Introduction
With the aging of the population and the
transformation of lifestyles, cardiovascular and cerebrovascular
diseases have become major public health problems threatening
individuals' lives and health. Research data show that the
prevalence of cardiovascular diseases is high (1). With the advancement of basic research
and clinical practice, related therapies and research have achieved
remarkable results. However, they still face numerous challenges,
including in-depth analysis of pathological mechanisms, exploration
of new therapeutic targets, and development of effective
therapeutic strategies.
In cardiovascular research, mitochondria act as a
‘power plant’ of cells, converting chemical energy in organic
matter into chemical energy in ATP through cellular respiration,
such as oxidative phosphorylation, providing direct energy for
cellular activities (2). This
function has attracted considerable attention in recent years.
Moreover, as the ‘processing workshop’ and ‘logistics system’ of
cells, the endoplasmic reticulum (ER) is not only involved in
protein synthesis, processing and transportation but is also
responsible for lipid synthesis, metabolism and detoxification of
foreign substances, which are essential for cell function
maintenance (3). However, studies
on the relationship between mitochondria and the ER remain
insufficient.
Electron microscopy revealed a narrow space of
~10–30 nm between mitochondria and the ER (4). Numerous protein interactions can
occur in this space to promote the orderly progress of biochemical
reactions. These proteins form the basis of the functions of
mitochondria and the ER, and this specific space is also
hypothesized to have special functions. It was mistaken for
artifacts owing to technical limitations (5); however, in previous years, this
structure has been objectively confirmed with the development of
detection technology, and mitochondria-associated membranes (MAMs)
may be this special place (6).
Research on MAMs as new targets for the treatment of
cardiovascular diseases is increasing. Interference with specific
protein complexes or lipid metabolism pathways in MAMs is expected
to restore cell Ca2+ homeostasis, lipid metabolism
balance and mitochondrial function and reduce apoptosis and
myocardial injury (7). In
addition, promoting MAM-mediated autophagy may help clear damaged
organelles and proteins and maintain cell homeostasis and survival.
Given the important role of MAMs in cardiovascular diseases,
regulating their function may provide a new way to improve the
prognosis and therapeutic effects of cardiovascular diseases.
Therefore, the present review aimed to elaborate on
the concept of MAMs, explore their structural characteristics,
comprehensively elaborate on the basic functions of MAMs based on
the proteins in the structure of MAMs and their interaction with
mitochondria and the ER, and highlight the application of MAMs in
cardiovascular diseases. The present study contributed to the
understanding of the relationship between MAMs and cardiovascular
diseases and provided new ideas and directions for the study of
cardiovascular diseases.
The present article is a literature review, with the
following key words being used for literature search: i) MAMs, ii)
Mitochondria and iii) ER. PubMed database (https://pubmed.ncbi.nlm.nih.gov/) was utilized and the
screening methods were: i) Summarizing the content of the article
by reading articles in the past 5 years and ii) eliminating
articles that were inconsistent with key words.
Special position of MAMs
Before understanding MAMs, it is necessary to
clarify that they are not immutable. Consistent with the highly
dynamic characteristics of mitochondria, the distance between MAMs,
mitochondria and the ER often changes according to the density and
difference in ribosomes. When the ribosome density near the smooth
ER is low, the distance between ribosomes and mitochondria is
usually 10–15 nm. The rough ER has a large number of ribosomes
attached to it, and the density of ribosomes is high; therefore,
the distance between ribosomes and mitochondria can increase to
20–30 nm (8). This feature also
indirectly indicates that MAMs have a relationship with
mitochondria. The specific reasons for how ribosomes control the
change in distance remain unknown, but it is sufficient to show
that the special structure of MAMs plays a specific role in
different types of organelles.
As a space shared by mitochondria and the ER, in
recent years, increasing evidence has proven the close relationship
between MAMs in mitochondria and the ER and it has gradually been
found that MAMs are involved in numerous cellular functions. Since
they can regulate functions closely related to mitochondria and the
ER, such as mitochondrial dynamics, lipid homeostasis (9) and Ca2+ homeostasis and as
the birthplace of numerous normal maintenance proteins that affect
numerous functions, MAMs can be vividly described as central
regulators. However, because of their unique location, the
organelles related to MAMs confirmed by current research are
limited to mitochondria and the ER. However, according to previous
studies, it is not difficult to hypothesize that the ER, as a place
for the initial processing of lipids and proteins in the Golgi
apparatus, is known as the ‘sorting station’ or ‘packaging
workshop’ of cells. It receives proteins and lipids from the ER and
further processes, sorting and packaging, in which Golgi bodies
modify proteins, such as through glycosylation, for specific
functions. The Golgi apparatus transports these processed
substances to specific parts of the cell, such as cell membranes,
lysosomes and mitochondria, or secretes them outside the cell
through exocytosis. The connection between the ER and Golgi
apparatus is also very close, suggesting that MAMs can indirectly
affect changes in the Golgi apparatus (10). Similarly, mitochondria must undergo
quality control processes such as fission, fusion and autophagy,
which are of great significance for cell metabolism, aging, disease
development, and therapeutic response. The process of autophagy
requires lysosomes to play the role of a ‘scavenger’. To protect
the integrity of mitochondrial function and stability of cells, and
prevent damaged mitochondria from causing damage to cells, damaged
mitochondria are specifically encapsulated in autophagosomes and
fused with lysosomes to complete the degradation of
mitochondrial-lysosomal complexes. This process is known as
mitophagy. Similarly, it is assumed that mitophagy-related signals
can be sent from MAMs, which are audited by mitochondria and
executed by lysosomes. This hypothesis has not yet been confirmed;
therefore, the specific mechanism of MAMs in various organelles
remains unclear.
Function of MAM structure
The structure of mitochondrial-ER membrane contact
sites is highly compatible with their function. Studies have shown
that there are numerous resident proteins on MAMs, and the
physiological functions of some of them have been elucidated.
However, due to the wide variety of proteins on MAMs, there are
some contradictions in the functional studies of different
proteins, leaving a broad space for future research.
Simultaneously, the effect of proteins on MAMs on mitochondria and
the ER is bidirectional, which also increases the complexity of
studying MAM function (Table
I).
 | Table I.Proteins related to MAMs. |
Table I.
Proteins related to MAMs.
| Functional
classification | Protein name | Functional
description |
|---|
| Calcium homeostasis
maintenance | Mitochondrial
calcium uniporter complex | It contains the
pore-forming subunit MCU and regulatory subunits, such as MICU1 and
MICU2, which are responsible for Ca2 entry into
mitochondria and regulate mitochondrial calcium uptake. MICU sets
the uptake threshold and regulates Ca2 exchange on both
sides of the intima through dimerization. |
|
| Mfn protein | Mfn2 makes MAMs
become a Ca2 transfer hub and participates in the
transport of Ca 2 between endoplasmic reticulum and mitochondria.
The control effect of Mfn2 on Ca 2 concentration is
controversial. |
|
| DJ-1 | It is expressed in
various tissues, located in the cytoplasm and membrane gap,
interacts with the IP3R3-Grp75-VDAC1 complex, regulates and
maintains the stability of MAMs, and participates in mitophagy and
homeostasis regulation. |
|
| TDP-43 | In physiology, it
is mainly in the nucleus, and in pathology, it is abnormally
aggregated. It is regulated by FUNDC1 and affects the stability of
MAMs. |
|
| CypD | Mitochondrial
cyclophilin regulates the opening and closing of mPTP, and abnormal
expression affects the structure and function of MAMs. |
|
| FUNDC1 | It is widely
distributed, concentrated in the heart, regulates mitosis and
mitochondrial dynamics, and its structural changes affect
mitochondrial morphology through different pathways, which is
critical for MAMs formation and disease occurrence. |
|
| SERCA | Endoplasmic
reticulum resides in the protein, actively pumps Ca2 Ω
into the ER to maintain normal ER Ca2 Ω level, and
mediates Ca2 Ω-related apoptosis. |
|
| IP3R1-GRP75-VDAC1
complex | MAMs play a
regulatory role, silencing GRP75 inhibits ER-mitochondrial calcium
transport and reduces mitochondrial oxidative stress. |
|
| ATAD3A | Endoplasmic
reticulum-mitochondrial interaction regulators in MAMs prevent
ISO-induced mitochondrial calcium accumulation and improve
mitochondrial dysfunction and ER stress. |
|
| Seipin | Endoplasmic
reticulum proteins, located in MAMs, interact with Ca2
storage regulators, control mitochondrial Ca2 input, and
regulate mitochondrial metabolism. |
| Lipid homeostasis
maintenance | PS
(phosphatidylserine) | The important
enzymes of endoplasmic reticulum, mainly in MAMs, are transported
to mitochondria and transformed into PE, and some PE return to
endoplasmic reticulum and transform into PC, and then distribute
other organelles. |
|
| Acetyl-CoA
cholesterol acyltransferase | It catalyzes
cholesterol to form cholesterol ester, regulates cell membrane
binding and cytoplasmic lipid storage, and is a marker of
MAMs. |
|
| MLCK | Phosphorylation of
MLC enhances its ability to recruit actin filaments to form tight
MAMs structure, which is involved in regulating lipid
homeostasis. |
|
| MLC | Under the action of
MLCK, it is involved in the formation of tight MAMs structure and
affects the storage and outflow of lipid cells. |
|
| Scavenger receptor
CD36 | The upregulated
expression of MAMs in metastasis-associated macrophages promotes
the uptake of lipid-rich extracellular vesicles by macrophages,
regulates lipid metabolism, drives M2 polarization, and promotes
tumor metastasis. |
|
| ORP5 and ORP8 | It is located or
enriched in MAMs, regulates PS transfer, maintains the normal
structure and function of mitochondria, or participates in seipin
recruitment to MAM-LD contact sites, and mediates the occurrence
and maintenance of LD. |
| Regulation of
mitochon-drial homeostasis | Mfn1 and Mfn2 | Mfn2 and its
heterodimer Mfn1 regulate mitochondrial outer membrane fusion. Mfn2
is important for mitochondrial morphology and function. |
|
| Drp1 | Motility-related
protein 1, which is involved in mitochondrial fission, interacts
with proteins such as FUNDC1 and is regulated by other proteins in
MAMs. |
|
| Mff | It is mainly
located in the outer membrane of mitochondria, containing
hydrophobic helix repeat sequence, transmembrane region and
N-terminal GTPase domain. It interacts with Drp1 and plays a key
role in mitochondrial division. |
|
| LonP1 | In cardiomyocytes,
MAMs are proteins that connect endoplasmic reticulum and
mitochondria. Deletion leads to decreased MAMs formation and
mitochondrial breakage. |
| Regulate
endoplasmic reticulum | VAPs | It forms a protein
complex with Mfn1/Mfn2, builds a physical connection between
mitochondria and endoplasmic reticulum, and promotes rapid exchange
of substances. |
| material exchange
and stress | ACAT | It plays an
important role in the transport of cholesterol from endoplasmic
reticulum to mitochondria, maintains membrane fluidity, prevents
abnormal cholesterol metabolism, and protects endoplasmic reticulum
integrity. |
|
| Bap31 | Endoplasmic
reticulum stress-related proteins affect ERS-related path ways to
ensure normal ERS. |
|
| PERK | Participate in
protein quality control, prevent protein misfolding and
aggregation, eliminate misassembled proteins, and maintain endo
plasmic reticulum function stability. |
|
| Fis1 | Mitochondrial
fission protein 1 homologues, with the participation of MAMs,
affect Drp1 recruitment, change mitochondrial morphology and
function, and participate in mitochondrial fission and endoplasmic
reticulum-induced apoptosis. |
Maintenance of Ca2+
homeostasis
The maintenance of Ca2+ homeostasis as a
key secondary messenger is crucial for cells. Numerous proteins on
MAMs endow them with unique functional properties, and mitochondria
play a key role in regulating Ca2+ homeostasis and
cytoplasmic Ca2+ signaling. Calcium flux across the
mitochondrial inner membrane is important for decoding cytoplasmic
Ca2+ signals and increasing ATP production.
