Introduction
Cyclin-dependent kinase 5 (CDK5) is a
serine/threonine kinase that, unlike other members of the CDK
family, is not directly involved in cell cycle regulation (1). Instead, CDK5 plays a crucial role in
various neuronal processes including brain development, synaptic
plasticity and neuronal migration (2–4).
Dysregulation of CDK5 activity has been linked to several
neurodegenerative disorders, highlighting its significance in
maintaining neural integrity and function. Studies have also begun
to elucidate the involvement of CDK5 in non-neuronal tissues and
its emerging role in cancer biology (1,5,6).
CDK5 is activated by binding to its neuron-specific activators, p35
and p39, which are essential for its kinase activity. In the
context of cancer, aberrant activation of CDK5 has been implicated
in promoting tumor progression and metastasis in various types of
cancer, influencing cellular processes such as migration, invasion
and angiogenesis, highlighting it as a potential target for
therapeutic intervention in cancer cells (1,6–15).
p21CIP1/WAF1, commonly referred to as
p21, is a critical regulator of the cell cycle, known for its role
as a CDK inhibitor (16). Encoded
by the CDKN1A gene, p21 primarily functions by binding to and
inhibiting the activity of cyclin-CDK2 or -CDK4 complexes, thus
playing a pivotal role in the G1 phase of the cell cycle
(17). This inhibition prevents
the phosphorylation of the retinoblastoma protein, effectively
halting cell cycle progression and allowing the cell time to repair
DNA damage or, if necessary, initiate apoptosis (18,19).
p21 is tightly regulated by the tumor suppressor protein p53, which
induces p21 expression in response to DNA damage and other stress
signals (16,20,21).
Through this mechanism, p21 acts as a safeguard against
uncontrolled cell proliferation, making it a key player in
maintaining genomic stability and preventing tumorigenesis
(22). Beyond its role in the cell
cycle, p21 is also involved in several other cellular processes,
including apoptosis, transcriptional regulation and cell senescence
(16). The dual role of p21 in
both cell cycle arrest and the promotion of cell survival makes it
a complex and context-dependent factor in cancer biology. While the
function of p21 as a tumor suppressor is well-established, its role
in cancer can be paradoxical. In certain contexts, particularly
when p53 is mutated, p21 can contribute to tumor progression by
promoting cell survival and resistance to chemotherapy (23). Understanding the multifaceted roles
of p21 in cancer, particularly its interactions with other cellular
proteins such as CDK5, is crucial for developing targeted therapies
that exploit these pathways for cancer treatment.
Thyroid cancer (TC) is a malignancy that originates
in the thyroid gland, a small, butterfly-shaped organ located at
the base of the neck (24). The
thyroid gland plays a crucial role in regulating the body's
metabolism through the production of hormones such as thyroxine and
triiodothyronine (25). Although
TC is relatively uncommon compared with other types of cancer, it
is the most prevalent endocrine malignancy and its incidence has
been increasing globally over the past few decades (26,27).
There are several distinct types of TC, each with unique
characteristics and prognoses. The most common form is papillary
(P)TC, which accounts for ~80% of all TC cases (28,29).
PTC generally has a favorable prognosis, with high survival rates,
especially when detected early (30). However, a subset of PTC cases can
be aggressive, leading to local invasion, recurrence and distant
metastasis (31–34). Other types of TC include follicular
TC (35), medullary TC (36) and the most aggressive form,
anaplastic TC (37), which is rare
but highly lethal. TC is often diagnosed through the detection of a
thyroid nodule, followed by further evaluation with ultrasound,
fine-needle aspiration biopsy and molecular testing (38). Treatment typically involves
surgical removal of the thyroid gland (thyroidectomy) (39), followed by radioactive iodine
therapy in certain cases (40).
While several patients respond well to these treatments, a subset
with aggressive disease or metastatic spread may require additional
therapies, including targeted therapies or external beam radiation
(41). Research into the molecular
mechanisms underlying TC has revealed a variety of genetic and
environmental factors that contribute to its development. Mutations
in genes such as BRAF, RAS and RET/PTC are commonly implicated in
TC (6,42), while alterations in the PI3K/AKT
and MAPK signaling pathways are frequently observed in more
aggressive forms of the disease (43–45).
