Introduction
Glioma, a common primary tumor in the human central
nervous system, is distinguished by its high invasiveness and
destructive characteristics. At present, the main clinical
treatments of glioma include surgical resection, chemotherapy and
radiation therapy (1). However,
due to the invasive growth pattern of glioma, the presence of
non-regenerative nerve tissue and the critical functions involved,
glioma is difficult to surgically resect and has a high recurrence
rate. In addition, the existence of blood-brain barrier leads to
poor efficacy of glioma chemotherapy. Apparently, traditional
treatments for glioma are inadequate in achieving a cure,
particularly for high-grade glioma with high nausea (2). Gene therapy and molecular targeted
therapy, which have become popular in recent years, can be used in
combination with traditional treatments to enhance the inhibitory
or killing effect on tumour tissue. This approach effectively
addresses metastasis and spread while overcoming the drawbacks of
traditional treatments, such as incomplete efficacy, ease of
metastasis and significant side effects. In addition to accurately
killing cancer cells, the combined treatment can also repair and
improve the patient's immune system (3,4).
Therefore, a thorough investigation of the cellular and molecular
mechanisms underlying glioma pathogenesis and prognosis is crucial
for identifying therapeutic targets and developing new treatment
strategies.
The occurrence of tumors is not only the result of
malignant cell proliferation, but also closely related to the
apoptosis disorder of tumor cells. The mitochondrial pathway
(intracellular apoptosis pathway) plays a crucial role in
apoptosis, serving as one of the key signal cascades that control
the apoptosis of tumor cells (5).
It is noteworthy that the apoptotic signaling cascade of abnormal
energy metabolism and mitochondrial dysfunction is often observed
in malignant gliomas (6,7). Therefore, mediating the mitochondrial
pathway to restart tumor cell apoptosis is an important strategy
for the treatment of glioma. Moreover, investigating mitochondrial
proteomic abnormalities may provide a new therapeutic target for
glioma. Silent information regulators (SIRTs), which have been
discovered recently, play a key role in simultaneously regulating
multiple downstream pathways. Silent Information regulators (SIRTs)
can also lead to changes in metabolic and enzyme pathways, or
regulate complex mechanisms that result in cell death in gliomas
(8). Among SIRTs family members,
SIRT1 is widely regarded as a key epigenetic regulator. It is
involved in a number of biological processes, including metabolism,
genome stability maintenance, reprogramming, autophagy, aging,
mitochondrial apoptosis and tumourgenesis (9). It has been confirmed that SIRT1 is
highly expressed in glioma tissues and silencing SIRT1 gene can
hinder the occurrence of tumors (10). The present study also noted that
SIRT1 can inactivate the phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (AKT) pathway in a deacetylase-dependent
manner (11). The PI3K/AKT
pathway, a known negative regulator of glioma (12,13),
plays an important role in blocking mitochondrial apoptosis.
Consequently, thoroughly examining how SIRT1 modulates the PI3K/AKT
pathway to mediate mitochondrial apoptosis in glioma cells could
offer a novel targeted approach for glioma therapy.
In recent years, the influence of anesthetics on the
prognosis of surgical cancer treatment has received extensive
attention. Sevoflurane (SEV) is a commonly used inhalation
anesthetic in clinics. In addition to its good anesthetic effect,
SEV is found to have anti-cancer effects in a variety of cancers,
including glioma (14–16). Specifically, SEV limits the
progression of glioma by regulating the balance between
proliferation and apoptosis of glioma cells (17). In other diseases, SEV has been
found to modulate tumor progression through the mitochondrial
apoptosis pathway (18). However,
few studies have investigated the effect of SEV-mediated
mitochondrial apoptosis pathway on apoptosis of glioma cells.
Furthermore, a previous study has shown that SEV inhibits the
development of glioma by regulating the circular RNA_0079593
(circ_0079593)/microRNA-633 (miR-633)/rho associated coiled-coil
containing protein kinase 1 axis (19). Additionally, SEV also suppresses
the malignant phenotype of glioma by regulating the
miR-146b-5p/nuclear factor I/B axis (20). Further research indicates that SEV
impedes glioma progression by modulating the
circ_0037655/miR-130a-5p/ribophorin II axis and the
circ_0000215/miR-1200/natural killer cell cytotoxicity receptor 3
ligand 1 axis (21). However, the
precise anti-cancer molecular mechanisms of SEV in glioma require
further elucidation. The important regulatory functions of miRNA in
tumors have been comprehensively studied. A previous study showed
that SEV can promote neuronal apoptosis by inducing miR-211-5p
expression (22). As a mature
sequence of miR-211, miR-211-5P is identified to be under-expressed
in gliomas and involved in the regulation of apoptosis pathways in
glioma cells (23). Based on this,
it was hypothesized that SEV might play an anticancer role in
glioma by inducing miR-211-5p expression. Importantly, the online
bioinformatics analysis databases StarBase, PITA and miRmap
predicted that miR-211-5p and SIRT1 have targeted binding sites and
miR-211-5p may target SIRT1 to regulate the PI3K/AKT pathway and
mediate the role of SEV in glioma progression.
Based on previous studies, the present study
hypothesised that SEV promotes apoptosis in glioma cells through
the induction of miR-211-5p, which in turn regulates the
SIRT1/PI3K/AKT pathway and mediates mitochondria-dependent
apoptosis. The present study identified the role and potential
molecular regulatory mechanisms of SEV in mitochondrial apoptosis
of glioma cells. Although the present study only examined
short-term effects, it may also open up new possibilities for SEV
as a potential treatment for glioma in the future.
Materials and methods
Cellular model
Human glioma cell line (U251, IM-H421, IMMOCELL) and
normal human astrocyte (NHA, IMP-H223, IMMOCELL) were obtained from
the American Type Culture Collection and cultured according to the
corresponding instructions. All cell lines were confirmed by short
tandem repeat analysis and mycoplasma contamination detection.
Experimental grouping and
administration
For SEV exposure, the U251 cells were seeded onto a
plate and cultured overnight at 37°C, then the cell plate was
placed in a closed glass chamber connected to the anesthesia
machine. SEV was fed into the chamber using an anesthetic
carburetor and the concentration of SEV was continuously monitored
by a gas monitor. Treated with SEV at different concentrations (0,
2, 4 and 6%), U251 cells were randomly divided into Control group,
2% SEV group, 4% SEV group and 6% SEV group. Upon treatment with
the optimal concentration of SEV at different times (0, 4, 6 and 8
h), U251 cells were randomly divided into the Control group, the 4
h group, the 6 h group and the 8 h group. According to cell
viability and apoptosis results, 6% sevoflurane (100 nM) and an
induction time of 8 h were selected as the optimal conditions for
subsequent experiments. miR-211-5p mimic (100 nM), miR-211-5p
inhibitor (in-miR-211-5p; 100 nM), SIRT1-targeted small interfering
(si)RNA (50 nM), SIRT1 overexpression plasmid (1 µg) and their
negative controls (NC; miR-NC, in-NC, si-NC and pcDNA 3.1) were
used at a concentration of 50 nM for siRNAs and 1 µg for plasmids,
obtained from Guangzhou Ruibo Co., Ltd. When the cells adhered to
the wall and the cell confluence reached 80%, the aforementioned
plasmids and RNA were transfected into U251 cells using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the experimental groups. The
transfection process was carried out at 37°C for 4 h. After
transfection, the cells were incubated in culture medium for 24 h
and then subjected to subsequent experiments. The negative controls
used in the experiments included scrambled or non-targeting
sequences for siRNA (si-NC); controls for miRNA mimics and
inhibitors (miR-NC, in-NC); and an empty vector for overexpression
plasmids (Tables I and II).
 | Table I.miR-211-5p transfection
sequences. |
Table I.
miR-211-5p transfection
sequences.
| Gene | Primer sequences
(5′-3′) |
|---|
|
miR-211-5p-mimic | Forward:
5′-UUCCCUUUGUCAUCCUUCGCCT-3′ |
|
| Reverse:
5′-GCGAAGGAUGACAAAGGGAANN-3′ |
| mimic-NC | Forward:
5′-UCACAACCUCCUAGAAAGAGUAGA-3′ |
|
| Reverse:
5′-UCUACUCUUUCUAGGAGGUUGUGA-3′ |
|
miR-211-5p-inhibitor |
AGGCGAAGGAUGACAAAGGG |
| inhibitor-NC |
UCUACUCUUUCUAGGAGGUUGUGA |
| Table II.SIRT1 transfection sequences. |
Table II.