Ca2+ ions enter mitochondria through the mitochondrial
Ca2+ uniporter complex (mtCU). The mtCU is not only a
Ca2+ channel, but it also decodes the dynamic
Ca2+ signal by reducing mitochondrial Ca2+
uptake at rest and increasing uptake when the physiological level
of Ca2+ increases. The large electrochemical gradient
generated by the electron transport chain on the mitochondrial
inner membrane drives mitochondrial Ca2+ uptake;
therefore, the mtCU requires a regulatory mechanism to prevent
continuous uptake and overload.
The mtCU is composed of multiple subunits, including
the pore-forming subunit, mitochondrial calcium uptake 1 (MCU), and
multiple regulatory subunits. The mitochondrial Ca2+
uptake protein (MICU), as the Ca2+ sensor of the MCU,
sets the threshold for mitochondrial Ca2+ uptake and
synergistically activates the MCU when the concentration of
Ca2+ increases (11).
The MICU regulates Ca2+ ion exchange on both sides of
the intima through dimerization to further regulate mitochondrial
Ca2+ concentration. MICU1 and MICU2 are important
members of the homologous protein family. They close the channel in
a low-Ca2+ environment through the MCU complex and
perform dimer rearrangement in a high-Ca2+ environment
to activate the MCU channel, thereby achieving precise regulation
of mitochondrial Ca2+ absorption (12).
In addition, the presence of mitofusim (Mfn) protein
makes MAMs key hubs for Ca2+ ion transfer. When
mitochondria absorb Ca2+ ions released from the ER to
MAMs through Mfn (13),
intracellular Ca2+ ions can enter the cytoplasm through
the Inositol-1,4,5-triphosphate receptor (IP3R) or RyR channels.
When the concentration of Ca2+ ions is low, the
absorption rate of Ca2+ by mitochondria decreases. When
the concentration of Ca2+ ions increases, the transfer
rate of Ca2+ ions between mitochondria and the ER
increases accordingly. Moreover, MAMs may also be involved in the
regulation of this process, but how Mfn regulates the movement of
Ca2+ ions in MAMs and the ER is inconclusive. Whether
Mfn2 is an important target for controlling Ca2+ ion
concentration in MAMs remains controversial (14).
In addition to the aforementioned important
proteins, MAMs also contain DJ-1, TDP-43, cyclophilin D (CypD), and
other proteins that are not directly involved in the structure of
MAMs but are essential for their regulatory functions. At present,
related research is gradually being carried out, among which the
study of FUN14 domain-containing protein 1 (FUNDC1) is the
clearest. FUNDC1 is widely distributed throughout the body,
particularly in the heart (15).
It consists of 155 amino acids, three transmembrane domains, and a
typical N-terminal leucine zipper transcription regulator motif
(5). This structure facilitates
the binding of microtubule-associated protein 1 light chain 3 to
regulate mitosis, which is more pronounced in hypoxic environments.
This is mainly due to decreased Src kinase activity and
phosphorylation of Tyr18, resulting in enhanced binding of
microtubule-associated protein 1 light chain 3 to FUNDC1 (16). In addition, nuclear respiratory
factor 1, a transcription factor, binds to promoter 186/176 of
FUNDC1 and activates FUNDC1 (17).
In addition to mitosis, when lysine 10 in FUNDC1 is
eliminated, mitochondria break, indicating that FUNDC1 has a
regulatory effect on mitochondrial dynamics (18). By contrast, FUNDC1 regulates
mitochondrial morphology. Inhibition of FUNDC1 in HeLa cells leads
to increased mitochondrial fusion. By contrast, promoting FUNDC1
expression inhibits mitochondrial fusion. This function achieves
mitochondrial fission by recruiting dynamin-related protein 1
(DRP1) to mitochondria (19). When
the S13 residue in FUNDC1 is phosphorylated, the connection between
FUNDC1 and DRP1 is weakened and the connection with optic atrophy
protein 1 is enhanced, synergistically regulating mitochondrial
dynamics (16). Thus, changes in
FUNDC1 structure affect mitochondrial morphology through different
pathways. Additionally, the upstream regulation of FUNDC1 and
changes in FUNDC1 expression play key roles in controlling the
formation of MAMs and the occurrence of different diseases.
Ca2+ ion flow is also regulated by the ER
resident protein SERCA, which maintains normal ER Ca2+
levels by actively pumping Ca2+ from the intracellular
space into the ER, and it also plays a crucial role in mediating
Ca2+-related apoptosis (20). In addition, the inositol
1,4,5-trisphosphate receptor type 3 (IP3R1)-heat shock protein
family A (Hsp70) member 9 (GRP75)-voltage-dependent anion channel
(VDAC1) complex is considered to play a regulatory role in MAMs.
Silencing GRP75 can inhibit ER-mitochondrial Ca2+
transport and reduce mitochondrial oxidative stress, thereby
preventing diabetes-induced atrial remodeling (21). Combot et al (22) reported that the ATPase family AAA
domain-containing protein 3A (ATAD3A) is an important regulator of
ER-mitochondrial interaction in MAMs. ATAD3A can prevent
isoproterenol-induced mitochondrial Ca2+ accumulation
and improve mitochondrial dysfunction and ER stress, thereby
protecting cardiac function and reducing cardiac hypertrophy. In
addition, studies have shown that the ER protein seipin is located
in MAMs and can control the input of mitochondrial Ca2+
ions by interacting with Ca2+ storage regulators, such
as SERCA2, IP3R, and VDAC1 in MAMs, thereby regulating the normal
operation of mitochondrial metabolism (22).
Maintenance of lipid homeostasis
MAMs are closely associated with lipid homeostasis
and interfere with lipid synthesis. The ER is the main synthesis
site of intracellular lipids. Lipids need to be synthesized by the
ER and delivered to other organelles, which requires the assistance
of mitochondria. Therefore, MAMs are crucial for maintaining lipid
homeostasis. In addition, MAMs are involved in the regulation of
cholesterol metabolism. When lipid levels are high, MAMs promote
cholesterol transport to the liver and accelerate its metabolism,
whereas when lipid levels are low, MAMs inhibit cholesterol
transport and help maintain intracellular cholesterol storage.
Phosphatidylserine is an important enzyme in the ER that is mainly
distributed on MAMs and transported to mitochondria for conversion
into phosphatidylethanolamine (6).
A small portion of PE is returned to the ER, converted into
phosphatidylcholine, and distributed to other organelles. MAMs are
also involved in the production of ceramides, regulation of cell
proliferation, inflammation, differentiation and apoptosis
(23,24). Acetyl-CoA cholesterol
acyltransferase plays an important role in lipid homeostasis by
catalyzing the formation of cholesterol esters and regulating cell
membrane binding and cytoplasmic lipid storage. It is often used as
a marker because of its abundance in MAMs. However, the mechanism
of action of lipid transfer proteins in MAMs remains to be
explored. Enzymes that maintain lipid homeostasis also play an
important role in maintaining the integrity of the mitochondrial
membrane, thereby strengthening the connection between the ER and
mitochondria. MAMs involved in the regulation of lipid homeostasis
are mainly composed of myosin light chain kinase and myosin light
chain protein. Myosin light chain kinase is responsible for
phosphorylating myosin light chain protein and enhancing its
ability to recruit actin filaments to form a tight MAM structure.
When the lipid level is high, the structure of MAMs is susceptible
to interference, the stability decreases, and fatty acids are more
likely to flow out of the cell; on the contrary, when the lipid
level is low, the structure of MAMs is stable, which is beneficial
to the storage of lipid in cells. Surprisingly, the expression of
the scavenger receptor CD36 is upregulated in the MAMs of
metastasis-associated macrophages. By promoting the uptake of
lipid-rich extracellular vesicles by macrophages, it regulates
macrophage lipid metabolism and drives M2 polarization to reshape
the tumor microenvironment and promote tumor metastasis (25).
Lipid droplets (LDs), the main organelles involved
in cellular lipid storage, originate from the ER (26). The ER resident protein seipin in
MAMs is a key regulator of LD biogenesis. Seipin mutations are
closely related to imbalances in intracellular lipid metabolism and
Ca2+ homeostasis (27).
Guyard et al (28) found
that oxysterol binding protein like (ORP)5 and ORP8 are also
localized or even enriched in specific ER subdomains in contact
with mitochondria, namely the MAM, and regulate the transfer of PS
to maintain the normal structure and function of mitochondria, or
are involved in the process of seipin recruitment to the MAM-LD
contact site, mediating the occurrence and maintenance of LDs.
Regulating mitochondrial
homeostasis
Mitochondria are a key source of power in mammalian
cells and are essential for the regulation of cell metabolism,
proliferation, survival and death (29–31).
It generates energy mainly through key metabolic pathways, such as
oxidative phosphorylation, tricarboxylic acid cycle, and fatty acid
β-oxidation, and produces most of the cellular reactive oxygen
species (ROS) through the electron transport chain.
MAMs mediate the exchange of different ions and
metabolites and play a key role in regulating mitochondrial
dynamics and Ca2+ homeostasis (32). Mitochondrial fission, fusion and
apoptosis constitute a complex dynamic network. In this process,
the Mfn2 protein and its heterodimer partner Mfn1 play a vital
role, and they jointly regulate the fusion process of the
mitochondrial outer membrane (33). MAMs are the initial sites of
mitochondrial fission (34).
Mitochondrial fission protein 1 (Fis1), mitochondrial fission
factor and mitochondrial dynamics proteins preferentially
accumulate in MAMs prior to mitochondrial fission (35). ATAD3A in MAMs can inhibit excessive
activation of mitochondrial fission, which is helpful in
maintaining the integrity of the mitochondrial ultrastructure
(36). Mfn2 is another resident
protein of MAMs that is present in the outer membrane of
mitochondria and is essential for mitochondrial morphology and
function. It is a key GTPase that is involved in mitochondrial
fusion. It has been found that it has the opposite function
(37). IP3R acts as a key
Ca2+ outflow channel on the ER surface, mediating the
flow of Ca2+ from the ER lumen to the cytoplasm. The
Ca2+ homeostasis in mitochondria is controlled by the
VDAC protein on the outer membrane of mitochondria, which controls
the inflow and outflow of Ca2+ to maintain
Ca2+ homeostasis. The two need to be connected by Grp75
in MAMs to maintain the structure of MAMs. The specific mechanism
of the interaction between the three remains to be further studied
(38). Other proteins such as DJ-1
are expressed in various tissues and are mainly located in the
cytoplasm and membrane. Under oxidative stress, DJ-1 mainly
regulates and maintains the stability of MAMs by interacting with
the IP3R3-Grp75-VDAC1 complex, which can be transferred to the
mitochondria to participate in mitophagy and regulate mitochondrial
homeostasis (39). TDP-43 is a
nucleic acid-binding protein encoded by TARDBP. It is mainly
localized to the nucleus under physiological conditions, whereas
abnormal aggregation occurs under pathological conditions. It has
been recently reported that the mitophagy receptor FUNDC1 can
regulate the structure and homeostasis of TDP-43 through its own
expression differences and interaction with different proteins, and
then participate in the quality control process of mitochondria and
affect the stability of MAMs (40). CypD is a cyclophilin located in
mitochondria, which can regulate the opening and closing of the
mitochondrial permeability transition pore (mPTP). Abnormal CypD
expression can affect the structure and function of MAMs, thereby
interfering with disease development (41).
Ca2+ homeostasis affects mitochondrial
dynamics. Mitochondrial Ca2+ and ROS synergistically
regulate the opening of mPTPs. Excessive ROS production may lead to
abnormal mitochondrial Ca2+ accumulation, which in turn
affects mitochondrial function. Under oxidative stress conditions,
MAMs produce a small increase in ROS, which maintains cell survival
by reducing the ER-mitochondrial Ca2+ transfer (42,43).