Understanding these molecular drivers has led to the development of
targeted therapies aimed at inhibiting these pathways, offering new
hope for patients with advanced TC. Despite advances in diagnosis
and treatment, challenges remain in managing aggressive and
recurrent TC cases. Continued research is essential to identify
novel therapeutic targets and improve outcomes for patients with
this complex and increasingly common disease.
In the present study, the biochemical interaction
between CDK5 and p21 in PTC cells was assessed and the functional
consequences of this interaction on cell proliferation and
malignancy were explored. Using advanced bioinformatic tools such
as AlphaFold 3.0 and Chimera X for protein-protein interaction
analysis alongside immunoprecipitation (IP) assays, it was shown
that CDK5 targeted p21 for ubiquitin-dependent degradation.
Furthermore, the findings were validated using clinical tumor
samples and data from The Cancer Genome Atlas (TCGA), highlighting
the clinical relevance of CDK5 and p21 in TC. The results not only
elucidated a novel regulatory mechanism involving CDK5 and p21 but
also suggested potential therapeutic strategies targeting this
pathway to curb TC progression. Given the emerging role of CDK5 in
cancer, these findings open novel avenues for research and
development of CDK5 inhibitors as anti-cancer agents.
Materials and methods
Ethical approval
The present study was approved by the Institutional
Review Board of Tungs' Taichung MetroHarbor Hospital, Taichung,
Taiwan. (approval no. 113003). All experimental procedures followed
the relevant ethical guidelines and informed consent was obtained
from all patients with TC included in the present study. Thyroid
tissue samples were collected from a cohort of these patients.
Patient samples
The patient samples were collected from Tungs'
Taichung MetroHarbor Hospital, Taichung Taiwan. Samples were
collected between 2024/05/01 and 2024/05/31. A total of 11 patients
participated in the study, five samples for each patient, and a
total of 55 samples were collected for analysis. Samples were
preserved and processed for immunohistochemical (IHC) analysis to
evaluate the expression of specific proteins. Clinical data on the
patients' age, sex and cancer staging were obtained from the
hospital's electronic medical records (Table I). All samples were anonymized to
ensure patient confidentiality in accordance with Institutional
Review Board (IRB) regulations.
 | Table I.Patients background and IHC
profile. |
Table I.
Patients background and IHC
profile.
| Patient | Age, years | Sex | Cancer stage | TNM staging | Malignancy
features | IHC profile | Expression profile
(%) |
|---|
| Patient 1 | 62 | Male | I | T1a, Nx, Mx | None | Moderate CDK5,
Moderate p21 | ~57 |
| Patient 2 | 60 | Male | II | T1b, N1b, Mx | None | High CDK5, Low
p21 | ~65 |
| Patient 3 | 61 | Male | III | T4a, N0, Mx | Recurrence | High CDK5, Low
p21 | ~70 |
| Patient 4 | 54 | Male | I | T1a, Nx, Mx | Double Cancer | High CDK5, Low
p21 | ~55 |
| Patient 5 | 42 | Female | I | T1a, N0, Mx | None | Moderate CDK5, High
p21 | ~46 |
| Patient 6 | 32 | Female | I | T1b, Nx, Mx | None | Moderate CDK5,
Moderate p21 | ~60 |
| Patient 7 | 24 | Female | I | T1b, N0, Mx | None | Moderate CDK5,
Moderate p21 | ~57 |
| Patient 8 | 70 | Female | I | T2, N0, Mx | None | Moderate CDK5,
Moderate p21 | ~58 |
| Patient 9 | 63 | Female | IVA | T1b, N1b, Mx | Recurrence | High CDK5, low
p21 | ~95 |
| Patient 10 | 58 | Female | II | T2, N1, Mx | None | Moderate CDK5,
Moderate p21 | ~58 |
| Patient 11 | 48 | Female | I | T1a, Nx, Mx | None | Moderate CDK5,
Moderate p21 | ~50 |
IHC staining
The expression of proteins was assessed using IHC on
formalin-fixed, paraffin-embedded tissue sections. The sections