SIRT1 transfection sequences.
| Gene | Primer sequences
(5′-3′) |
|---|
| SIRT1 siRNA-1 | Forward:
5′-TTAAAAGTGGTTTTTTGTGTTTTCAAGAGAAACACAAAAAACCACTTTTAA-3′ |
|
| Reverse:
5′-CACAAAAAACCACTTTTAAATAAGTTCTCTATTTAAAAGTGGTTTTTTGTG-3′ |
| SIRT1 siRNA-2 | Forward:
5′-ATTTAAAAGTGGTTTTTTGTGTTCAAGAGACACAAAAAACCACTTTTAAAT-3′ |
|
| Reverse:
5′-CAAAAAACCACTTTTAAATGGAAGTTCTCTCCATTTAAAAGTGGTTTTTTG-3′ |
| SIRT1 siRNA-3 | Forward:
5′-AAACATAAATGTTTAGTCCGTTTCAAGAGAACGGACTAAACATTTATGTTT-3′ |
|
| Reverse:
5′-GGACTAAACATTTATGTTTCAAAGTTCTCTTGAAACATAAATGTTTAGTCC-3′ |
| si-NC | Forward:
5′-CACCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTG-3′ |
|
| Reverse:
5′-GATCCAAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAC-3′ |
Cells in logarithmic growth stages were selected for
experimental intervention and grouping. They were divided into 10
groups: the Control group (normal control group), the SEV group
(cells treated with SEV), the SEV + in-NC group (cells treated with
SEV + transfection inhibitor negative control), the SEV +
in-miR-211-5p group (cells treated with SEV + transfection
miR-211-5p inhibitor), the SEV + pcDNA 3.1 group (cells were
treated with SEV + transfection of blank overexpressed plasmid),
the SEV + pcDNA-SIRT1 group (cells treated with SEV + transfection
of SIRT1 targeted overexpressed plasmid), the si-NC group
(transfection of si-NC), the si-SIRT1 group (transfection of
SIRT1-targeted siRNA), the si-SIRT1 + in-NC group (negative control
transfected with SIRT1-targeted siRNA + inhibitors) and the
si-SIRT1 + in-miR-211-5p (transfection with SIRT1-targeted siRNA +
miR-211-5p inhibitors).
Reverse transcription-quantitative
(RT-q) PCR
The RNA extraction was performed using the RNA
extraction buffer (Vazyme Biotech Co., Ltd.) with a cell density of
1×106 cells/well. The concentration and purity of RNA
was determined using a NanoDrop spectrophotometer (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
HiScript III 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co.,
Ltd.) was used for RT according to the manufacturer's protocol.
qPCR was conducted using Taq Pro Universal SYBR qPCR Master Mix
(Vazyme Biotech Co., Ltd.) under the following conditions: 95°C for
5 min (pre-denaturation), followed by 40 cycles at 95°C for 10 sec
(denaturation) and 60°C for 1 min (annealing/extension). U6 and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as internal
controls for miR-211-5p and messenger RNA (mRNA) normalization,
respectively. The primer sequences are listed in Table III. The relative transcript
levels were analyzed by the 2−ΔΔCq method (24).
| Table III.Primer sequences. |
Table III.
Primer sequences.
| Gene | Primer sequences
(5′-3′) |
|---|
| hsa-miR-211-5p | Forward:
5′-CGCGTTCCCTTTGTCATCCT-3′ |
|
| Reverse:
5′-AGTGCAGGGTCCGAGGTATT-3′ |
| SIRT1 | Forward:
5′-TAGCCTTGTCAGATAAGGAAGGA-3′ |
|
| Reverse:
5′-ACAGCTTCACAGTCAACTTTGT-3′ |
| U6 | Forward:
5′-CGACAAGACGATCCGGGTAAA-3′ |
|
| Reverse:
5′-GGTTGAGGAGTGGGTCGAAG-3′ |
| GAPDH | Forward:
5′-ACAACTTTGGTATCGTGGAAGG-3′ |
|
| Reverse:
5′-GCCATCACGCCACAGTTTC-3′ |
Western blotting
Protein was extracted from cell precipitates using a
radioimmunoprecipitation assay lysis buffer containing protease
inhibitors (Beyotime Institute of Biotechnology). Protein
concentration was determined using a BCA Protein Assay Kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. A total of 30 µg protein was loaded per lane and
separated by sodium dodecyl sulfate polyacrylamide gel
electrophoresis (NCM Biotech) on 10% gels. The separated proteins
were then transferred onto a polyvinylidene fluoride membrane. The
membrane was blocked with 5% skimmed milk at room temperature for 2
h and then incubated with primary antibodies at 4°C overnight.
Subsequently, the membrane was incubated with secondary antibodies
(1:10,000; cat. no. bs-0295G-HRP; BIOSS) at room temperature for 2
h. Protein bands were visualized using enhanced chemiluminescence
(NCM Biotech) and semi-quantified using ImageJ software (version
1.53; National Institutes of Health). GAPDH was used as a loading
control. Primary antibodies were: B-cell lymphoma-2
(Bcl-2)-associated X protein (Bax) (cat. no. 50599-2-Ig; 1:8,000;
Wuhan Sanying Biotechnology); cytochrome c (cat. no.
10993-1-AP, 1:4,000; Wuhan Sanying Biotechnology);
cleaved-caspase-3 (cat. no. ab32042, 1:2,000, Abcam); caspase-3
(cat. no. ab184787; 1:2,000, Abcam); caspase-9 (cat. no. ab184786;
1:2,000, Abcam); caspase-9 (cleaved Asp330) antibody (cat. no.
PA5-105272; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.);
Bcl2 antibody (cat. no. 12789-1-AP; 1:9,000; Wuhan Sanying
Biotechnology); microtubule-associated protein light chain 3 (LC3)
(cat. no. 14600-1-AP; 1:2,500; Wuhan Sanying Biotechnology); P62
(cat. no. 18420-1-AP; 1:10,000; Wuhan Sanying Biotechnology); SIRT1
(cat. no. 13161-1-AP; 1:3,000; Wuhan Sanying Biotechnology); PI3K
antibody (cat. no. 20584-1-AP; 1:300; Wuhan Sanying Biotechnology);
phosphorylated (p-)PI3K p85/p55 (Tyr458, Tyr199) (cat. no.
PA5-17387; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.);
AKT (cat. no. 10176-2-AP; 1:1,000; Wuhan Sanying Biotechnology);
and p-AKT (Ser473) (cat. no. ab81283; 1:2,000; Abcam).
MTT assay
The cells in logarithmic growth stage were seeded
into a 96-well plate with 2,000 cells per well and 100 µl cell
suspension was added to each well. Thereafter, MTT solution (5
mg/ml) was added to each well and incubated in the incubator at
37°C for 4 h. The supernatant was then removed and 150 µl of
dimethyl sulfoxide was added to each well. The plate was oscillated
at room temperature for 10 min to fully dissolve the crystals. The
optical density value at a wavelength of 570 nm was measured using
a microplate reader.
Flow cytometry
The cells were seeded into a 6-well plate
(5×105 cells/well) and administered or transfected as
described aforementioned. After 48 h, staining was carried out in
the dark at 25°C with 5 µl Annexin V-FITC and 10 µl propidium
iodide for 5 min. The apoptotic cells were analyzed using a flow
cytometer (BD FACSCanto II; BD Biosciences) and the data were
processed with FlowJo software (version 10.8.1; BD Biosciences).
The apoptotic rate was calculated as the percentage of early
apoptotic cells plus late apoptotic cells.
Mitochondrial membrane potential (MMP)
determination. Referring to previous studies, cells
(2×105 cells/well) were seeded into a 6-well microplate,
cultured at 37°C for 24 h and treated with SEV. After 48 h, the
staining was performed with JC-1 (Beyotime Institute of
Biotechnology) at 37°C and 5% CO2 for 30 min.
Fluorescence intensity was monitored with an EVOS M5000
fluorescence microscope (Thermo Fisher Scientific, Inc.) and MMP
levels were assessed by measuring the reduction in the red/green
fluorescence intensity ratio.
Reactive oxygen species (ROS)
determination
Stably transfected cells were uniformly seeded into
a 24-well plate with 5×104 cells per well and cultured
for 16–18 h. MitoSOX™ Red stock solution (5 mmol/l; Thermo Fisher
Scientific, Inc.) was prepared in DMSO under light protection, then
diluted in phosphate buffered saline buffer (1:1,000) to produce a
MitoSOX™ Red working solution (5 µmol/l). Next, the MitoSOX™ Red
working solution was added to the well at 300 µl/well and incubated
at 37°C for 10 min in the dark. The culture solution was aspirated
and the cells were washed three times. Then, 500 µl preheated
phosphate buffered saline buffer was added and the plate was
immediately placed under a fluorescence microscope (EVOS M5000;
Thermo Fisher Scientific, Inc.) for observation and image
capture.
Dual luciferase reporter (DLR)
analysis
In the DLR assay, the binding site between
miR-211-5p and SIRT1 was predicted using bioinformatics analysis
tools, including StarBase (https://rnasysu.com/encori/), PITA (https://tools4mirs.org/software/target_prediction/pita/)
and miRmap (https://mirmap.ezlab.org/) databases.