Excessive Ca2+ induces excessive production of
mitochondrial ROS through MAM transfer (32). Inhibition of FUNDC1 and reduction
in MAM formation can improve mitochondrial ROS overproduction
(44). Ca2+ channel
modulators in MAMs play a key role in the regulation of ROS. ERO1α
and ERp44 are highly enriched enzymes in MAMs. ERO1α contains a
redox domain responsible for catalyzing the formation and
isomerization of disulfide bonds. It is mainly located in the ER
and participates in oxidative folding of proteins, indirectly
affecting the structure and function of MAMs. ERp44 also exhibits
disulfide isomerase activity and can repair misfolded proteins.
Although it is not directly localized to MAMs, it can circulate
between the ER and Golgi apparatus, indirectly affecting MAM
function (45). Similarly, INF2,
an ER-localized protein, aggregates with Drp1 and initiates the
initial mitochondrial fission process (45). ERO1α causes the separation of ERp44
and IP3R by oxidizing IP3R, thereby exacerbating the transfer of
Ca2+ from the ER to mitochondria and leading to
excessive ROS production (46).
It has been shown that mtDNA replication,
mitochondrial transport, and mitochondrial fission occur in MAMs
(9). Notably, MAMs have been
recently regarded as the starting point of mitochondrial fission,
where Drp1, Fis1 and Mff are present in large numbers to
participate in this process (45).
Mff is mainly located in the outer membrane of mitochondria and
participates in the localization process. Its N-terminus also
contains a GTPase domain that plays a critical role in
mitochondrial division by interacting with Drp1 (47), thereby maintaining mitochondrial
health and normal function. In cardiomyocytes, LonP1 connects the
ER and mitochondria in MAMs, and its deletion leads to decreased
MAM formation, which in turn causes mitochondrial rupture (45). Thus, MAMs play a role in the
survival of mitochondria not only from their own proteins, but also
through the ER, an organelle.
MAMs regulate ER material exchange and
stress
As a key link between mitochondria and the ER, MAMs
are essential for cell activity (48). MAMs are important platforms for
substance exchange, allowing for the effective exchange of key
molecules and substances, such as Ca2+ ions,
phospholipids and cholesterol, between two organelles to maintain
the normal function and metabolic stability of related organelles.
MAMs form protein complexes through Mfn1/Mfn2 and amine oxidase
copper containing 3 (VAP) to build a direct physical connection
between mitochondria and the ER, thereby promoting the rapid
exchange of Ca2+ ions, phospholipids and other
metabolites. MAMs are rich in cholesterol acyltransferase and play
an important role in cholesterol transport from the ER to
mitochondria. After synthesis of the ER, it is transported to other
organelles, such as mitochondria, through lipid rafts to meet the
structural and functional requirements of target organelles,
maintain fluidity between mitochondria and the ER membrane, prevent
metabolic abnormalities of cholesterol, and protect the integrity
of the ER. In this process, MAMs may be involved in the formation
and maintenance of lipid rafts, which are of great significance for
maintaining their function (49,50).
MAMs are also important for the regulation of ER stress (ERS)
(51). MAMs are rich in the
Ca2+ release channel protein IP3R as the receptor of IP3
in the ER and have the pore protein VDAC1 of the mitochondrial
outer membrane, which is connected to form a complex through the
molecular chaperone GRP75, thereby promoting Ca2+ ion
signal transmission between the ER and mitochondria to maintain the
balance and stability of Ca2+ ions (52), which is crucial for the regulation
of ERS. Because Ca2+ fluctuations can activate the
unfolded protein response, and proteins in the ER must be correctly
folded to perform corresponding functions, MAMs participate in the
quality control process of proteins through their own proteins,
such as eukaryotic translation initiation factor 2α kinase 3
(PERK), to prevent misfolding of proteins and excessive aggregation
of misfolded proteins, while removing misassembled proteins to
maintain the stability of ER function. There are also some
ERS-related proteins on MAMs (53), such as B cell receptor-associated
protein 31 (BAP31) and Mfn2, which can directly or indirectly
affect the related pathways of ERS and ensure the normal progress
of ERS.
In addition to these functions, MAMs participate in
the regulation of autophagy, apoptosis, and numerous other
biological processes. For example, MAMs affect cell apoptosis and
signal transduction through specific proteins (53), such as IP3R, VDAC1 and PERK, and
thus play a key role in maintaining the stability of the
intracellular environment and coordination between organelles. For
example, Fis1 and BAP31 are resident proteins on MAMs. Fis1 has an
effect on the recruitment of Drp1 under the participation of MAMs,
as well as the morphological and functional changes in
mitochondria, which have an important impact on mitochondrial
fission, and participates in ER-induced apoptosis (54), thereby maintaining cell
homeostasis.
In summary, MAMs play a vital role in cells. As a
bridge between the ER and mitochondria, they are involved in
several cellular physiological and pathological processes. MAM
dysfunction is also associated with various diseases, including
neurodegenerative (53),
cardiovascular and metabolic diseases (55). Therefore, studying MAMs is of great
significance for the diagnosis and treatment of cardiovascular
diseases (Fig. 1).
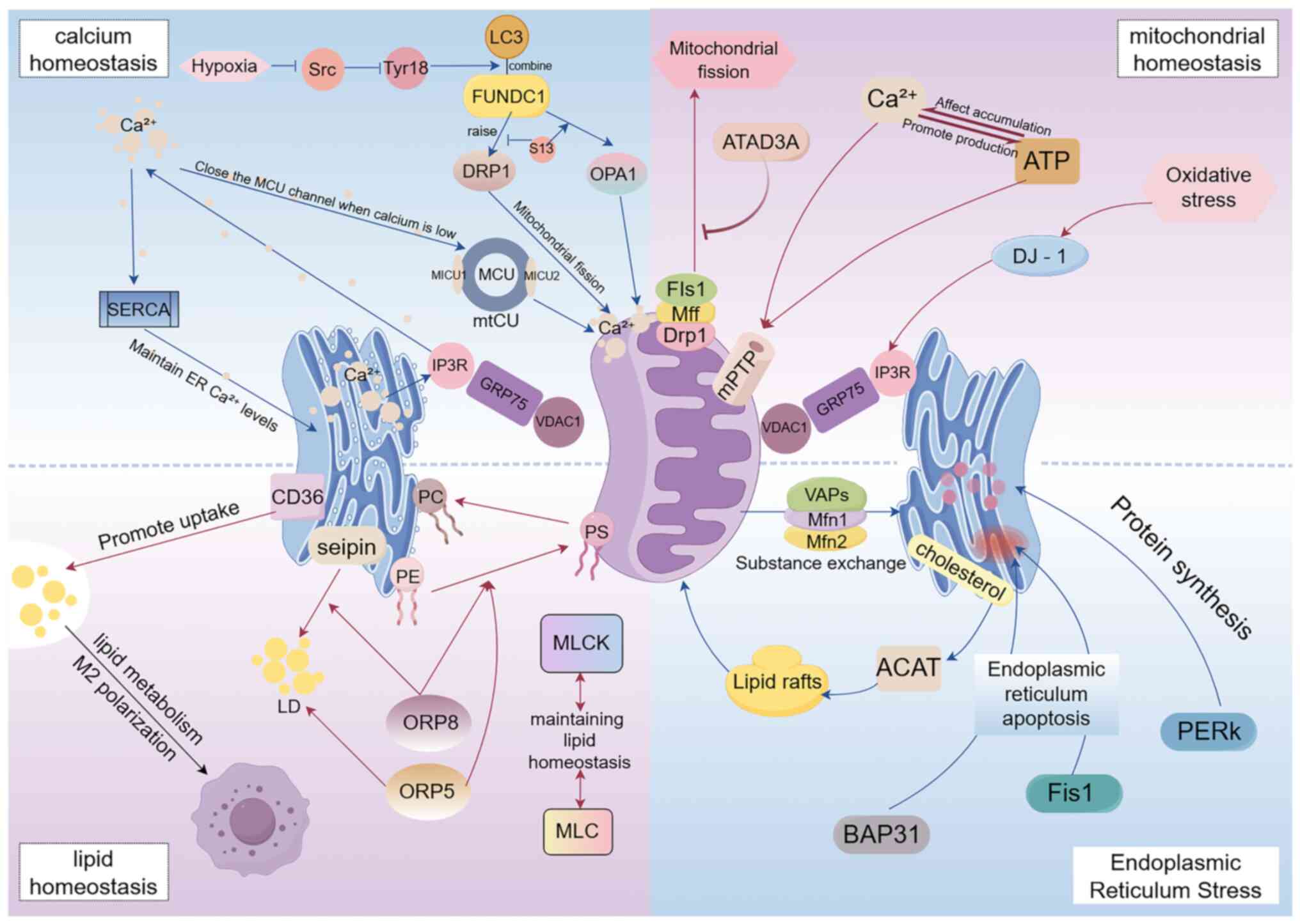 | Figure 1.Summary diagram of the functions
created by the structure of MAMs. MAMs, mitochondria-associated
membranes; FUNDC1, FUN14 domain-containing protein 1; Drp1,
recruiting dynamin-related protein 1; MICU, mitochondrial
Ca2+ uptake protein; MCU, mitochondrial calcium uptake
1; MTCU, mitochondrial Ca2+ uniporter complex; SERCA,
ATPase sarcoplasmic/endoplasmic reticulum Ca2+
transporting 2; IP3R1, inositol 1,4,5-trisphosphate receptor type
3; GRP75, heat shock protein family A (Hsp70) member 9; VDAC1,
voltage-dependent anion channel; mff, mitochondrial fission factor;
DJ-1, parkinsonism associated deglycase; VAPs, amine oxidase copper
containing 3; Mfn, mitofusim; ACAT, cholesterol acyltransferase;
PERK, eukaryotic translation initiation factor 2 α kinase 3; Fis1,
mitochondrial fission protein 1; Bap31, B cell receptor-associated
protein 31; MLCK, myosin light chain kinase; MLC, myosin light
chain protein; ORP, oxysterol binding protein like; KLF4, KLF
transcription factor 4. |
MAMs and cardiovascular diseases
Diabetic cardiomyopathy (DCM)
DCM is a unique complication of diabetes and a major
cause of death. Its pathological features include cardiomyocyte
hypertrophy, interstitial fibrosis and impaired coronary
microvascular perfusion (56). DCM
usually has no clinical symptoms in the early stages; however,
patients generally show abnormal cardiac diastolic function.
Subsequently, in the absence of dyslipidemia, hypertension, or
coronary heart disease, cardiac diastolic or systolic dysfunction
occurs, which eventually leads to heart failure (HF) and is one of
the main causes of death in patients with diabetes (57). Studies have shown that, compared
with non-diabetic patients of the same age, the incidence of HF in
male patients with diabetes increased by 2.4 times, whereas that in
female patients increased by 5 times. However, owing to the lack of
large amount of data on different populations with diabetes, the
incidence of DCM remains unclear. Other studies have reported that
the prevalence of diastolic dysfunction in patients with type 2
diabetes is high, reaching 40–60% (58,59).
Presently, it is considered that comorbid factors such as chronic
systemic hypertension and vascular atherosclerosis (AS) can promote
the occurrence of cardiovascular diseases in patients with
diabetes, but insulin resistance and hyperglycemia are undoubtedly
the basis and core of the occurrence and development of DCM
(60). The potential molecular
mechanism underlying DCM is not fully understood and involves
multiple factors and complex pathophysiological processes,
including oxidative stress, neurohormone activation,
Ca2+ homeostasis damage and mitochondrial damage
(16). Abnormal glucose and lipid
metabolism can lead to inflammatory responses, oxidative stress and
ERS, which promote each other and form a vicious circle, leading to
myocardial death, hypertrophy, fibrosis, microcirculation
disorders, and ultimately, HF (61). Energy metabolism remodeling is the
core hub connecting diabetes and HF. As the most energy-consuming
organ of the human body, the heart has very little energy reserve
and requires a continuous ATP supply to maintain normal operation.