were cut into 4 µm thick slices and mounted on positively charged
glass slides. The slides were deparaffinized in xylene and
rehydrated using a decreasing series of graded alcohol solutions,
followed by a rinse in distilled water. For antigen retrieval,
heat-induced epitope retrieval using a citrate buffer (pH 6.0) was
performed for 20 min in a pressure cooker. To block endogenous
peroxidase activity, sections were incubated with 3% hydrogen
peroxide (H2O2) in methanol for 10 min at
room temperature, followed by washing in PBS. After retrieval, the
slides were allowed to cool to room temperature and then washed in
PBS. To block non-specific binding, the slides were incubated with
5% normal goat serum (Vector Laboratories, Inc. cat. no. ZH083)
with blocking solution for 30 min at room temperature. The tissue
sections were incubated overnight at 4°C with the following primary
antibodies: Mouse monoclonal anti-CDK5 (cat. no. sc-249; Santa Cruz
Biotechnology, Inc.; 1:20) or rabbit polyclonal anti-p21 (cat. no.
2947; Cell Signaling Technology, Inc.; 1:50). After washing the
slides in PBS, they were incubated with appropriate biotinylated
secondary antibodies for 30 min at room temperature (Goat
anti-mouse IgG, cat. no. BA-9200 or Goat anti-rabbit IgG, cat. no.
BA-1000; Vector Laboratories, Inc.; dilution 1:200), followed by a
wash in PBS. Signals were visualized using a horseradish peroxidase
(HRP)-conjugated streptavidin detection system (Vectastain ABC kit,
Vector Laboratories, Inc.). The slides were developed using
3,3′-diaminobenzidine (DAB; Dako) as the chromogen for 5 min at
room temperature and counterstained with hematoxylin to visualize
nuclei for 2 min at room temperature. Finally, the sections were
dehydrated, cleared in xylene and a coverslip was placed over them
using a permanent mounting medium. Positive control slides were
included for each staining run. Negative controls were performed by
omitting the primary antibodies to assess non-specific binding of
the secondary antibody.
Imaging and analysis
Stained slides were scanned using a high-resolution
digital microscope (Zeiss Imager. Z2; Carl Zeiss AG) and images
were captured for analysis. Protein expression was
semi-quantitatively evaluated based on staining intensity and the
percentage of positive cells using TissueFAXS viewer version 7.1.6.
supplied by TissueGnostics GmbH. The results were classified into
low, moderate, or high expression levels. Statistical analysis was
performed to evaluate the correlation between protein expression
and clinical outcomes in patients with TC.
Cell culture and transfection
Human TC cell lines BCPAP and TPC-1 were purchased
from the Food Industry Research and Development Institute in
Taiwan. The two TC cell lines were provided by collaborators Dr
Chen-Kai Chou and Dr Hong-Yo Kang in Kaohsiung Chang Gung Hospital
(Taiwan) (46). Cells were
maintained in RPMI-1640 culture medium (Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), penicillin-streptomycin at concentrations of 100
IU/ml and 100 µg/ml, respectively (MilliporeSigma), along with 2 mM
L-glutamine, 1.5 g/l sodium bicarbonate (NaHCO3)
(MilliporeSigma), 10 mM HEPES (MilliporeSigma) and 1 mM sodium
pyruvate (MilliporeSigma). For cell transfection, cells were
transfected with either short hairpin (sh)RNA targeting CDK5
(shCDK5) or a scrambled shRNA control (shCtrl) using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) as the
transfection reagent for 6 h at 37°C. Overexpression plasmids were
transfected in the same manner. Transfections were performed in
serum-free medium according to the manufacturer's protocol. After 6
h of incubation, the transfection medium was replaced with fresh
complete medium and cells were cultured for an additional 24–48 h
before proceeding with downstream analyses. Plasmids used for