Based on the predicted binding site, the target gene
SIRT1-wild-type (SIRT1-WT) vector and mutant (SIRT1-MUT) vector
with miR-211-5p binding sites were designed through an online
bioinformation analysis website. The DLR vector plasmid was
constructed by Sangon Biotech Co., Ltd. Cells were transfected with
the aforementioned vectors and miR-211-5p mimic/mimic-NC using
Lipofectamine as the transfection reagent for 24 h at 37°C for 24
h. Luciferase activity was measured using the
Dual-Luciferase® Reporter Assay System (Promega
Corporation) according to the manufacturer's instructions.
Renilla luciferase activity was used for normalization.
Statistical analysis
Data were analyzed and mapped using GraphpadPrism9
(version 9.5.0; Dotmatics). The diagrams were made using Photoshop
(Adobe Systems, Inc.). All data were presented as means ± standard
deviation; independent samples t-test was used for pairwise
comparisons and a one-way analysis of variance followed by Tukey's
post hoc test was used for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
Sevoflurane exposure hinders the
viability of glioma cells and induces apoptosis
To explore whether the influences of SEV on glioma
cells were concentration-dependent and time-dependent, the present
study examined the influences of SEV treatment on glioma cells at
different concentrations and times. The results of MTT and flow
cytometry revealed that SEV reduced U251 cell viability and
promoted apoptosis in a concentration-dependent manner (Fig. 1A and B; P<0.001). Also, SEV
decreased U251 cell viability and accelerated apoptosis (Fig. 1C and D) in a time-dependent manner
(P<0.001). Therefore, the present study selected the optimal
concentration of 6% in the concentration gradient and the optimal
induction time of 8 h in the time gradient as the test
concentration for subsequent experiments.
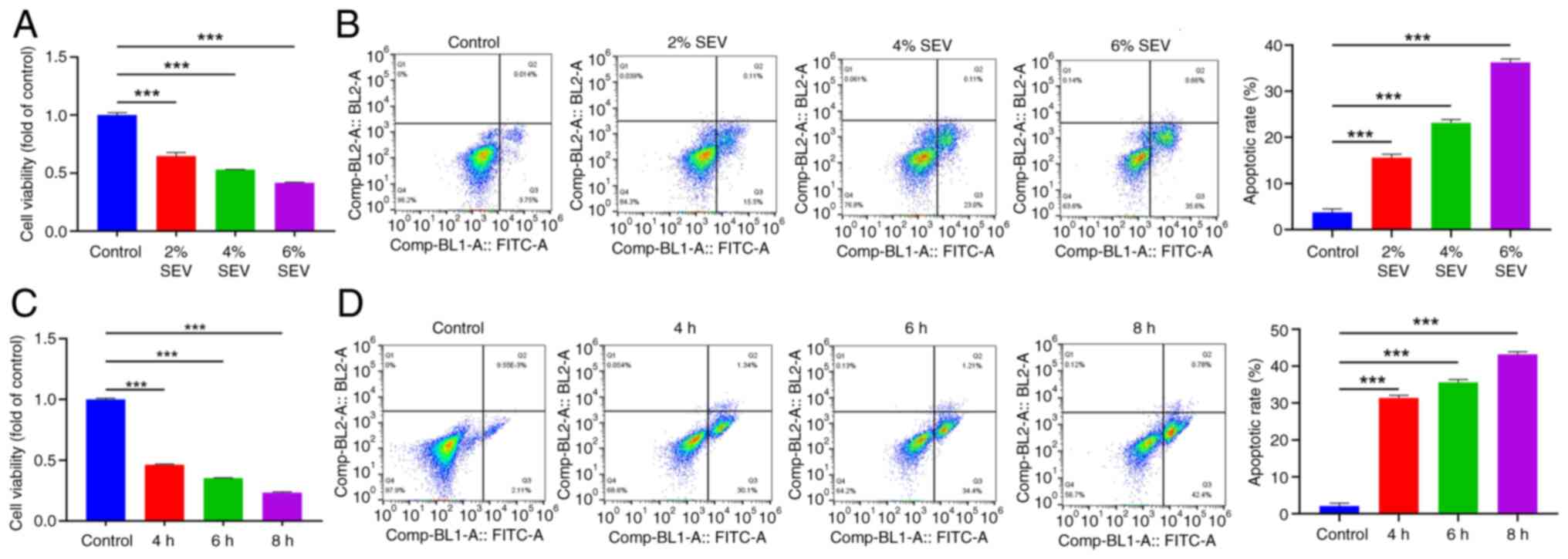 | Figure 1.SEV exposure hinders the viability of
glioma cells and induces apoptosis. (A) MTT assay was performed to
detect SEV at different concentrations (0, 2, 4 and 6%) on the U251
cell viability. (B) Flow cytometry was performed to detect SEV at
different concentrations (0, 2, 4 and 6%) on apoptosis of U251
cells. (C) MTT assay was performed to detect SEV at different times
(0, 4, 6 and 8 h) on the U251 cells viability. (D) Flow cytometry
was performed to detect SEV at different times (0, 4, 6 and 8 h) on
apoptosis of U251 cells. ***P<0.001, n=3. SEV, sevoflurane. |
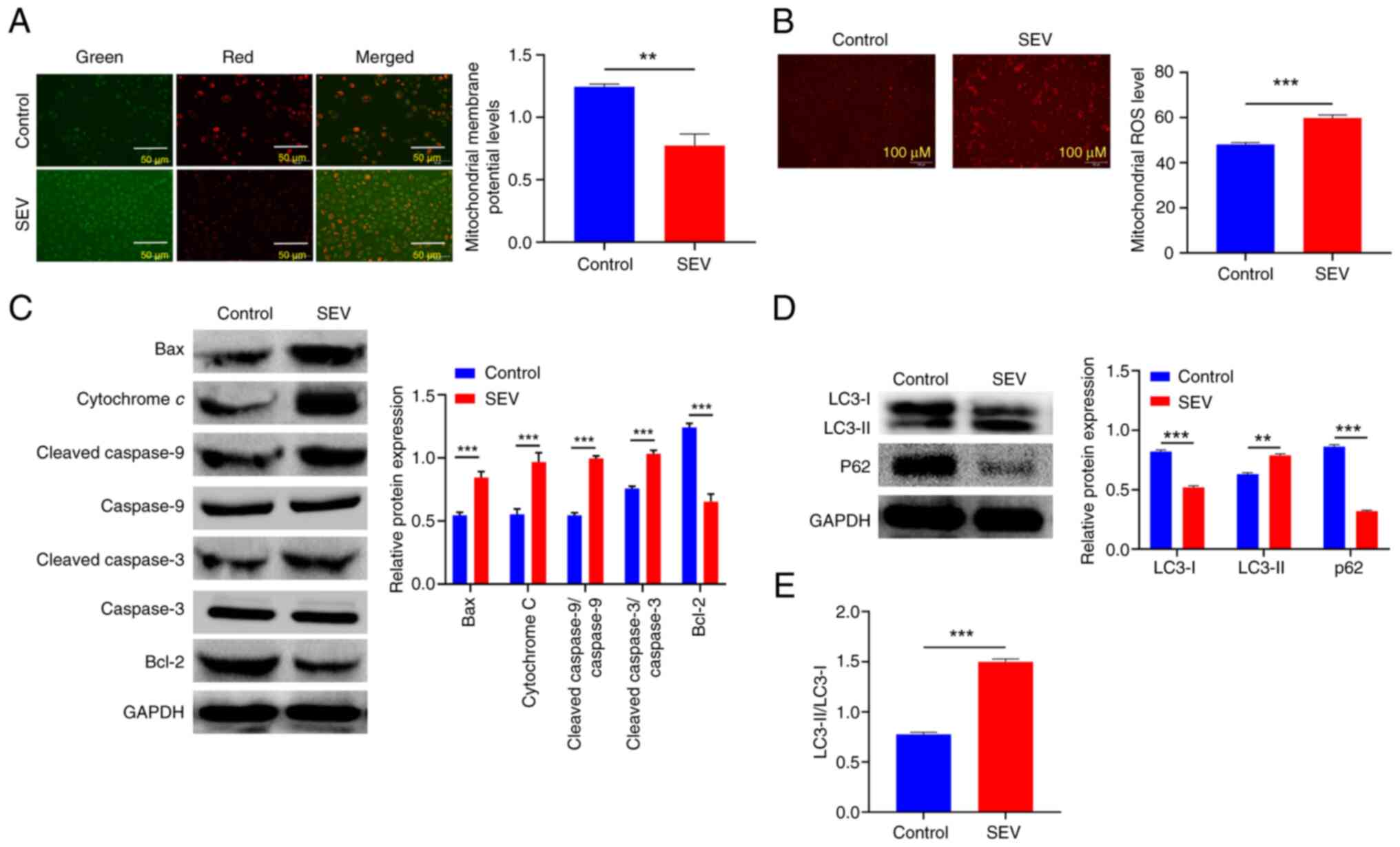
Sevoflurane induces apoptosis of
glioma cells by mitochondrial apoptosis pathway
Further, MMP levels were detected to determine
whether SEV can accelerate apoptosis by mediating the mitochondrial
apoptosis pathway. As a key marker of mitochondrial function, MMP
is a critical driver of ATP synthesis and a decline in MMP can lead
to reduced mitochondrial productivity and even cell death. The
present study results showed that after treating U251 cells with
SEV, the red fluorescence intensity, representing high MMP, was
weakened. Conversely, the green fluorescence intensity,
representing low MMP, was enhanced under the same fluorescence
intensity (P<0.01). This indicated that SEV interfered with and
destroyed MMP, thereby activating the intrinsic apoptosis pathway
(Fig. 2A). Since ROS is a major
mediator of the mitochondria-mediated apoptosis pathway (19), the present study further
investigated mitochondrial ROS levels in U251 cells. This revealed
enhanced red fluorescence intensity (P<0.001) after treating
U251 cells with SEV, suggesting that SEV increased mitochondrial
ROS levels (Fig. 2B). Afterwards,
exploration was further performed on the expression of proteins
associated with the mitochondrial apoptosis pathway. The results
showed that the expression levels of Bax, cytochrome c,
cleaved-caspase-9/caspase-9 and cleaved-caspase-3/caspase-3 were
significantly increased after the treatment of U251 cells with SEV
(P<0.01). The expression level of Bcl-2 was significantly
decreased, indicating that SEV promoted mitochondrial apoptosis by
regulating the proteins associated with the mitochondrial apoptosis
pathway (Fig. 2C). Since the
mitochondrial apoptosis pathway was closely related to autophagy,
the expression of autophagy-related proteins was further examined.