In diabetic HF, the two core energy ‘reactors’ of the heart are
disordered and toxic; one is the imbalance of fatty acid oxidation
and utilization; the supply and uptake of fatty acids in
cardiomyocytes are increased, but the ability of mitochondrial
oxidation and utilization is decreased, resulting in myocardial
lipid accumulation and lipotoxic substances; second, the glucose
supply and uptake of cardiomyocytes are increased, but the aerobic
oxidative phosphorylation of myocardium is decreased, anaerobic
glycolysis is increased, and glucotoxic substances are produced
(62–64). Additionally, DCM cardiomyocytes
lose the ability to randomly select substrates and rely on fatty
acid utilization to supply energy. This contradiction forces the
heart to compensate for high energy consumption, high oxygen
consumption, low supply, and low efficiency. Long-term overload
eventually causes the myocardium to enter a state of decompensation
and failure (65).
In previous years, studies have indicated that MAMs
play an important role in DCM. In a mouse model of type 2 diabetes,
the connection between the ER and mitochondria was enhanced in the
early stages (66,67). Wu et al (68) confirmed this in high
glucose-treated mouse cardiomyocytes and mice carrying the insulin
2 gene; however, the specific mechanism has not been elucidated.
Changes in FUNDC1 levels in cardiac tissues of patients with
diabetes are significant. Decreased FUNDC1 expression inhibits
abnormal cardiac tissue in patients with diabetes, which may be
closely related to the Ca2+ transfer function of MAMs.
Wang et al (69) detected
mitochondrial Ca2+ overload in patients with high
glucose levels. Under normal circumstances, MAMs ensure the
stability of mitochondrial and ER contact points and play an
important role in maintaining intracellular Ca2+ balance
and lipid metabolism. In the case of diabetes, when the level of
FUNDC1 increases, it interacts with IP3R on MAMs, promotes MAM
formation, increases Ca2+ concentration in mitochondria,
hinders the normal energy metabolism function of mitochondria, and
affects the production process and redox state of cardiomyocytes,
thus affecting the function of the whole heart. In response to this
situation, Xie et al (70)
found that metformin improved cardiac structure by reducing FUNDC1
levels by inactivating AMPK, suggesting that MAMs are potential
targets for metformin to improve blood vessels. FUNDC1 interacts
with IP3R2 to promote Ca2+ transfer in DCM. When glucose
concentration increases, the activation of FUNDC1 promotes the
binding of cyclic AMP response element-binding protein to the Fis1
promoter, thereby promoting MAM formation. Thus, FUNDC1 has a
maintenance effect on MAMs. Inhibition of FUNDC1 leads to the
formation of a large number of MAMs, changes in mitochondrial
morphology and function, initiation of apoptotic procedures, and
abnormal death of numerous mitochondria, resulting in reduced
mitochondrial synthesis and failure of the respiratory chain to
operate normally. In response to this problem, Ding et al
(71) reduced the formation of
MAMs using ferulic acid and metformin, enhanced the expression of
FUNDC1, and successfully ameliorated the symptoms of cardiomyopathy
caused by diabetes.
In the context of DCM, the function of phosphofurin
acidic cluster sorting protein 2 (PACS-2) has also received
significant attention. As a multifunctional sorting protein that
plays a role in MAMs, PACS-2 plays a key role in maintaining the
homeostasis of mitochondria, ER and lysosomes (72). For patients with diabetes, the
decreased level of PACS-2 in MAMs in vivo may lead to the
destruction of the structure and function of MAMs, which may be
related to the IP3R-Grp75-VDAC1 complex regulating Ca2+
homeostasis and mitochondrial autophagy dysfunction (73). The significant decrease in the
complex IP3R-VDAC1 detected in the subjects confirmed this
conclusion (66). In a diabetic
mouse model, SIRT3 inhibited the excessive proximity of Mito-ER and
increased ROS by limiting the formation of abnormal MAMs, while
reducing the levels of the pro-apoptotic protein Bax, activating
caspase 3, and increasing the level of the anti-apoptotic protein
Bcl2, thereby inhibiting apoptosis. SIRT3 can also promote the
deacetylation of VDAC1 and reduce the enhancement of the
interaction between IP3R, GRP75 and VDAC1 in MAMs, thereby reducing
the formation of MAMs and protecting neurons from damage (74). Salin et al (75) found that under high glucose
conditions, the upregulation of phosphofurin acidic cluster sorting
protein 2 (PACS2) promotes the formation of MAMs, while
reducing mitochondrial biosynthesis and oxidative phosphorylation,
and driving mitochondrial apoptosis, which is crucial in the
molecular pathogenesis of DCM. Basic helix loop helix ARNT like 1
(Bmal1) plays a key role in heart health, and changes in its
expression directly affect the heart function. Downregulation of
Bmal1 aggravates cardiac hypertrophy and DCM, whereas its
upregulation improves symptoms. Bmal1 reduces mitochondrial
Ca2+ overload and apoptosis in MAMs by regulating the
Bcl2/IP3R pathway, thereby providing a new direction for DCM
treatment (76).
AS
AS is a progressive chronic vascular disease. It
refers to a lesion on the inner wall of large and medium arteries.
It mainly manifests as lipid deposition in the intima of the artery
and gradually accumulates to form atherosclerotic plaques,
eventually leading to stenosis or occlusion of the arterial lumen.
It is an important branch of cardiovascular and cerebrovascular
diseases. The common risk factors for AS include hypertension,
smoking, hyperlipidemia, diabetes and obesity (77). AS mortality, morbidity and
disability rates are high and show a trend toward a younger age.
One reason why AS has these characteristics is that plaques are
more likely to rupture during the evolution of the disease,
resulting in more dangerous clinical events secondary to
thrombosis, such as stroke and acute coronary syndrome. Currently,
clinical diagnosis mainly relies on ultrasound, magnetic resonance
imaging and CT imaging, and early identification and prognostic
judgment are still lacking (78).
Widely used clinical treatment methods include best medical
treatment (BMT) and surgical treatment, but long-term BMT treatment
undoubtedly increases the liver burden for patients and may also
cause adverse reactions such as gastrointestinal discomfort and
cardiac rhythm disorders (79).
Surgical treatment may lead to postoperative bleeding, recurrence,
and other consequences in patients with a high preoperative status.
Therefore, in recent years, the diagnosis, treatment and late
detection of thrombotic plaques in AS have gradually become the
focus of global AS prevention and treatment.
Inflammation is generally considered to be a key
factor in all stages of AS, involving macrophages, lymphocytes,
dendritic cells, endothelial cells, vascular smooth muscle cells,
and various cytokines and adhesion factors. Oxidized low-density
lipoprotein triggers NF-κB through the CD36 receptor, thereby
triggering an inflammatory response that eventually leads to
vascular wall destruction and thrombosis (80). In recent years, the role of immune
factors in AS progression has attracted attention, and researchers
have explored AS treatment strategies that target the immune
response. Nanomedicine has shown potential in the diagnosis and
treatment of AS, but it also faces biosafety challenges (78). The effect of gut microbiota on
cardiovascular diseases provides a new approach for the targeted
treatment of AS. Intestinal flora may be involved in the
development of AS by affecting the inflammatory response and
endothelial cell function, as well as being associated with changes
in blood pressure. The gut and oral microbes found in
atherosclerotic plaques, as well as the association between plaque
stability and Roseburia fecal levels, further confirmed this
hypothesis (81).
A number of studies have shown that MAMs can
crosstalk with inflammation in cardiovascular diseases, including
in the field of AS (82–85). For example, studies have shown that
oxidized low-density lipoprotein can increase the expression of
MAM-related PACS-2 protein in cells, thereby mediating MAM
formation by promoting ER-mitochondrial contact (85). Chen et al (85) found that PACS-2 is an essential
factor for normal mitophagy in vascular smooth muscle cells. Its
downregulation inhibits mitophagy and reduces the assembly of the
inflammasome NLR family pyrin domain containing 3 (NLRP3), which in
turn enhances cell death (85). In
addition, MAMs, as a platform for the assembly of the inflammasome
NLRP3, can also affect the release of the chronic inflammatory
factors IL-1β and IL-18 (82). The
dissociation of hexokinase 2 and VDAC triggers the activation of
IP3R, which mediates the production of ER-mitochondrial
Ca2+ current, leading to VDAC oligomerization and the
formation of NLRP3 inflammasome complex (86). Therefore, targeting MAM formation
or NLRP3 and its upstream steps is a potential strategy for
treating AS.
From the current research status, Mfn2 protein is
also a promising therapeutic target for AS. Studies have confirmed
that Mfn2 can inhibit calcification of blood vessels in AS by
activating the RAS-RAF-ERK1/2 pathway (87). Yimai granules regulate mitophagy
mediated by the PTEN induced kinase 1 (Pink1)-Mfn2-Parkin pathway
through miRNA-125a-5p and regulate pro-inflammatory factors and
blood lipids to inhibit AS (88).
In addition to miRNA-125a-5p, MiR-93 acts on Mfn2 protein to
regulate the proliferation and migration of smooth muscle cells
(89). The ER resident protein
RTN4 has the potential to regulate MAM formation, and NUS1
dehydrodolichyl diphosphate synthase subunit (Nogo-B) is an
important member of this family. Nogo-B is highly expressed in
vascular endothelial cells and smooth muscle cells (7). Clinical studies have shown that
Nogo-B expression is upregulated in human atherosclerotic plaques.
Zhang et al (90) further
explored its mechanism of action, proving that Nogo-B mediates
inflammation by triggering mitochondrial ROS production and p38-p65
signaling, thereby affecting the normal function of vascular
endothelial cells.
Phosphorylation of the Ca2+ channel IP3R
directly affects the recruitment of Ca2+ from the ER to
the cytoplasm and regulates platelet activation and aggregation in
AS. Artesunate induces the phosphorylation of IP3R by increasing
cAMP content in platelets and indirectly downregulating the
recruitment of Ca2+ ions in platelets, platelet
aggregation and thrombosis, thereby improving the prognosis of
patients with cardiovascular disease (91). Targeting the Ca2+ ion
flow during platelet activation is an effective treatment approach.
The relationship between the mitophagy downstream mediator DJ-1 and
AS can be observed in the regulation of vascular smooth muscle cell
surface conversion and plaque stability. The underlying mechanism
is that DJ-1 inhibits the KLF transcription factor 4 pathway,
thereby affecting the phenotypic transformation of Lamin A/C (VSMC)
(92). In addition, downregulation
of DJ-1 significantly increases plaque vulnerability by affecting
matrix proteins and metalloproteinases. Thus, DJ-1 also plays an
important role in plaque stability (92).
HF
HF, the end stage of various cardiovascular
diseases, is a complex clinical syndrome. As the heart cannot pump
enough blood and oxygen to support the metabolic needs of other
organs, it often manifests clinically as dyspnea, lower-limb edema
and fatigue. Owing to the rapid increase in morbidity and mortality
rates, it has become a serious public health problem.
A total of ~64 million individuals worldwide suffer
from HF, and this number is increasing annually owing to factors
such as an aging population (41).
According to statistics from the European Society of Cardiology,
>15 million/~1 billion individuals have HF (23). In addition, the National Hospital
Discharge Survey in the United States showed that the number of
patients over 65 years of age diagnosed with congestive HF
increased by 48.5% from 1970 to 1985. Epidemiological studies in
China have found that ~8.9 million individuals are currently
affected, an increase of 44% compared with that in 2000 (24,25).
According to research, the prevalence of HF in China has exceeded
2–3% (35). It can be observed
that HF has become a serious public health problem, despite the
diversity of current treatments. However, hospitalization and
mortality rates remain high (14).
