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee IC, Chang TM, Liang JS and Li SY:
KCNQ2 mutations in childhood nonlesional epilepsy: Variable
phenotypes and a novel mutation in a case series. Mol Genet Genomic
Med. 7:e008162019. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Orhan G, Bock M, Schepers D, Ilina EI,
Reichel SN, Löffler H, Jezutkovic N, Weckhuysen S, Mandelstam S,
Suls A, et al: Dominant-negative effects of KCNQ2 mutations are
associated with epileptic encephalopathy. Ann Neurol. 75:382–394.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maljevic S, Naros G, Yalçin Ö, Blazevic D,
Loeffler H, Cağlayan H, Steinlein OK and Lerche H: Temperature and
pharmacological rescue of a folding-defective, dominant-negative KV
7.2 mutation associated with neonatal seizures. Hum Mutat.
32:E2283–E2293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miceli F, Soldovieri MV, Ambrosino P,
Barrese V, Migliore M, Cilio MR and Taglialatela M:
Genotype-phenotype correlations in neonatal epilepsies caused by
mutations in the voltage sensor of K(v)7.2 potassium channel
subunits. Proc Natl Acad Sci USA. 110:4386–4391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang X, Pan G, Li WH, Zhang LM, Wu BB,
Wang HJ, Zhang P and Zhou SZ: Analysis of gene mutation of early
onset epileptic spasm with unknown reason. Zhonghua Er Ke Za Zhi.
55:813–817. 2017.(In Chinese). PubMed/NCBI
|
|
31
|
Zeng Q, Yang X, Zhang J, Liu A, Yang Z,
Liu X, Wu Y, Wu X, Wei L and Zhang Y: Genetic analysis of benign
familial epilepsies in the first year of life in a Chinese cohort.
J Hum Genet. 63:9–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang L, Chen X, Liu X, Dong X, Ye C, Deng
D, Lu Y, Lin Y and Zhou W: Clinical features and underlying genetic
causes in neonatal encephalopathy: A large cohort study. Clin
Genet. 98:365–373. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang ZX, Zhang M, Xie LL, Jiang L, Hong
SQ, Li XJ, Hu Y and Chen J: KCNQ2 related early-onset epileptic
encephalopathies in Chinese children. J Neurol. 266:2224–2232.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie
H, Wu Y, Zhang Y, Wang J, Gao F, et al: Gene mutation analysis in
253 Chinese children with unexplained epilepsy and
intellectual/developmental disabilities. PLoS One. 10:e01417822015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Lu Y, Dong X, Lu G, Cheng G, Qian
Y, Ni Q, Zhang P, Yang L, Wu B and Zhou W: Optimized trio genome
sequencing (OTGS) as a first-tier genetic test in critically ill
infants: Practice in China. Hum Genet. 139:473–482. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fung CW, Kwong AK and Wong VC: Gene panel
analysis for nonsyndromic cryptogenic neonatal/infantile epileptic
encephalopathy. Epilepsia Open. 2:236–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou P, He N, Zhang JW, Lin ZJ, Wang J,
Yan LM, Meng H, Tang B, Li BM, Liu XR, et al: Novel mutations and
phenotypes of epilepsy-associated genes in epileptic
encephalopathies. Genes Brain Behav. 17:e124562018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu N, Schoch K, Luo X, Pena LDM, Bhavana
VH, Kukolich MK, Stringer S, Powis Z, Radtke K, Mroske C, et al:
Functional variants in TBX2 are associated with a syndromic
cardiovascular and skeletal developmental disorder. Hum Mol Genet.
27:2454–2465. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He M, Tang BS, Li N, Mao X, Li J, Zhang
JG, Xiao JJ, Wang J, Jiang H, Shen L, et al: Using a combination of
whole-exome sequencing and homozygosity mapping to identify a novel
mutation of SCARB2. Clin Genet. 86:598–600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang B, Li H, Xia K, Jiang H, Pan Q, Shen
L, Long Z, Zhao G and Cai F: A novel mutation in KCNQ2 gene causes
benign familial neonatal convulsions in a Chinese family. J Neurol
Sci. 221:31–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu T, Gong X, Bei F, Ma L, Chen Y, Zhang
Y, Wang X, Sun J, Wang J, Qiu G, et al: Application of
next-generation sequencing for genetic diagnosis in neonatal
intensive care units: Results of a multicenter study in China.
Front Genet. 11:5650782020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ambrosino P, Alaimo A, Bartollino S,
Manocchio L, De Maria M, Mosca I, Gomis-Perez C, Alberdi A, Scambia
G, Lesca G, et al: Epilepsy-causing mutations in Kv7.2 C-terminus
affect binding and functional modulation by calmodulin. Biochim
Biophys Acta. 1852:1856–1866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Miceli F, Soldovieri MV, Iannotti FA,
Barrese V, Ambrosino P, Martire M, Cilio MR and Taglialatela M: The
voltage-sensing domain of K(v)7.2 channels as a molecular target
for epilepsy-causing mutations and anticonvulsants. Front
Pharmacol. 2:22011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li P, Chen Z, Xu H, Sun H, Li H, Liu H,
Yang H, Gao Z, Jiang H and Li M: The gating charge pathway of an
epilepsy-associated potassium channel accommodates chemical
ligands. Cell Res. 23:1106–1118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miceli F, Vargas E, Bezanilla F and
Taglialatela M: Gating currents from Kv7 channels carrying neuronal
hyperexcitability mutations in the voltage-sensing domain. Biophys
J. 102:1372–1382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Watanabe H, Nagata E, Kosakai A, Nakamura
M, Yokoyama M, Tanaka K and Sasai H: Disruption of the epilepsy
KCNQ2 gene results in neural hyperexcitability. J Neurochem.
75:28–33. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ackermann S, Quandt D, Hagenbuch N, Niesse
O, Christmann M, Knirsch W and Kretschmar O: Transcatheter atrial
septal defect closure in children with and without fluoroscopy: A
comparison. J Interv Cardiol. 2019:65986372019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
van der Linde D, Konings EE, Slager MA,
Witsenburg M, Helbing WA, Takkenberg JJ and Roos-Hesselink JW:
Birth prevalence of congenital heart disease worldwide: A
systematic review and meta-analysis. J Am Coll Cardiol.
58:2241–2247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arvanitaki A, Giannakoulas G, Baumgartner
H and Lammers AE: Eisenmenger syndrome: Diagnosis, prognosis and
clinical management. Heart. 106:1638–1645. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Constantine A, Dimopoulos K and Opotowsky
AR: Congenital heart disease and pulmonary hypertension. Cardiol
Clin. 38:445–456. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Penaloza D and Arias-Stella J: The heart
and pulmonary circulation at high altitudes: Healthy highlanders
and chronic mountain sickness. Circulation. 115:1132–1146. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mirrakhimov AE and Strohl KP:
High-altitude pulmonary hypertension: An update on disease
pathogenesis and management. Open Cardiovasc Med J. 10:19–27. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang N, Cheng W, Wang B, Liu Q and Zhou C:
Geomorphological regionalization theory system and division
methodology of China. J Geog Sci. 30:212–232. 2020. View Article : Google Scholar
|