According to the data, U251 cells treated by SEV showed higher
LC3-II levels, lower p62 and LC3-I levels. Additionally, the
conversion rate of LC3-I to LC3-II was increased (Fig. 2D and E; P<0.01). These results
suggested that SEV could induce autophagy of glioma cells. Taken
together, these results supported the hypothesis that SEV could
induce apoptosis of U251 cells through a mitochondria-dependent
apoptosis pathway.
Sevoflurane exposure promotes
apoptosis of glioma cells through upregulation of miR-211-5p
expression, mediating mitochondrial apoptosis pathway
To further clarify whether SEV could mediate the
role of miR-211-5p in glioma, RT-qPCR was used to detect the
regulatory influences of SEV on miR-211-5p in glioma cells. The
results revealed that SEV upregulated miR-211-5p expression.
Subsequently, miR-211-5p expression was interfered with in the
presence of SEV exposure and transfection efficiency was confirmed
by RT-qPCR (Fig. 3A; P<0.01).
The results of MTT and flow cytometry assays showed that
downregulation of miR-211-5p reversed the inhibitory influence of
SEV on the viability of glioma cells and the promotion of apoptosis
(Fig. 3B and C; P<0.001). In
addition, downregulation of miR-211-5p reversed the influence of
SEV on mitochondrial apoptotic pathway-associated proteins, MMP,
ROS and autophagy in glioma cells (Fig. 3D-H; P<0.01). To sum up, these
data showed that SEV played a role in the malignant phenotype of
glioma cells by inducing miR-211-5p.
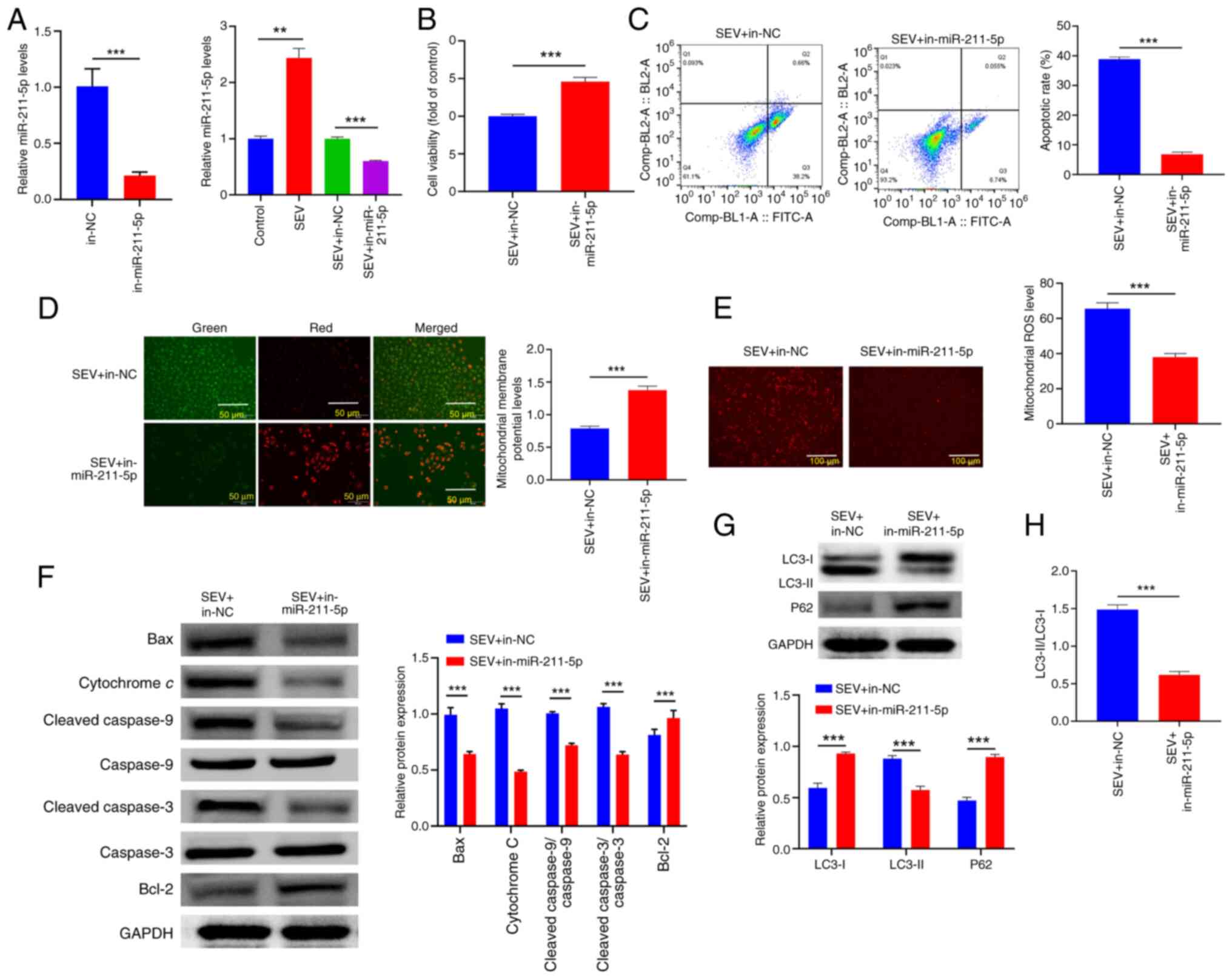 | Figure 3.SEV exposure promotes apoptosis of
glioma cells through upregulation of miR-211-5p expression,
mediating mitochondrial apoptosis pathway. (A) U251 cells were
treated with SEV and miR-211-5p mimic/mimic NC, followed by reverse
transcription-quantitative PCR was performed to detect miR-211-5p
in cells. (B) MTT method was performed to evaluate cell viability,
while (C) flow cytometry was performed to detect cell apoptosis.
(D) JC-1 staining was performed to detect MMP level. (E) MitoSOX™
Red fluorescence staining was used to detect mitochondrial reactive
oxygen species levels. (F) Western blotting was used to detect the
expression of Bcl-2, Bax, cytochrome c,
cleaved-caspase-9/caspase-9 and cleaved-caspase-3/caspase-3
proteins. (G) Western blotting was used to detect the expression of
LC3-I, LC3-II and p62 proteins. (H) LC3-II/I ratio. **P<0.01,
***P<0.001, n=3. SEV, sevoflurane; miR, microRNA; NC, negative
control; MMP, mitochondrial membrane potential; ROS, reactive
oxygen species; Bcl-2, B-cell lymphoma-2; Bax, bcl-2-associated X
protein. |
SIRT1 serves as the target of
miR-211-5p
In addition, the present study searched for possible
downstream mRNA of miR-211-5p. The binding site (Fig. 4A) between miR-211-5p and SIRT1 was
predicted by the StarBase, PITA and miRmap databases. This finding
was further confirmed by a DLR gene assay. The data showed that the
upregulation of miR-211-5p could depress the luciferase activity of
SIRT1-WT, while the activity of SIRT1-MUT was almost unaffected
under the same conditions (Fig.
4B) (P<0.001). In addition, SIRT1 was highly expressed in
U251 cells compared to normal cells (Fig. 4C and D; P<0.01). Notably,
overexpression of miR-211-5p or SEV depressed SIRT1 expression in
U251 cells (Fig. 4D). Furthermore,
downregulation of miR-211-5p eliminated the negative influence of
SEV on SIRT1 expression (Fig. 4E;
P<0.01). In summary, SIRT1 was the direct target of miR-211-5p
and SEV could depress SIRT1 by upregulating miR-211-5p.