Currently, it is considered that the
pathophysiological mechanism of HF is extremely complex. It is
generally considered that myocardial cell mitochondria, one of the
main areas of cell aerobic respiration and energy metabolism, are
widely involved in cell proliferation, differentiation, apoptosis,
information transmission, energy metabolism, and other
pathophysiological processes. Their dysfunction is closely
associated with the occurrence and development of HF (93). It is necessary to conduct more
in-depth and detailed prevention and control studies in this
field.
Patients with myocardial infarction (MI), myocardial
injury, and even necrosis are prone to HF. By examining mice after
MI, it was found that the SR Ca2+ of MAMs was damaged
through the RyR2 channel, resulting in a large accumulation of
Ca2+ in mitochondria, which changed mitochondrial
activity and increased the probability of MI. In addition, FUNDC1
can affect the connections between SR Ca2+ channels.
Under normal conditions, FUNDC1 interacts with FBXL2 to degrade
IP3R3, thereby maintaining balanced mitochondrial mass. However,
the lack of FUNDC1 in mice fed a high-fat diet disrupted this
balance (68,94), causing myocardial cell remodeling
and HF. The Sig-IR pathway is also present in mice fed a high-fat
diet (95). Under normal
conditions, Sig-IR prevents the death of mice by inhibiting the
destruction of myocardial cells caused by oxidative stress, whereas
the Sig-IR pathway in mice fed a high-fat diet is reversed by
mitochondrial Ca2+ overload.
Mfn2 also plays an important role in mice with HF.
The upregulation of Mfn2 reverses the production of ROS and
depolarization of mitochondria, thus avoiding changes in the
hypertrophic phenotypes of cardiomyocytes (96). This was also confirmed by the
decrease in Mfn2 levels in mice with HF (97). The expression levels of Fis1 and
DRP1 in mice with HF after myocardial ischemia-reperfusion were
downregulated by drugs, while the expression level of Mfn2 was
upregulated; this effectively prevented abnormal division and loss
of mitochondria and protected myocardial function. During this
process, the expression of Mfn1 increased (98).
BAP31 is a membrane protein on MAMs that is
involved in apoptosis and autophagy (99). The BAP31-Fis1 complex is involved
in the regulation of Ca2+ release from the ER, thereby
activating mitochondria and leading to apoptosis. There is a close
relationship between PACS2 and BAP31. When PACS2 concentration
decreases, BAP31 is cleaved into p20, resulting in the release of
Ca2+ from the ER. Studies have shown that myocardial
injury is aggravated after reduced PACS2 concentration. This result
suggests that BAP31 on MAMs is associated with HF (100).
In addition, decreased FUNDC1 expression has been
detected in both patients with HF and mouse cardiomyocytes
(101,102). Further studies have shown that
FUNDC1 mainly damages mitochondrial function and cardiomyocytes.
There is an inevitable link between mitochondrial dysfunction and
MAM destruction (102). Similar
to metformin, α-lipoic acid reduces the phenotypic changes in
cardiomyocytes by inhibiting FUNDC1 (103). In addition, FUNDC1 can induce
mitochondrial autophagy, rapidly induce apoptosis, and lead to MI
(104).
Myocardial hypertrophy is an independent risk
factor for cardiovascular diseases and is closely associated with
the development of HF. Treatment of rats with myocardial
hypertrophy with difluoro-methyl-ornithine showed that
difluoro-methyl-ornithine downregulated the expression of GRP75,
VDAC1 and CypD in MAMs, improved myocardial hypertrophy, and
regulated apoptosis. The present study also showed that MAMs
pathway may play a key role in myocardial hypertrophy (105). Luan et al (106) evaluated protein expression in
cardiomyocytes of rats with TAC-induced cardiac hypertrophy and
found that genes encoding MAM-related proteins were preferentially
expressed in cardiac hypertrophy samples. Notably, Fis1, Hspa9, Mfn
1, and Mfn2 expression increased 2 weeks after TAC treatment but
decreased at 8 and 11 weeks (HF period). These two experiments
showed that different proteins in MAMs have different regulatory
effects on the development of cardiac hypertrophy.
Myocardial ischemia reperfusion
Ischemic heart disease is one of the four leading
causes of death worldwide with a high mortality rate (107) and prevalence in men. In addition,
with increasing age, the incidence of ischemic heart disease shows
an increasing trend annually (108). Ischemic heart disease includes
the pathological process of myocardial ischemia, and myocardial
ischemia-reperfusion is generally the main treatment plan for
restoring the blood supply after MI. If the blood supply to the
coronary artery of the heart cannot be restored in time, the
myocardium will die due to ischemia, hypoxia, necrosis, formation
of fibrous scars on the myocardium, and finally, HF. However, this
treatment inevitably causes the original ischemic heart to undergo
inflammatory reactions and myocardial damage. Myocardial
ischemia-reperfusion injury increases mortality associated with MI
and the difficulty of perfusion therapy (109). Animal experiments confirmed that
in early ischemia-reperfusion, myocardial cells change from edema
to inflammation, and the phosphorylation levels of VEGF and Src are
increased (110). Myocardial
necrosis and dissolution, increased mitochondrial damage, and
oxidative stress-induced autophagosome accumulation were
correspondingly increased. Sirt1 may play an important role in
autophagosome clearance by upregulating Rab7 in MI/R (1,111).
In addition, MI/R can affect a series of organelles, such as the ER
and mitochondrial stress response, resulting in ROS aggregation.
The mPTP is opened, and matrix-resident CypD is responsible for
regulating the mPTP, which ultimately damages cardiomyocytes and
leads to HF (112).
Mitochondrial Ca2+ overload is observed
during myocardial ischemia-reperfusion, and MAMs are used as a
medium for Ca2+ transport. When mitochondria and the ER
are separated due to certain factors, such as the destruction of
protein tyrosine phosphatase interacting protein 51-vesicular
associated membrane protein-associated protein B, it leads to
Ca2+ ion transport and energy metabolism disorders
(112). The role of MAMs in
myocardial ischemia-reperfusion has been previously confirmed
(86). Gomez et al
(113) found that during
ischemia-reperfusion, increased activity of glycogen synthase
kinase-3β in the SR/ER and MAM, which is localized in the heart,
interacts specifically with the IP3Rs Ca2+ channel
complex and increases the phosphorylation level of IP3Rs, which
promotes the transfer of Ca2+ from the SR/ER to
mitochondria, thereby triggering Ca2+ overload in the
cytoplasm and mitochondria. Kirshenbaum et al (114) found that the interaction between
cytoplasmic recombination diaphanous homologue 1 and Mfn2 leads to
a direct correlation between the shortening the mito-ER distance.
Activation of AGE carboxymethyl lysine in the heart may increase
the interaction between diaphanous homologue 1 and Mfn2, shorten
the mito-ER distance, lead to abnormal Ca2+ metabolism,
and render the heart more vulnerable to damage during I/R injury.
Researchers have used Cl-intracellular channel protein as a
specific Cl-channel in myocardial cell MAM, which can regulate
Ca2+ homeostasis, mitochondrial membrane potential
balance and oxidative stress, thereby protecting the heart from
ischemic reperfusion injury (115). Second, Nox4 enhances the
Akt-dependent phosphorylation of InsP3R (a Ca2+ release
channel) and reduces the activity of InsP3R on the ER by reducing
the contact between the ER and mitochondria in cardiomyocytes and
ischemic mouse hearts, thereby avoiding excessive accumulation of
Ca2+ ions in mitochondria. It has a significant
protective effect on the heart from mPT-dependent cell necrosis
during ischemia-reperfusion injury (43).
There are also numerous autophagy-related proteins
in MAMs, such as the mitophagy receptor FUNDC1. Studies have shown
that empagliflozin can activate AMPKα1 and enhance FUNDC1-dependent
mitophagy after ULK1 phosphorylation. Mitochondrial autophagy
clears damaged mitochondria and ultimately reduces MI/R injury;
however, FUNDC1 also interacts with other MAM proteins to promote
Ca2+ transfer. Therefore, it is certain that FUNDC1 is a
double-edged sword in I/R injury (11,116)(Table
II).
| Table II.Application of MAMs in cardiovascular
diseases. |
Table II.
Application of MAMs in cardiovascular
diseases.
| Disease | First author/s,
year | Achievement | (Refs.) |
|---|
| Diabetic
cardiomyopathy | Tan et al,
2020 | Abnormal glucose
and lipid metabolism cause inflammatory response, oxidative stress
and endoplasmic reticulum stress, leading to heart failure. | (61) |
|
| Chong et al,
2017 | Remodeling of
energy metabolism is the ‘promoter’ of myocardial injury. | (62) |
|
| Opie et al,
2009 | The imbalance of
fatty acid oxidation and utilization in cardiomyocytes leads to the
production of lipotoxic substances. | (63) |
|
| Kolwicz et
al, 2013 | The imbalance of
glucose oxidation and utilization in cardiomyocytes leads to the
production of glucotoxic substances. | (64) |
|
| Wu et al,
2019 | Mitochondrial
calcium overload in patients with high glucose leads to
mitochondrial dysfunction. | (68) |
|
| Xie et al,
2011 | Metformin can
inactivate AMPK and reduce FUNDC1 to improve cardiac
structure. | (70) |
|
| Wang et al,
2021 | The reduction of
FUNDC1 leads to cardiac tissue abnormalities in diabetic patients
by affecting intracellular calcium balance and lipid
metabolism. | (69) |
|
| Meng et al,
2023 | The
IP3R-Grp75-VDAC1 complex regulates calcium homeostasis and
mitophagy dysfunction. The decreased level of PACS-2 on MAMs in
diabetic patients affects the function of MAMs. | (73) |
|
| Tubbs et al,
2018 | The significant
decrease of IP3R-VDAC1 complex in patients with diabetic
cardiomyopathy can be detected by experiments. | (66) |
|
| Chang et al,
2023 | SIRT3 inhibits
apoptosis, promotes the deacetylation of VDAC1 and reduces its
interaction with MAM, thereby reducing the formation of MAMs. | (74) |
|
| Salin et al,
2023 | Under the action of
high glucose, the up-regulation of PACS2 promotes the formation of
MAMs, reduces mitochondrial biosynthesis and oxidative
phosphorylation, and drives mitochondrial apoptosis. | (75) |
|
| Zhang et al,
2023 | Downregulation of
Bmal1 expression exacerbates cardiac hypertrophy and Diabetic
cardiomyopathy, while up-regulation helps to improve symptoms by
regulating the Bcl2/IP3R pathway to maintain the homeostasis of
MAMs. | (76) |
|
Atherosclerosis | Chen et al,
2024 | The downregulation
of PACS-2 protein inhibits mitophagy and reduces the assembly of
inflammasome NLRP3, which in turn enhances cell death. | (85) |
|
| Moulis et
al, 2019 | As a platform for
the assembly of inflammasome NLRP3, MAMs can also affect the
release of chronic inflammatory factors IL-1β and IL-18. | (82) |
|
| Baik et al,
2023 | The dissociation of
hexokinase 2 and VDAC triggers the activation of IP3R, which leads
to VDAC oligomerization and the formation of NLRP3 inflammasome
complex. | (86) |
|
| Zhang et al,
2022 | Studies have
confirmed that Mfn2 can inhibit vascular calcification in
atherosclerosis by activating RAS-RAF-ERK1/2 pathway. | (87) |
|
| Kong et al,
2024 | Yimai Granule
regulates mitophagy mediated by Pink1-Mfn2-Parkin pathway through
miRNA-125a-5p, and regulates pro-inflammatory factors and blood
lipids to inhibit atherosclerosis. | (88) |
|
| Feng et al,
2019 | MiR-93 has also
been shown to act on Mfn2 protein to regulate the proliferation and
migration of smooth muscle cells. | (89) |
|
| Yang et al,
2019 | The protein Nogo-B
has the potential to regulate the formation of MAMs, mainly
expressed in vascular endothelial cells and smooth muscle
cells. | (7) |
|
| Zhang et al,
2023 | It is proved that
Nogo-B mediates inflammation as a trigger of mitochondrial ROS
production and p38-p65 signal, thus affecting the normal function
of vascular endothelial cells. | (90) |
|
| Yoon et al,
2022 | Artesunate induces
IP3R phosphorylation by increasing cAMP content in platelets, which
indirectly improves the prognosis of patients with cardiovascular
disease. | (91) |
|
| Wang et al,
2021 | DJ-1 affects the
phenotypic transformation of VSMC by inhibiting the KLF4 pathway,
and its down-regulation can also significantly increase plaque
vulnerability. | (92) |
| Heart failure | Li et al,
2022 | Mitochondrial
dysfunction in cardiomyocytes is closely related to the occurrence
and development of heart failure. | (93) |
|
| Zhou et al,
2019 | The lack of FUNDC1
in mice with high-fat diet breaks the balance between FUNDC1 and
FBXL2. | (94) |
|
| Wang et al,
2018 | Sig-IR pathway was
reversed by mitochondrial Ca2+ overload in mice fed with
high-fat diet. | (95) |
|
| Xu et al,
2021 | The upregulation of
Mfn2 reversed ROS production and mitochondrial depolarization. | (96) |
|
| Dong et al,
2022 | The downregulation
of FIS1, DRP1 and Mfn2 expression levels can protect myocardial
function. | (98) |
|
| Zhao et al,
2021 | DFMO can
down-regulate the expression of GRP75, VDAC1 and CypD in MAMs,
improve myocardial hypertrophy and regulate apoptosis. | (105) |
|
| Luan et al,
2023 | The genes of
MAMs-related proteins are preferentially expressed in cardiac
hypertrophy samples. | (106) |
|
| Ding et al,
2024 | The reduction of
PACS2 will lead to the cleavage of BAP31 into p20, which will
aggravate myocardial injury. | (100) |
| Myocardial ischemia
reperfusion | Huang et al,
2023 | Sirt1 plays an
important role in autophagosome clearance by up-regulating Rab7 in
MI/R. | (111) |
|
| Gong et al,
2021 | MI/RI can cause the
accumulation of reactive oxygen species, make mPTP open, and
ultimately damage myocardial cells and lead to heart failure. | (112) |
|
| Gomez et al,
2015 | The activity of
GSK3β increases and interacts with the Ca2+ channel
complex of IP3Rs to promote the transfer of calcium, thereby
triggering calcium overload. | (113) |
|
| Ponnalagu et
al, 2022 | CLIC4 can regulate
Ca2+ homeostasis, mitochondrial membrane potential
balance and oxidative stress, thereby protecting the heart from
ischemic reperfusion injury. | (115) |
|
| Beretta et
al, 2020 | Nox4 can avoid
excessive accumulation of calcium ions in mitochondria and protect
the heart from ischemia-reperfusion injury. | (43) |
|
| Cai et al,
2022 | FUNDC1 can reduce
MI/R injury, but can also interact with other MAMs proteins to
promote Ca2 + transfer. | (116) |
Therefore, MAMs play an important role in
myocardial ischemia-reperfusion injury, and their dysfunction may
lead to Ca2+ ion transport disorders, energy metabolism
disorders and organelle dysfunction, which aggravates myocardial
injury. This therapeutic strategy for MAMs may provide a new
therapeutic approach for myocardial ischemia-reperfusion injury
(Fig. 2).