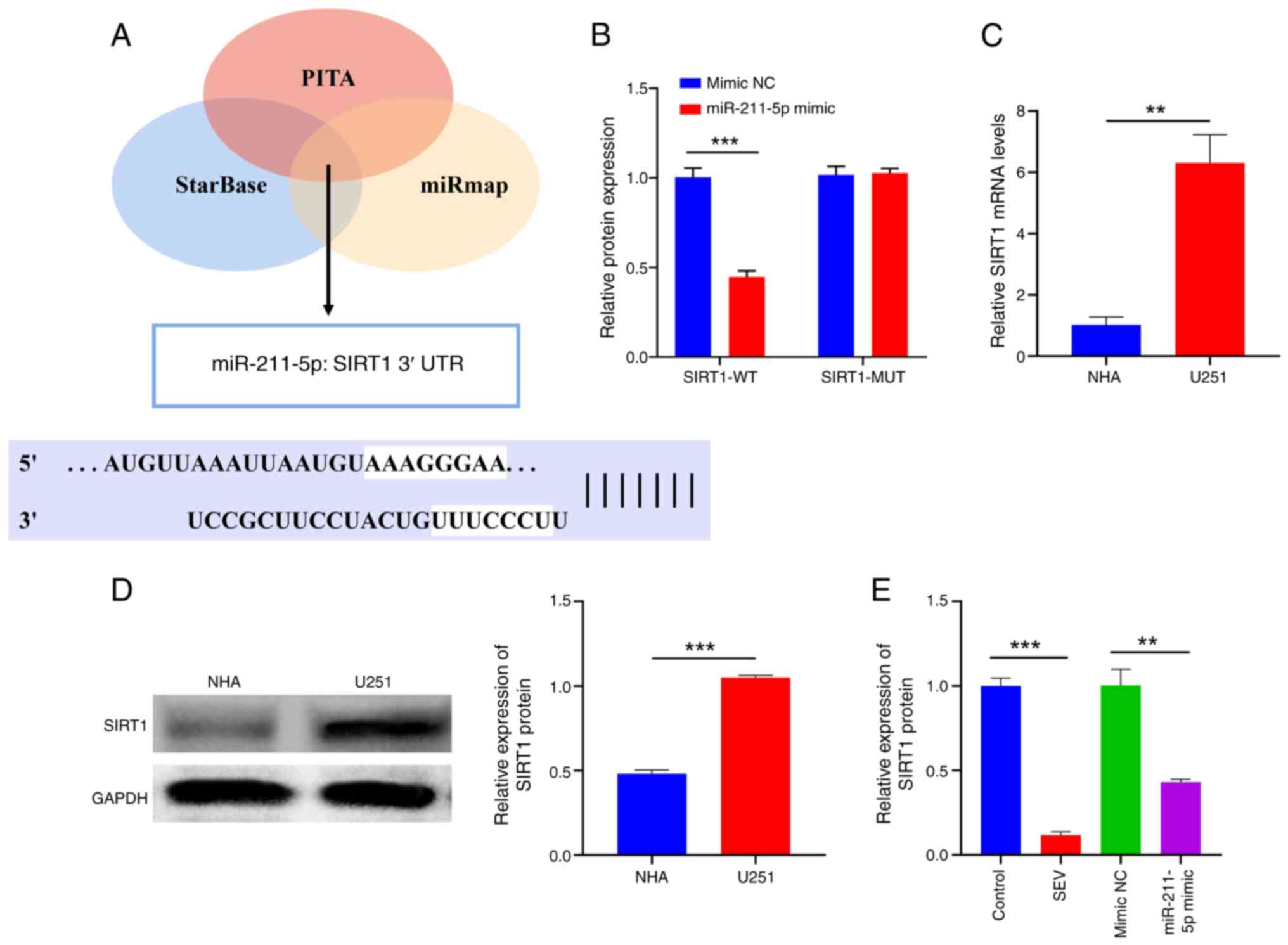 | Figure 4.SIRT1 acts as the target of
miR-211-5p. (A) Bioinformatics tools, including StarBase, PITA and
miRmap, were used to predict the binding site between miR-211-5p
and SIRT1. (B) The luciferase activity was used to evaluate the
interaction between miR-211-5p and SIRT1. (C) Reverse
transcription-quantitative PCR and (D) Western blotting showed
SIRT1 expression in NHA and U251 cells. (E) Reverse
transcription-quantitative PCR showed SIRT1 expression in U251
cells under different treatments. **P<0.01, ***P<0.001, n=3.
SIRT1, silent information regulator 1; miR, microRNA; NHA, normal
human astrocytes; WT, wild type; MUT, mutant; NC, negative
control. |
SIRT1 mediates SEV
mitochondria-dependent apoptosis pathway to induce the apoptosis of
glioma cells
To clarify whether SIRT1 mediated the effect of SEV
on glioma cells, the present study upregulated SIRT1 expression in
the presence of SEV and the transfection efficiency was proved by
RT-qPCR (Fig. 5A). The effects of
SIRT1 on the proliferation and apoptosis of glioma cells were then
observed. The data demonstrated that upregulation of SIRT1 reversed
the effects of SEV on the viability and apoptosis of glioma cells
(Fig. 5B and C; P<0.01).
Additionally, upregulation of SIRT1 reversed the effects of SEV on
mitochondrial apoptotic pathway-associated proteins, MMP, ROS and
autophagy in glioma cells (Fig.
5D-G; P<0.05). These results suggested that the role of SEV
in glioma may depend on the miR-211-5p/SIRT1 signal axis
activation.
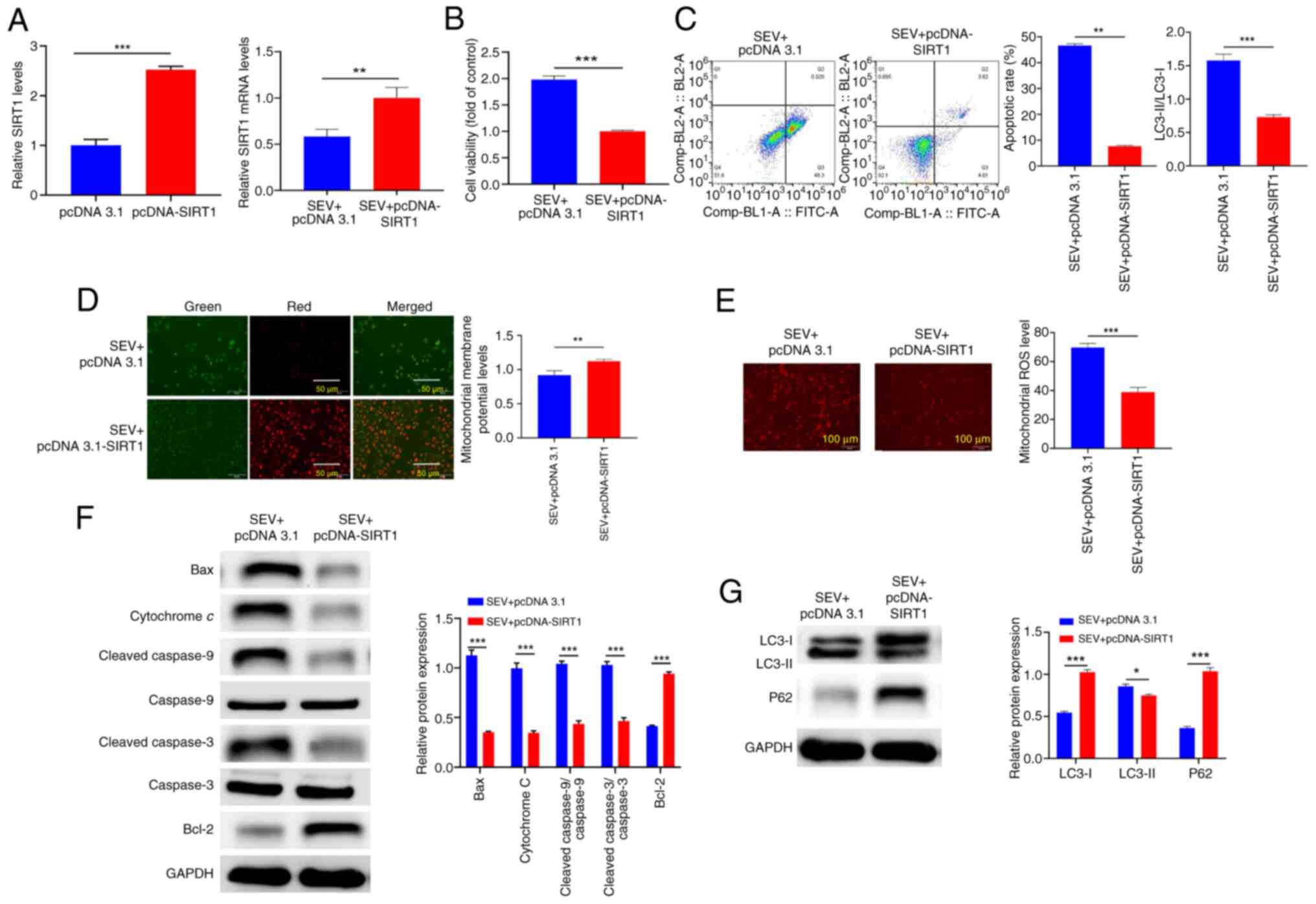 | Figure 5.SIRT1 mediates sevoflurane
mitochondria-dependent apoptosis pathway to induce the apoptosis of
glioma cells. (A) The transfection efficiency of SIRT1 in U251
cells was elevated by reverse transcription-quantitative PCR. (B)
Cell viability was assessed using the MTT method, and (C) flow
cytometry detection of cell apoptosis. (D) JC-1 staining to detect
MMP level. (E) MitoSOX™ Red fluorescence staining was used to
detect mitochondrial reactive oxygen species levels. (F) Western
blotting detection of Bcl-2, Bax, cytochrome c,
cleaved-caspase-9/caspase-9, cleaved-caspase-3/caspase-3, and Bcl-2
protein expression. (G) Western blotting detection of LC3-I, LC3-II
and p62 protein expression. *P<0.05, **P<0.01, ***P<0.001,
n=3. SIRT1, silent information regulator 1; MMP, mitochondrial
membrane potential; Bcl-2, B-cell lymphoma-2; Bax, bcl-2-associated
X protein. |
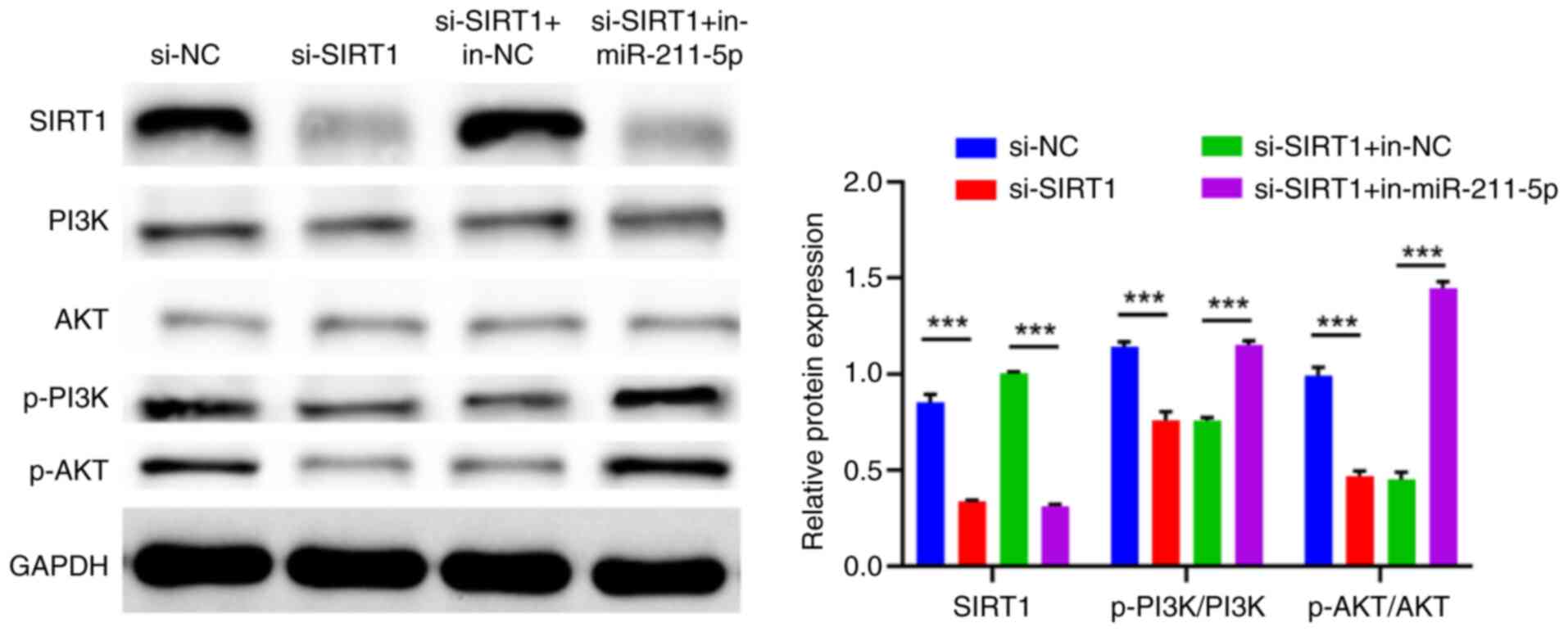
miR-211-5p inactivates the PI3K/AKT
pathway by targeting SIRT1
According to previous studies, the driver of the
PI3K/AKT signaling pathway was significantly associated with
inhibition of the endogenous apoptotic pathway in glioma cells
(12,25). The experimental results of the
present study showed that after SIRT1 expression was knocked down,
p-PI3K/PI3K and p-AKT/AKT protein were decreased in U251 cells
(P<0.001), suggesting that the downregulation of SIRT1 inhibited
the PI3K/AKT pathway activation in U251 cells (Fig. 6). In addition, inhibition of
miR-211-5p could promote SIRT1 expression (P<0.001) and then
activate the PI3K/AKT pathway (Fig.
6). In summary, miR-211-5p could depress the PI3K/AKT pathway
via targeting SIRT1.
Discussion
SEV is one of the most commonly used inhaled
anesthetics in clinical practice and its anti-cancer benefits have
been reported. Recently, there have been a number of studies on how
SEV can treat glioma (20,21,26).
As reported by an earlier study, SEV inhibited the malignant
phenotype of glioma cells by regulating the miR-218-5p/DEK
proto-oncogene/β-Catenin axis (27). However, until now, only a few
articles have addressed the effects and molecular regulatory
mechanisms of SEV on mitochondrial apoptosis. Therefore, the
present study focused on how SEV affected the induction of
apoptosis in glioma cells based on the mitochondrial apoptosis
pathway.
The regulatory balance between cancer cell viability
and apoptosis is the basis of tumorigenesis and the imbalance of
apoptosis is the terminal marker of cancer occurrence and
progression (28). Researchers
have indicated that SEV can induce apoptosis of a number of tumor
cells such as lung cancer cells (29), colon cancer cells (30) and glioma cells (27). However, there is no uniform
concentration standard and time standard for SEV induction in these
researches. Therefore, the present study examined the impact of SEV
on glioma cells using varying concentration and time gradients.
Consistent with a study on the function of SEV on breast cancer
cells (14), SEV significantly
reduced cell viability and increased apoptosis in a dose-dependent
and time-dependent manner. The optimal induction concentration and
time were 6% and 8 h, respectively, indicating the protective
effect of SEV on glioma.
Mitochondria play a central role in the process of
apoptosis because both intrinsic and extrinsic pathways can
converge at the mitochondrial level (31). A number of studies identify a close
correlation between mitochondrial dysfunction and the apoptosis of
glioma cells (32). For example,
Wang et al (33) confirmed
that Embelin induced apoptosis and cell cycle arrest of brain
glioma cells through the mitochondrial pathway. Gu et al
(6) verified that Jinsofenol
promoted apoptosis by activating the mitochondrial apoptotic
pathway in glioma. It is noteworthy that SEV can initiate
mitochondrial apoptotic pathway, induce cytotoxicity and cause cell
apoptosis (18,19). Therefore, the present study
hypothesized that SEV could also initiate mitochondrial apoptotic
pathway in glioma, thereby causing cell apoptosis. The intrinsic
mitochondria-mediated pathway is characterized by depolarization of
MMP. MMP, an indicator of mitochondrial membrane permeability, is
reduced in early apoptosis, leading to the activation of caspase 9
(7). In the present study, SEV
interfered with and destroyed MMP. Some studies show that
mitochondria are the main producers of ROS. Notably, excessive ROS
can lead to the loss of respiratory chain functional complexes,
resulting in the reduction of biological energy, ultimately leading
to cell death (34,35). In the present study, SEV increased
ROS accumulation in mitochondria. Mitochondria-mediated apoptosis
has been reported to be associated with apoptotic pathway-related
proteins. When mitochondria are damaged, cytochrome c in the
inter-membrane space will be released into the cytoplasm.
Cytochrome c interacts with apoptotic protease activating
factor-1, which binds and activates caspase 9, promoting the
formation of apoptosome and ultimately leading to cell apoptosis
(31). In addition, the
mitochondrial apoptotic pathway is directly and strictly regulated
by the balance of pro-apoptotic and anti-apoptotic Bcl-2 family
proteins. Following apoptosis stimulation, Bax will specifically
translocate to the mitochondrial membrane to form homologous
oligomers with other Bax proteins there. This process damages the
MMP, increases the permeability of the mitochondrial outer membrane
and promotes the release of apoptosis factors into the cytoplasm.