 | Figure 2.Mechanistic diagram of MAMs in
cardiovascular diseases. MAMs, mitochondria-associated membranes;
PACS2, phosphofurin acidic cluster sorting protein 2; IP3R1,
inositol 1,4,5-trisphosphate receptor type 3; GRP75, heat shock
protein family A (Hsp70) member 9; VDAC1, voltage-dependent anion
channel; FUNDC1, FUN14 domain-containing protein 1; Bmal1,basic
helix loop helix ARNT like 1; SIRT, sirtuin; Mfn, mitofusim;
Nogo-B, NUS1 dehydrodolichyl diphosphate synthase subunit; DJ-1,
parkinsonism associated deglycase; KLF4, KLF transcription factor
4; NLRP3, NLR family pyrin domain containing 3; Bap31, B cell
receptor-associated protein 31; Fis1, mitochondrial fission protein
1; VAP, amine oxidase copper containing 3; CLIC4, chloride
intracellular channel 4; Nox4, NADPH oxidase 4. |
Outlook
As a special connection area between the ER and
mitochondria, the MAM determines the two important organelles in
cells, mitochondria and the ER, and shows excellent therapeutic
potential in cardiovascular and other diseases. However, how it
affects the activity and function of mitochondria and the ER
requires further exploration. Molecular targets related to heart
diseases in MAMs provide a theoretical basis for the development of
new drugs for the treatment of heart diseases. To translate the
research results of MAMs into clinical applications, clinical
trials should be conducted; the effectiveness and safety of
MAM-related drugs in the treatment of heart disease should be
verified; and interdisciplinary cooperation in biology, medicine,
pharmacy, and other fields should be strengthened to jointly
promote the progress of MAMs in the field of heart disease
research. There are only a few clinical studies on how MAMs can
improve diseases. Therefore, the performance and function of MAMs
under different disease conditions must be extensively studied.
With an in-depth study of the role of MAMs in heart disease,
development of more effective treatments for heart diseases in the
future is expected. However, research in this field still faces
numerous challenges, such as the complex regulatory mechanisms of
MAMs, diversity of heart diseases, and difficulty in clinical
transformation. Therefore, it is necessary to work together to
promote the research and application of MAMs in the field of heart
disease treatment. However, based on the existing literature, MAMs
have far-reaching therapeutic significance in diseases, and the
role of MAMs in heart diseases has broad research prospects and
clinical application potential. In-depth research and exploration
are expected to provide new strategies and ideas for the treatment
of heart diseases. In-depth research on MAMs may reveal the key
mechanisms of certain diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Hunan Provincial Natural
Science Foundation (grant no. 2022JJ70112), the second batch of key
talent project in traditional Chinese medicine of Hunan during the
‘14th Five-Year Plan’ period [grant no. Xiang Zhong Yi Yao (2024)
3] and the Undergraduate Scientific Research and Innovation Fund
Project of Hunan University of Chinese Medicine in 2024 (grant no.
2024BKS007).
Availability of data and materials
Not applicable.
Authors' contributions
JZ conceptualized the study, performed the
methodology, wrote the first draft of the manuscript, and wrote,
reviewed and edited the manuscript. JT conceptualized the study,
wrote the first draft of the manuscript, and wrote, reviewed and
edited the manuscript. ZZ wrote the first draft of the manuscript,
and wrote, reviewed and edited the manuscript. WP performed
visualization (Fig. 1), and wrote,
reviewed and edited the manuscript. YX performed visualization
(Fig. 2), wrote figure legends and
assisted in writing and editing the manuscript. YG prepared
Table I and assisted in writing
and editing the manuscript. JG prepared Table II and assisted in writing and
editing the manuscript. CJ and LD translated the manuscript from
Chinese, edited the language and assisted in writing and editing
the manuscript. CT and GZ wrote, reviewed and edited the
manuscript. Data authentication is not applicable. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang J, Cui J, Zhao F, Yang L, Xu X, Shi
Y and Wei B: Cardioprotective effect of MLN4924 on ameliorating
autophagic flux impairment in myocardial ischemia-reperfusion
injury by Sirt1. Redox Biol. 46:1021142021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abdellatif M, Sedej S, Carmona-Gutierrez
D, Madeo F and Kroemer G: Autophagy in Cardiovascular Aging. Circ
Res. 123:803–824. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oakes SA and Papa FR: The role of
endoplasmic reticulum stress in human pathology. Annu Rev Pathol.
10:173–194. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Csordás G, Weaver D and Hajnóczky G:
Endoplasmic reticulum-mitochondrial contactology: Structure and
signaling functions. Trends Cell Biol. 28:523–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu
H, Huang L, Zhou C, Cai X, Fu C, et al: A regulatory signaling loop
comprising the PGAM5 phosphatase and CK2 controls receptor-mediated
mitophagy. Mol Cell. 54:362–377. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu S, Yang Y and Zheng Z: Summary of'!
Chinese cardiovascular disease report 2018. Chin J Circ.
34:209–220. 2019.
|
|
7
|
Yang YD, Li MM, Xu G, Feng L, Zhang EL,
Chen J, Chen DW and Gao YQ: Nogo-B receptor directs
mitochondria-associated membranes to regulate vascular smooth
muscle cell proliferation. Int J Mol Sci. 20:23192019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giacomello M and Pellegrini L: The coming
of age of the mitochondria-ER contact: A matter of thickness. Cell
Death Differ. 23:1417–1427. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li L, Wang Q and Cai C: Effects of
ischemia-reperfusion on cerebral mitochondrial phospholipids, free
fatty acids and respiration. J Stroke Neurol Dis. 14:2–4. 1997.
|
|
10
|
An G, Park J, Song J, Hong T, Song G and
Lim W: Relevance of the endoplasmic reticulum-mitochondria axis in
cancer diagnosis and therapy. Exp Mol Med. 56:40–50. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bai X, Zhang Z, Li X, Yang Y and Ding S:
FUNDC1: An emerging mitochondrial and MAMs protein for
mitochondrial quality control in heart diseases. Int J Mol Sci.
24:91512023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaye SD, Goyani S and Tomar D: MICU1′s
calcium sensing beyond mitochondrial calcium uptake. Biochim
Biophys Acta Mol Cell Res. 1871:1197142024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tomasoni D, Adamo M, Lombardi CM and Metra
M: Highlights in heart failure. ESC Heart Fail. 6:1105–1127. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kurmani S and Squire I: Acute heart
failure: Definition, classification and epidemiology. Curr Heart
Fail Rep. 14:385–392. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang W, Siraj S, Zhang R and Chen Q:
Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and
protects the heart from I/R injury. Autophagy. 13:1080–1081. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuang Y, Ma K, Zhou C, Ding P, Zhu Y, Chen
Q and Xia B: Structural basis for the phosphorylation of FUNDC1 LIR
as a molecular switch of mitophagy. Autophagy. 12:2363–2373. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu L, Li Y, Wang J, Zhang D, Wu H, Li W,
Wei H, Ta N, Fan Y, Liu Y, et al: Mitophagy receptor FUNDC1 is
regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis.
EMBO Rep. 22:e506292021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li
Y, Han Z, Chen L, Gao R, Liu L and Chen Q: Mitophagy receptor
FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy.
12:689–702. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu W, Li W, Chen H, Jiang L, Zhu R and
Feng D: FUNDC1 is a novel mitochondrial-associated-membrane (MAM)
protein required for hypoxia-induced mitochondrial fission and
mitophagy. Autophagy. 12:1675–1676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marchi S, Patergnani S, Missiroli S,
Morciano G, Rimessi A, Wieckowski MR, Giorgi C and Pinton P:
Mitochondrial and endoplasmic reticulum calcium homeostasis and
cell death. Cell Calcium. 69:62–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thoudam T, Ha CM, Leem J, Chanda D, Park
JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, et al: PDK4
augments er-mitochondria contact to dampen skeletal muscle insulin
signaling during obesity. Diabetes. 68:571–586. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Combot Y, Salo VT, Chadeuf G, Hölttä M,
Ven K, Pulli I, Ducheix S, Pecqueur C, Renoult O, Lak B, et al:
Seipin localizes at endoplasmic-reticulum-mitochondria contact
sites to control mitochondrial calcium import and metabolism in
adipocytes. Cell Rep. 38:1102132022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dickstein K, Cohen-Solal A, Filippatos G,
McMurray JJ, Ponikowski P, Poole-Wilson PA, Strömberg A, van
Veldhuisen DJ, Atar D, Hoes AW, et al: ESC guidelines for the
diagnosis and treatment of acute and chronic heart failure 2008:
The Task Force for the diagnosis and treatment of acute and chronic
heart failure 2008 of the European society of cardiology. developed
in collaboration with the Heart Failure Association of the ESC
(HFA) and endorsed by the European society of intensive care
medicine (ESICM). Eur J Heart Fail. 10:933–989. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hao G, Wang X, Chen Z, Zhang L, Zhang Y,
Wei B, Zheng C, Kang Y, Jiang L, Zhu Z, et al: Prevalence of heart
failure and left ventricular dysfunction in China: The China
hypertension survey, 2012–2015. Eur J Heart Fail. 21:1329–1337.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng
H, Su C, Luo X, Lu Q, Liao M, et al: CD36-mediated metabolic
crosstalk between tumor cells and macrophages affects liver
metastasis. Nat Commun. 13:57822022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Olzmann JA and Carvalho P: Dynamics and
functions of lipid droplets. Nat Rev Mol Cell Biol. 20:137–155.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rao MJ and Goodman JM: Seipin: Harvesting
fat and keeping adipocytes healthy. Trends Cell Biol. 31:912–923.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guyard V, Monteiro-Cardoso VF, Omrane M,
Sauvanet C, Houcine A, Boulogne C, Ben Mbarek K, Vitale N, Faklaris
O, El Khallouki N, et al: ORP5 and ORP8 orchestrate lipid droplet
biogenesis and maintenance at ER-mitochondria contact sites. J Cell
Biol. 221:e2021121072022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bock FJ and Tait SWG: Mitochondria as
multifaceted regulators of cell death. Nat Rev Mol Cell Biol.
21:85–100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Javadov S, Kozlov AV and Camara AKS:
Mitochondria in health and diseases. Cells. 9:11772020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kummer E and Ban N: Mechanisms and
regulation of protein synthesis in mitochondria. Nat Rev Mol Cell
Biol. 22:307–325. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang P, Konja D, Zhang Y and Wang Y:
Communications between mitochondria and endoplasmic reticulum in
the regulation of metabolic homeostasis. Cells. 10:21952021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen H, Detmer SA, Ewald AJ, Griffin EE,
Fraser SE and Chan DC: Mitofusins Mfn1 and Mfn2 coordinately
regulate mitochondrial fusion and are essential for embryonic
development. J Cell Biol. 160:189–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chinese Cardiovascular Health and Disease
Reporting Group, . Chinese cardiovascular health and disease report
2019 summary. Chin J Circ. 35:833–854. 2020.
|
|
35
|
Huang J: Epidemiological characteristics
and prevention strategies of heart failure in China. Chin Heart
Rhythm Electron J. 3:2–3. 2015.
|
|
36
|
Li Z, Hu O, Xu S, Lin C, Yu W, Ma D, Lu J
and Liu P: The SIRT3-ATAD3A axis regulates MAM dynamics and
mitochondrial calcium homeostasis in cardiac hypertrophy. Int J
Biol Sci. 20:831–847. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han S, Zhao F, Hsia J, Ma X, Liu Y, Torres
S, Fujioka H and Zhu X: The role of Mfn2 in the structure and
function of endoplasmic reticulum-mitochondrial tethering in vivo.
J Cell Sci. 134:jcs2534432021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Atakpa-Adaji P, Ivanova A, Kujawa K and
Taylor CW: IP3R at ER-mitochondrial contact sites: Beyond the
IP3R-GRP75-VDAC1 Ca2+ funnel. Contact. 2:93–101. 2023.PubMed/NCBI
|
|
39
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ma J, Liu L, Song L, Liu J, Yang L, Chen
Q, Wu JY and Zhu L: Integration of FUNDC1-associated mitochondrial
protein import and mitochondrial quality control contributes to
TDP-43 degradation. Cell Death Dis. 14:7352023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mao H, Chen W, Chen L and Li L: Potential
role of mitochondria-associated endoplasmic reticulum membrane
proteins in diseases. Biochem Pharmacol. 199:1150112022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Andreadou I, Schulz R, Papapetropoulos A,
Turan B, Ytrehus K, Ferdinandy P, Daiber A and Di Lisa F: The role
of mitochondrial reactive oxygen species, NO and H2 S in
ischaemia/reperfusion injury and cardioprotection. J Cell Mol Med.
24:6510–6522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beretta M, Santos CX, Molenaar C, Hafstad
AD, Miller CC, Revazian A, Betteridge K, Schröder K,
Streckfuß-Bömeke K, Doroshow JH, et al: Nox4 regulates
InsP3 receptor-dependent Ca2+ release into
mitochondria to promote cell survival. EMBO J. 39:e1035302020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wei X, Wei X, Lu Z, Li L, Hu Y, Sun F,
Jiang Y, Ma H, Zheng H, Yang G, et al: Activation of TRPV1 channel
antagonizes diabetic nephropathy through inhibiting endoplasmic
reticulum-mitochondria contact in podocytes. Metabolism.
105:1541822020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Huang D, Jia L, Shangguan F, Gong S,
Lan L, Song Z, Xu J, Yan C, Chen T, et al: LonP1 links
mitochondria-ER interaction to regulate heart function. Research
(Wash D C). 6:01752023.PubMed/NCBI
|
|
46
|
Marchi S, Corricelli M, Branchini A, Vitto
VAM, Missiroli S, Morciano G, Perrone M, Ferrarese M, Giorgi C,
Pinotti M, et al: Akt-mediated phosphorylation of MICU1 regulates
mitochondrial Ca2+ levels and tumor growth. EMBO J.
38:e994352019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Courchet J, Lewis TL Jr and Polleux F:
MFF-dependent mitochondrial fission regulates presynaptic release
and axon branching by limiting axonal mitochondria size. Nat
Commun. 9:49162018.
|
|
48
|
Wang N, Wang C, Zhao H, He Y, Lan B, Sun L
and Gao Y: The MAMs structure and its role in cell death. Cells.
10:6572021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun Z and Feng H: Effects of
mitochondria-associated endoplasmic reticulum membrane on
mitochondrial function. Chin J Nat. 45:127–138. 2023.
|
|
50
|
Rieusset J: Mitochondria-associated
membranes (MAMs): An emerging platform connecting energy and immune
sensing to metabolic flexibility. Biochem Biophys Res Commun.
500:35–44. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lan B, He Y, Sun H, Zheng X, Gao Y and Li
N: The roles of mitochondria-associated membranes in mitochondrial
quality control under endoplasmic reticulum stress. Life Sci.
231:1165872019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gao P, Yang M and Sun L:
Mitochondria-associated endoplasmic reticulum (MAM) calcium
transport and regulatory proteins mediate mitochondrial calcium
homeostasis. Chin J Cell Biol. 40:585–593. 2018.(In Chinese).
|
|
53
|
He Q, Qu M, Shen T, Su J, Xu Y, Xu C,
Barkat MQ, Cai J, Zhu H, Zeng LH and Wu X: Control of
mitochondria-associated endoplasmic reticulum membranes by protein
S-palmitoylation: Novel therapeutic targets for neurodegenerative
diseases. Ageing Res Rev. 87:1019202023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xian H, Yang Q, Xiao L, Shen HM and Liou
YC: Author Correction: STX17 dynamically regulated by Fis1 induces
mitophagy via hierarchical macroautophagic mechanism. Nat Commun.
12:67822021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang Y, Zhao X, Xie W and Zhang Y:
Research progress of mitochondria-associated endoplasmic reticulum
membranes (MAMs) in aging-related cardiovascular diseases. Acta
Physiologica Sinica. 75:799–816. 2023.(In Chinese). PubMed/NCBI
|
|
56
|
Jia GH, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhou Y and Tang Y: The pathogenesis of
diabetic cardiomyopathy and the effect of glucagon-like brain 1
receptor agonists on it. Chin J Cardiovasc Dis. 51:440–442.
2023.(In Chinese).
|
|
58
|
Kannel WB, Hjortland M and Castelli WP:
Role of diabetes in congestive heart failure: The Framingham study.
Am J Cardiol. 34:29–34. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sun L, Yu M, Zhou T, Zhang S, He G, Wang G
and Gang X: Current advances in the study of diabetic
cardiomyopathy: From clinicopathological features to molecular
therapeutics (Review). Mol Med Rep. 20:2051–2062. 2019.PubMed/NCBI
|
|
60
|
Minciună IA, Hilda Orășan O, Minciună I,
Lazar AL, Sitar-Tăut AV, Oltean M, Tomoaia R, Puiu M, Sitar-Tăut
DA, Pop D and Cozma A: Assessment of subclinical diabetic
cardiomyopathy by speckle - tracking imaging. Eur J Clin Invest.
51:e134752021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tan Y, Zhang Z, Zheng C, Wintergerst KA,
Keller BB and Cai L: Mechanisms of diabetic cardiomyopathy and
potential therapeutic strategies: Preclinical and clinical
evidence. Nat Rev Cardiol. 17:585–607. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chong CR, Clarke K and Levelt E: Metabolic
remodeling in diabetic cardiomyopathy. Cardiovasc Res. 113:422–430.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Opie LH and Knuuti J: The adrenergic-fatty
acid load in heart failure. J Am Coll Cardiol. 54:1637–1646. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kolwicz SC Jr, Purohit S and Tian R:
Cardiac metabolism and its interactions with contraction, growth,
and survival of cardiomyocytes. Circ Res. 113:603–616. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Herrero P, Peterson LR, McGill JB, Matthew
S, Lesniak D, Dence C and Gropler RJ: Increased myocardial fatty
acid metabolism in patients with type 1 diabetes mellitus. J Am
Coll Cardiol. 47:598–604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tubbs E, Chanon S, Robert M, Bendridi N,
Bidaux G, Chauvin MA, Ji-Cao J, Durand C, Gauvrit-Ramette D, Vidal
H, et al: Disruption of mitochondria-associated endoplasmic
reticulum membrane (MAM) integrity contributes to muscle insulin
resistance in mice and humans. Diabetes. 67:636–650. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Thivolet C, Vial G, Cassel R, Rieusset J
and Madec AM: Reduction of endoplasmic reticulum-mitochondria
interactions in beta cells from patients with type 2 diabetes. PLoS
One. 12:e01820272017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P,
Mao X, Huang K, Xie Z and Zou MH: Hyperglycemia-driven inhibition
of AMP-activated protein kinase α2 induces diabetic cardiomyopathy
by promoting mitochondria-associated endoplasmic reticulum
membranes in vivo. Circulation. 139:1913–1936. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang C, Dai X, Wu S, Xu W, Song P, Huang K
and Zou MH: FUNDC1-dependent mitochondria-associated endoplasmic
reticulum membranes are involved in angiogenesis and
neoangiogenesis. Nat Commun. 12:26162021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xie Z, Lau K, Eby B, Lozano P, He C,
Pennington B, Li H, Rathi S, Dong Y, Tian R, et al: Improvement of
cardiac functions by chronic metformin treatment is associated with
enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes.