Accordingly, the effects of SEV on endogenous mitochondria-mediated
pathway-associated proteins were examined in the present study. The
examination results showed that SEV could increase the expressions
of apoptotic proteins (Bax, cytochrome c,
Cleaved-caspase-9/caspase-9, Cleaved-caspase-3/caspase-3). In
addition, the anti-apoptotic protein (Bcl-2) was decreased,
suggesting that SEV promoted mitochondrial apoptosis by regulating
proteins associated with the mitochondrial apoptosis pathway. These
findings imply that SEV may cause the apoptosis of glioma cells by
initiating the mitochondrial apoptosis pathway.
The homeostasis of mitochondria seems to have
nothing to do with autophagy, but in fact they are inextricably
linked. The relationship between the two is crucial for the state
and survival of mitochondria and tumor cells (5). The autophagy mechanism enables cells
to selectively eliminate excess or redundant mitochondria, thereby
preserving mitochondrial function and maintaining cellular
homeostasis (36). In the process
of autophagy activation, LC3-I is transformed into LC3-II.
Thereafter, LC3-II is connected with p62, integrated into the
autophagosome and finally degraded into the autophagolysosome
(37). In the present study,
glioma cells treated with SEV showed higher LC3-II levels and lower
p62 and LC3-I levels. Also, the conversion rate of LC3-I to LC3-II
increased significantly, suggesting that SEV induced autophagy
activity in glioma cells. Overall, SEV can induce apoptosis through
a series of mechanisms, including the loss of MMP, the production
of ROS, increased expression of apoptotic proteins, decreased
expression of anti-apoptotic proteins, as well as the blocking of
mitochondrial autophagy.
With the advances in genomics and proteomics, more
and more miRNAs have been confirmed to be abnormally expressed
during the development of glioma, such as miR-19 (38), miR-21 and miR-26a (39). They contribute to the pathological
process of glioma by regulating the biological function of glioma
cells. It is noteworthy that SEV plays a role in tumor progression
through regulation of miRNA (20,26).
Based on this, the present study hypothesized that SEV may be
involved in the progression of glioma through regulation of miRNA,
which encouraged the further exploration of SEV and related miRNA.
As a mature sequence of miR-211, miR-211-5P has been identified to
be under-expressed in gliomas and involved in the regulation of
apoptosis pathways in glioma cells (11). Notably, the present study indicated
that SEV could promote miR-211-5p expression. In addition,
downregulation of miR-211-5p reversed the effects of SEV on glioma
cells, suggesting that SEV was involved in the progression of
glioma through regulation of miR-211-5p expression.
A previous study reveales that miRNA, as part of the
RNA-induced silencing complex, can regulate the expression of a
number of genes by partially supplementing the 3′ untranslated
region of the target mRNA (40).
In the present study, SIRT1 was confirmed as a direct target gene
of miR-211-5p through bioinformatics websites and DLR gene assays.
SIRT1 has been widely recognized as a key epigenetic regulator
involved in a number of biological processes, including metabolic
reprogramming, maintenance of genomic stability, autophagy,
senescence, mitochondrial apoptosis and tumorigenesis (9). It has been shown that SIRT1 highly
expresses in glioma tissues and cell lines and that patients with
higher SIRT1 expression have a poorer prognosis (41). Moreover, silencing SIRT1 gene can
significantly promote the apoptosis of glioma cells and inhibit the
proliferation, invasion and metastasis of glioma cells (10). However, whether SEV is involved in
the progression of glioma cells through the miR-211-5p/SIRT1 axis
is unclear. Therefore, the present study conducted a functional
rescue experiment and discovered that SEV inhibited the expression
of SIRT1 by upregulating miR-211-5p. SEV then mediated the
mitochondrial apoptosis pathway to induce apoptosis of glioma
cells. More importantly, the present study also validated that
miR-211-5p inactivated the PI3K/AKT signaling pathway by targeting
SIRT1. According to a previous study, SIRT1 may be a promoter of
cancer cell death mediated by mitochondrial apoptosis through the
PI3K/AKT signaling pathway (42).
Hence, it was hypothesized that SEV was involved in the malignant
progression of tumor cells through the mitochondrial apoptosis
pathway, which was closely related to the regulation of
miR-211-5p/SIRT1/PI3K/AKT signaling axis.
Furthermore, the present study also focused on the
role of autophagy in SEV-induced apoptosis of glioma cells.
Autophagy is a process by which cells maintain intracellular
balance by breaking down their own organelles and proteins. Its
regulation mainly involves a series of genes and proteins,
including mTOR, Beclin 1 and LC3. Inhibition of mTOR activity can
promote the autophagy process of cells and then foster the
occurrence of apoptosis. AKT is reported to activate Rheb protein
by inhibiting the function of the tuberous sclerosis complex
1/tuberous sclerosis complex 2. The activated Rheb protein further
enables mTOR to form the active mTOR complex 1, which mediates cell
growth and metabolic responses (43,44).
Therefore, the present study hypothesized that SEV may promote
autophagy and eventually induce apoptosis by inhibiting the
activity of the mTOR signaling pathway.
The present study confirmed that SEV induced
apoptosis in glioma cells via the mitochondrial pathway,
highlighting its potential role in cancer treatment beyond surgery.
Clinically, SEV can be repurposed as an adjuvant therapy for
glioma, inducing apoptosis through the miR-211-5p/SIRT1/PI3K/AKT
axis. miR-211-5p can also serve as a biomarker for treatment
response. However, further research is still needed. Future studies
need to validate these results in animal models and clinical trials
to establish safety and efficacy. Additionally, exploring the
effects of SEV on tumor migration, invasion and immune response can
provide a comprehensive understanding of anti-cancer potential of
SEV. In the future, the present study will consider including
glioma samples of different grades to more fully evaluate the role
of SIRT1 in glioma development and its feasibility as a potential
therapeutic target. The present study hypothesized this will
provide greater insight into our research and the field of glioma
therapy. While the findings of the present study provided a
scientific basis for the potential application of SEV in the
treatment of glioma, more research and evaluation is needed before
it can be translated into clinical practice. This includes ensuring
that the dosing strategy is safe, effective and takes into account
individual patient differences and the need for a combination
treatment regimen. In addition, the findings were acquired based on
cell experiments and further validation is necessary in animal
models.
The present study revealed that SEV could induce
apoptosis through a series of events, including the loss of MMP,
the production of ROS, the activation of the mitochondrial
apoptosis pathway and the blocking of mitochondrial autophagy.
Furthermore, it also revealed that SEV could induce apoptosis of
glioma cells through the mitochondrial apoptosis pathway and was
related to the regulation of miR-211-5p/SIRT1/PI3K/AKT signaling
axis. The results may enrich the application function of SEV and
provide some new evidences for the efficacy of SEV in the treatment
of glioma. However, there are certain limitations in the present
study. The research was conducted on glioma cell lines, which may
not fully reflect the tumor microenvironment in vivo. The
long-term effects of SEV were not assessed, and the lack of
validation in animal models limits its clinical relevance.
Additionally, other glioma-related pathways and tumor behaviors,
such as migration and invasion, were not explored. Future studies
should address these aspects to provide a more comprehensive
understanding.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Henan Province Science
and Technology Research Project (grant no. 232102310211).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HW and HP contributed to conception and design; GC,
SZ and HQ made contributions to acquisition of data; XZ, AY and XS
analysed and interpretated data; HW and HP were involved in
drafting the manuscript and revising it critically for important
intellectual content. HW, GC and HP confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SIRT1
|
silent Information regulator 1
|
|
SEV
|
sevoflurane
|
|
miRNA
|
microRNA
|
|
U251
|
human glioma cell line
|
|
NHA
|
normal human astrocytes
|
|
ECL
|
enhanced chemiluminescence
|
|
SIRT1-WT
|
SIRT1-wild-type
|
|
SIRT1-MUT
|
mutant vector
|
|
RISC
|
RNA-induced silencing complex
|
References
|
1
|
Xu S, Tang L, Li X, Fan F and Liu Z:
Immunotherapy for glioma: Current management and future
application. Cancer Lett. 476:1–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bush NA, Chang SM and Berger MS: Current
and future strategies for treatment of glioma. Neurosurg Rev.
40:1–14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kurozumi K, Koizumi S and Otani Y: Gene
therapy and viral therapy for malignant glioma. No Shinkei Geka.
49:608–616. 2021.(In Japanese). PubMed/NCBI
|
|
4
|
Natsume A and Yoshida J: Gene therapy for
high-grade glioma: Current approaches and future directions. Cell
Adh Migr. 2:186–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guntuku L, Naidu VG and Yerra VG:
Mitochondrial dysfunction in gliomas: Pharmacotherapeutic potential
of natural compounds. Curr Neuropharmacol. 14:567–583. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu J, Rauniyar S, Wang Y, Zhan W, Ye C, Ji
S and Liu G: Chrysophanol induced glioma cells apoptosis via
activation of mitochondrial apoptosis pathway. Bioengineered.