60:1770–1778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ding M, Feng N, Tang D, Feng J, Li Z, Jia
M, Liu Z, Gu X, Wang Y, Fu F and Pei J: Melatonin prevents
Drp1-mediated mitochondrial fission in diabetic hearts through
SIRT1-PGC1α pathway. J Pineal Res. 65:e124912018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li C, Li L, Yang M, Zeng L and Sun L:
PACS-2: A key regulator of mitochondria-associated membranes
(MAMs). Pharmacol Res. 160:1050802020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Meng M, Jiang Y, Wang Y, Huo R, Ma N, Shen
X and Chang G: β-carotene targets IP3R/GRP75/VDAC1-MCU axis to
renovate LPS-induced mitochondrial oxidative damage by regulating
STIM1. Free Radic Biol Med. 205:25–46. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chang Y, Wang C, Zhu J, Zheng S, Sun S, Wu
Y, Jiang X, Li L, Ma R and Li G: SIRT3 ameliorates
diabetes-associated cognitive dysfunction via regulating
mitochondria-associated ER membranes. J Transl Med. 21:4942023.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Salin Raj P, Nair A, Preetha Rani MR,
Rajankutty K, Ranjith S and Raghu KG: Ferulic acid attenuates high
glucose-induced MAM alterations via PACS2/IP3R2/FUNDC1/VDAC1
pathway activating proapoptotic proteins and ameliorates
cardiomyopathy in diabetic rats. Int J Cardiol. 372:101–109. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang N, Yu H, Liu T, Zhou Z, Feng B, Wang
Y, Qian Z, Hou X and Zou J: Bmal1 downregulation leads to diabetic
cardiomyopathy by promoting Bcl2/IP3R-mediated mitochondrial
Ca2+ overload. Redox Biol. 64:1027882023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Herrington W, Lacey B, Sherliker P,
Armitage J and Lewington S: Epidemiology of atherosclerosis and the
potential to reduce the global burden of atherothrombotic disease.
Circ Res. 118:535–546. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cheng J, Huang H, Chen Y and Wu R:
Nanomedicine for diagnosis and treatment of atherosclerosis. Adv
Sci (Weinh). 10:e23042942023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Newman CB, Preiss D, Tobert JA, Jacobson
TA, Page RL II, Goldstein LB, Chin C, Tannock LR, Miller M,
Raghuveer G, et al: Statin safety and Associated adverse events: A
scientific statement from the American heart association.
Arterioscler Thromb Vasc Biol. 39:e38–e81. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han
X, Tang D and Chen R: Research progress on the relationship between
atherosclerosis and inflammation. Biomolecules. 8:802018.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sanchez-Rodriguez E, Egea-Zorrilla A,
Plaza-Díaz J, Aragón-Vela J, Muñoz-Quezada S, Tercedor-Sánchez L
and Abadia-Molina F: The gut microbiota and its implication in the
development of atherosclerosis and related cardiovascular diseases.
Nutrients. 12:6052020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Moulis M, Grousset E, Faccini J, Richetin
K, Thomas G and Vindis C: The multifunctional sorting protein
PACS-2 controls mitophagosome formation in human vascular smooth
muscle cells through mitochondria-ER contact sites. Cells.
8:6382019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wang Z, Sun W, Zhang K, Ke X and Wang Z:
New insights into the relationship of mitochondrial metabolism and
atherosclerosis. Cell Signal. 127:1115802025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chen C, Dong X, Zhang W, Chang X and Gao
W: Dialogue between mitochondria and endoplasmic
reticulum-potential therapeutic targets for age-related
cardiovascular diseases. Front Pharmacol. 15:13892022024.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen X, Yang Y, Zhou Z, Yu H, Zhang S,
Huang S, Wei Z, Ren K and Jin Y: Unraveling the complex interplay
between mitochondria-associated membranes (MAMs) and cardiovascular
Inflammation: Molecular mechanisms and therapeutic implications.
Int Immunopharmacol. 141:1129302024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Baik SH, Ramanujan VK, Becker C, Fett S,
Underhill DM and Wolf AJ: Hexokinase dissociation from mitochondria
promotes oligomerization of VDAC that facilitates NLRP3
inflammasome assembly and activation. Sci Immunol. 8:eade76522023.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang WB, Feng SY, Xiao ZX, Qi YF, Zeng ZF
and Chen H: Down-regulating of MFN2 promotes vascular calcification
via regulating RAS-RAF-ERK1/2 pathway. Int J Cardiol. 366:11–18.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kong Z, Sun P, Lu Y, Yang Y, Min DY, Zheng
SC, Yang Y, Zhang Z, Yang GL and Jiang JW: Yi Mai granule improve
energy supply of endothelial cells in atherosclerosis via
miRNA-125a-5p regulating mitochondrial autophagy through
Pink1-Mfn2-Parkin pathway. J Ethnopharmacol. 319:1171142024.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Feng S, Gao L, Zhang D, Tian X, Kong L,
Shi H, Wu L, Huang Z, Du B, Liang C, et al: MiR-93 regulates
vascular smooth muscle cell proliferation, and neointimal formation
through targeting Mfn2. Int J Biol Sci. 15:2615–2626. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhang Y, Li JJ, Xu R, Wang XP, Zhao XY,
Fang Y, Chen YP, Ma S, Di XH, Wu W, et al: Nogo-B mediates
endothelial oxidative stress and inflammation to promote coronary
atherosclerosis in pressure-overloaded mouse hearts. Redox Biol.
68:1029442023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yoon SS, Kwon HW, Shin JH, Rhee MH, Park
CE and Lee DH: Anti-thrombotic effects of artesunate through
regulation of cAMP and PI3K/MAPK pathway on human platelets. Int J
Mol Sci. 23:15862022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang ZY, Cheng J, Liu B, Xie F, Li CL,
Qiao W, Lu QH, Wang Y and Zhang MX: Protein deglycase DJ-1
deficiency induces phenotypic switching in vascular smooth muscle
cells and exacerbates atherosclerotic plaque instability. J Cell
Mol Med. 25:2816–2827. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li B, Liu P and Liu C: Research progress
on the relationship between mitochondrial dysfunction and heart
failure. Transformative Med. 9:126–129. 2020.
|
|
94
|
Zhou H, Zhu P, Wang J, Toan S and Ren J:
DNA-PKcs promotes alcohol-related liver disease by activating
Drp1-related mitochondrial fission and repressing FUNDC1-required
mitophagy. Signal Transduct Target Ther. 4:562019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang JP, Chi RF, Liu J, Deng YZ, Han XB,
Qin FZ and Li B: The role of endogenous reactive oxygen species in
cardiac myocyte autophagy. Physiol Res. 67:31–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Xu X, Su YL, Shi JY, Lu Q and Chen C:
MicroRNA-17-5p promotes cardiac hypertrophy by targeting Mfn2 to
inhibit autophagy. Cardiovasc Toxicol. 21:759–771. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu Q, Zhang H, Gutiérrez Cortés N, Wu D,
Wang P, Zhang J, Mattison JA, Smith E, Bettcher LF, Wang M, et al:
Increased Drp1 acetylation by lipid overload induces cardiomyocyte
death and heart dysfunction. Circ Res. 126:456–470. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dong X, Chen Z, Jiang J, Lin Z, Guan Z, Li
X, Wang L, Fang H and Xian S: Effect and mechanism of Nuanxinkang
on cardiac function in mice with heart failure based on
mitochondrial dynamics. CJTCMP. 37:3538–3542. 2022.(In
Chinese).
|
|
99
|
Jiang X, Li G, Zhu B, Yang J, Cui S, Jiang
R and Wang B: p20BAP31 induces autophagy in colorectal cancer cells
by promoting PERK-Mediated ER stress. Int J Mol Sci. 25:51012024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ding Y, Liu N, Zhang D, Guo L, Shang Q,
Liu Y, Ren G and Ma X: Mitochondria-associated endoplasmic
reticulum membranes as a therapeutic target for cardiovascular
diseases. Front Pharmacol. 15:13983812024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang XL, Feng ST, Wang YT, Yuan YH, Li ZP,
Chen NH, Wang ZZ and Zhang Y: Mitophagy, a form of selective
autophagy, plays an essential role in mitochondrial dynamics of
Parkinson's disease. Cell Mol Neurobiol. 42:1321–1339. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gong Y, Luo Y, Liu S, Ma J, Liu F, Fang Y,
Cao F, Wang L, Pei Z and Ren J: Pentacyclic triterpene oleanolic
acid protects against cardiac aging through regulation of mitophagy
and mitochondrial integrity. Biochim Biophys Acta Mol Basis Dis.
1868:1664022022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Li W, Yin L, Sun X, Wu J, Dong Z, Hu K,
Sun A and Ge J: Alpha-lipoic acid protects against pressure
overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1
signaling. Cell Death Dis. 11:5992020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu
S, Ren J and Chen Y: NR4A1 aggravates the cardiac microvascular
ischemia reperfusion injury through suppressing FUNDC1-mediated
mitophagy and promoting Mff-required mitochondrial fission by CK2α.
Basic Res Cardiol. 113:232018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhao Y, Jia WW, Ren S, Xiao W, Li GW, Jin
L and Lin Y: Difluoromethylornithine attenuates
isoproterenol-induced cardiac hypertrophy by regulating apoptosis,
autophagy and the mitochondria-associated membranes pathway. Exp
Ther Med. 22:8702021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Luan Y, Guo G, Luan Y, Yang Y and Yuan R:
Single-cell transcriptional profiling of hearts during cardiac
hypertrophy reveals the role of MAMs in cardiomyocyte subtype
switching. Sci Rep. 13:83392023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
GBD 2021 Causes of Death Collaborators, .
Global burden of 288 causes of death and life expectancy
decomposition in 204 countries and territories and 811 subnational
locations, 1990–2021: A systematic analysis for the Global Burden
of Disease Study 2021. Lancet. 403:2100–2132. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Khan MA, Hashim MJ, Mustafa H, Baniyas MY,
Al Suwaidi SKBM, AlKatheeri R, Alblooshi FMK, Almatrooshi MEAH,
Alzaabi MEH, Al Darmaki RS and Lootah SNAH: Global epidemiology of
ischemic heart disease: Results from the global burden of disease
study. Cureus. 12:e93492020.PubMed/NCBI
|
|
109
|
Sánchez-Hernández CD, Torres-Alarcón LA,
González-Cortés A and Peón AN: Ischemia/reperfusion injury:
Pathophysiology, current clinical management, and potential
preventive approaches. Mediators Inflamm. 2020:84053702020.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wu Q, Xu R, Zhang K, Sun R, Yang M, Li K,
Liu H, Xue Y, Xu H and Guo Y: Characterization of early myocardial
inflammation in ischemia-reperfusion injury. Front Immunol.
13:10817192023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Huang KY, Que JQ, Hu ZS, Yu YW, Zhou YY,
Wang L, Xue YJ, Ji KT and Zhang XM: Metformin suppresses
inflammation and apoptosis of myocardiocytes by inhibiting
autophagy in a model of ischemia-reperfusion injury. Int J Biol
Sci. 16:2559–2579. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Gong Y, Lin J, Ma Z, Yu M, Wang M, Lai D
and Fu G: Mitochondria-associated membrane-modulated
Ca2+ transfer: A potential treatment target in cardiac
ischemia reperfusion injury and heart failure. Life Sci.
278:1195112021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gomez L, Thiebaut PA, Paillard M, Ducreux
S, Abrial M, Crola Da Silva C, Durand A, Alam MR, Van Coppenolle F,
Sheu SS and Ovize M: The SR/ER-mitochondria calcium crosstalk is
regulated by GSK3β during reperfusion injury. Cell Death Differ.
22:18902015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kirshenbaum LA, Dhingra R, Bravo-Sagua R
and Lavandero S: IAPH1-MFN2 interaction decreases the endoplasmic
reticulum-mitochondrial distance and promotes cardiac injury
following myocardial ischemia. Nat Commun. 15:14692024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ponnalagu D, Hamilton S, Sanghvi S, Antelo
D, Schwieterman N, Hansra I, Xu X, Gao E, Edwards JC, Bansal SS, et
al: CLIC4 localizes to mitochondrial-associated membranes and
mediates cardioprotection. Sci Adv. 8:eabo12442022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cai C, Guo Z, Chang X, Li Z, Wu F, He J,
Cao T, Wang K, Shi N, Zhou H, et al: Empagliflozin attenuates
cardiac microvascular ischemia/reperfusion through activating the
AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 63:1027382022.
View Article : Google Scholar : PubMed/NCBI
|