12:6855–6868. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao H, Liu Z, Xu W, Wang Q, Zhang C, Ding
Y, Nie W, Lai J, Chen Y and Huang H: Pterostilbene promotes
mitochondrial apoptosis and inhibits proliferation in glioma cells.
Sci Rep. 11:63812021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kunadis E and Piperi C: Exploring the
multi-faceted role of sirtuins in glioblastoma pathogenesis and
targeting options. Int J Mol Sci. 23:128892022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alves-Fernandes DK and Jasiulionis MG: The
role of SIRT1 on DNA damage response and epigenetic alterations in
cancer. Int J Mol Sci. 20:31532019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Chen X, Cui Y, Wei Q, Chen S and
Wang X: Effects of SIRT1 silencing on viability, invasion and
metastasis of human glioma cell lines. Oncol Lett. 17:3701–3708.
2019.PubMed/NCBI
|
|
11
|
Qu Y, Zhang J, Wu S, Li B, Liu S and Cheng
J: SIRT1 promotes proliferation and inhibits apoptosis of human
malignant glioma cell lines. Neurosci Lett. 525:168–172. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wongkularb S, Limboonreung T, Tuchinda P
and Chongthammakun S: Suppression of PI3K/Akt/mTOR pathway in
chrysoeriol-induced apoptosis of rat C6 glioma cells. In Vitro Cell
Dev Biol Anim. 58:29–36. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou C, Liang A, Zhang J, Leng J, Xi B,
Zhou B, Yang Y, Zhu R, Zhong L, Jiang X and Wan D: Depleting ANTXR1
suppresses glioma growth via deactivating PI3K/AKT pathway. Cell
Cycle. 22:2097–2112. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng X, Vipani M, Liang G, Gouda D, Wang B
and Wei H: Sevoflurane modulates breast cancer cell survival via
modulation of intracellular calcium homeostasis. BMC Anesthesiol.
20:2532020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yun S, Kim K, Shin K, Park H, Lee S, Shin
Y, Paing AS, Choi S and Lim C: Effect of sevoflurane on the
proliferation of A549 lung cancer cells. Medicina (Kaunas).
59:6132023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu F and Bai T: Sevoflurane activates the
IL-6/HO-1 pathway to promote macrophage M2 polarization and
prostate cancer lung metastasis. Int Immunopharmacol.
113:1093802022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao Z, Gao B, Zong X and Gao R:
Sevoflurane impedes glioma progression via regulating
circ_0000215/miR-1200/NCR3LG1 axis. Metab Brain Dis. 36:2003–2014.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Loop T, Dovi-Akue D, Frick M, Roesslein M,
Egger L, Humar M, Hoetzel A, Schmidt R, Borner C, Pahl HL, et al:
Volatile anesthetics induce caspase-dependent,
mitochondria-mediated apoptosis in human T lymphocytes in vitro.
Anesthesiology. 102:1147–1157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng S and Cheng J: Sevoflurane
suppresses glioma tumorigenesis via regulating
circ_0079593/miR-633/ROCK1 axis. Brain Res. 1767:1475432021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Cheng G, Quan L, Qu H, Yang A, Ye
J, Feng Y, Li X, Shi X and Pan H: Sevoflurane inhibits the
malignant phenotypes of glioma through regulating miR-146b-5p/NFIB
axis. Metab Brain Dis. 37:1373–1386. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu J, Xie B, Huang G, Li Y and Liu Z:
Sevoflurane represses the progression of glioma by the regulation
of circ_0037655/miR-130a-5p/RPN2 axis. Metab Brain Dis. 37:787–799.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen Y, Zhou T, Liu X, Liu Y, Li Y, Zeng
D, Zhong W and Zhang M: Sevoflurane-induced miR-211-5p promotes
neuronal apoptosis by inhibiting Efemp2. ASN Neuro.
13:175909142110350362021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Q, Zheng D, Li Y, Zhang Y, Sui R,
Chen Y, Liang H, Shi J, Pan R, Xu X and Sun D: Circular RNA
circ_0001588 sponges miR-211-5p to facilitate the progression of
glioblastoma via up-regulating YY1 expression. J Gene Med.
23:e33712021. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao C, Shen J, Meng ZX and He XF:
Sevoflurane inhibits glioma cells proliferation and metastasis
through miRNA-124-3p/ROCK1 axis. Pathol Oncol Res. 26:947–954.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Edgunlu TG, Avci CB, Ozates NP, Bagca BG,
Celik SK, Boluk A and Ugur B: In vitro effects of propofol on
cytotoxic, apoptotic and PI3K-akt signaling pathway genes on brain
cancer cells. Anticancer Agents Med Chem. 22:356–361. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhan X, Lei C and Yang L: Sevoflurane
inhibits cell proliferation and migration of glioma by targeting
the miR-27b/VEGF axis. Mol Med Rep. 23:4082021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi Y, Guo L, Liu Y, Zhao T, Liu X and
Zhang Y: Sevoflurane Limits glioma progression by regulating cell
proliferation, apoptosis, migration, and invasion via
miR-218-5p/DEK/β-catenin axis in glioma. Cancer Manag Res.
13:2057–2069. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su G, Yan Z and Deng M: Sevoflurane
inhibits proliferation, invasion, but enhances apoptosis of lung
cancer cells by Wnt/β-catenin signaling via regulating lncRNA
PCAT6/miR-326 axis. Open Life Sci. 15:159–172. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang X, Zheng YT and Rong W: Sevoflurane
induces apoptosis and inhibits the growth and motility of colon
cancer in vitro and in vivo via inactivating Ras/Raf/MEK/ERK
signaling. Life Sci. 239:1169162019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim R, Emi M and Tanabe K: Role of
mitochondria as the gardens of cell death. Cancer Chemother
Pharmacol. 57:545–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ordys BB, Launay S, Deighton RF, McCulloch
J and Whittle IR: The role of mitochondria in glioma
pathophysiology. Mol Neurobiol. 42:64–75. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang A, Zhang B, Zhang J and Wu W and Wu
W: Embelin-induced brain glioma cell apoptosis and cell cycle
arrest via the mitochondrial pathway. Oncol Rep. 29:2473–2478.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng X, Li J, Li H, Chen X, Liu D and Li
R: Bioactive C21 steroidal glycosides from euphorbia kansui
promoted HepG2 cell apoptosis via the degradation of ATP1A1 and
inhibited macrophage polarization under co-cultivation. Molecules.
28:28302023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xue YY, Lu YY, Sun GQ, Fang F, Ji YQ, Tang
HF, Qiu PC and Cheng G: CN-3 induces mitochondrial apoptosis in
glioma via Ros-mediated PI3K/AKT pathway. Pharmazie. 76:208–214.
2021.PubMed/NCBI
|
|
36
|
Ney PA: Mitochondrial autophagy: Origins,
significance, and role of BNIP3 and NIX. Biochim Biophys Acta.
1853:2775–2783. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bisulli F, Tinuper P, Marini C, Avoni P,
Carraro G and Nobile C: Partial epilepsy with prominent auditory
symptoms not linked to chromosome 10q. Epileptic Disord. 4:183–187.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang W, Zhang A, Hao Y, Wang G and Jia Z:
The emerging role of miR-19 in glioma. J Cell Mol Med.
22:4611–4616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
ParvizHamidi M, Haddad G, Ostadrahimi S,
Ostadrahimi N, Sadeghi S, Fayaz S and Fard-Esfahani P: Circulating
miR-26a and miR-21 as biomarkers for glioblastoma multiform.
Biotechnol Appl Biochem. 66:261–265. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ali Syeda Z, Langden SSS, Munkhzul C, Lee
M and Song SJ: Regulatory mechanism of MicroRNA expression in
cancer. Int J Mol Sci. 21:17232020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen H, Lin R, Zhang Z, Wei Q, Zhong Z,
Huang J and Xu Y: Sirtuin 1 knockdown inhibits glioma cell
proliferation and potentiates temozolomide toxicity via
facilitation of reactive oxygen species generation. Oncol Lett.
17:5343–5350. 2019.PubMed/NCBI
|
|
42
|
Wang G, Wang JJ, To TS, Zhao HF and Wang
J: Role of SIRT1-mediated mitochondrial and Akt pathways in
glioblastoma cell death induced by Cotinus coggygria flavonoid
nanoliposomes. Int J Nanomedicine. 10:5005–5023. 2015.PubMed/NCBI
|
|
43
|
Peng Y, Wang Y, Zhou C, Mei W and Zeng C:
PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we
making headway? Front Oncol. 12:8191282022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G,
Liu J and Zhang J: Targeting PI3K/AKT/mTOR-mediated autophagy for
tumor therapy. Appl Microbiol Biotechnol. 104:575–587. 2020.
View Article : Google Scholar : PubMed/NCBI
|