transfection were obtained from the National RNAi Core Facility
(Academia Sinica). Specifically, the pLKO.1 plasmid harboring shRNA
sequence targeting CDK5 or scrambled control (shCtrl) were used at
a concentration of 2 µg/ml. The target sequences of shGFP and
shCDK5 were: TRCN0000072192 (shGFP): 5′-GAACGGCATCAAGGTGAACTT-3′,
TRCN0000021466 (shcdk5 #1): 5′-TGTCCAGCGTATCTCAGCAGA-3′,
TRCN0000199652 (shcdk5 #2): 5′-GTGAACGTCGTGCCCAAACTC-3′,
TRCN0000194974 (shcdk5 #3): 5′-CCTGAGATTGTAAAGTCATTC-3′.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from cultured cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's instructions. RNA quality and
concentration were assessed by spectrophotometry. Reverse
transcription was performed using 1 µg of total RNA with the
High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. qPCR was performed using SYBR Green (Roche Applied
Science) on a real-time PCR system by using Applied Biosystems
StepOnePlus (Applied Biosystems; Thermo Fisher Scientific, Inc.) as
described previously (22). Gene
expression levels were normalized to the housekeeping gene, and
relative quantification was calculated using the 2−ΔΔCq
method as described previously by Livak and Schmittgen (47). The thermocycling conditions were as
follows: initial denaturation at 95°C for 10 min, followed by 40
cycles of denaturation at 95°C for 15 sec, annealing and extension
at 60°C for 1 min. Each experiment was performed in triplicate and
independently replicated at least three times. The following
primers were used to amplify the cDNA: CDKN1A
(5′-ACCCTAGTTCTACCTCAGGC-3′ and 5′-AAGATCTACTCCCCCATCAT-3′), CDK5
(5′-TCTTTTTCCCGGCAATGAT-3′ and 5′-TCTGGCAGCTTGGTCATAGA-3′), and
actin (5′-TTGCCGACAGGATGCAGAA-3′ and
5′-GCCGATCCACACGGAGTACT-3′).
Western blotting
Cells were lysed at approximately 80–90% confluency
using lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40,
0.5% sodium deoxycholate, 0.1% SDS] supplemented with a protease
inhibitor cocktail (MilliporeSigma). Protein concentrations were
measured using a BCA Protein Assay Kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of protein (30 µg per lane) were
separated by electrophoresis on 10% SDS-polyacrylamide gels
(SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF)
membranes (MilliporeSigma). Membranes were blocked in 5% non-fat
dry milk (MilliporeSigma) in TBS containing 0.1% Tween-20 (TBST)
for 1 h at room temperature. After blocking, membranes were
incubated overnight at 4°C with primary antibodies diluted in
blocking buffer: anti-CDK5 (cat. no. ab40773; Abcam; 1:1,000) and
anti-p21 (cat. no. 2947; Cell Signaling Technology, Inc.; 1:1,000).
After incubation, membranes were washed three times (5 min each) in
TBST (Tris-buffered saline containing 0.1% Tween-20). Subsequently,
membranes were incubated with HRP-conjugated secondary antibodies
(goat anti-rabbit IgG-HRP, cat. no. 7074, 1:5,000; goat anti-mouse
IgG-HRP, cat. no. 7076, 1:5,000; Cell Signaling Technology, Inc.)
at room temperature for 1 h. Following incubation, membranes were
washed three times (5 min each) in TBST. Proteins were visualized
using enhanced chemiluminescence (ECL) detection reagent (Pierce
ECL Western Blotting Substrate; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Signal intensities were
captured using a chemiluminescent imaging system (Bio-Rad
Laboratories, Inc.). β-actin (cat. no. 4970; Cell Signaling
Technology, Inc.; dilution 1:2,000) was used as the loading
control. Each experiment was independently repeated at least three
times.
IP
IP was performed as previously described (48). Briefly, BCPAP and TPC-1 cells were
seeded into 10-cm culture dishes at a density of ~1×106
cells per dish and cultured until reaching 80–90% confluency. BCPAP
and TPC-1 cells were lysed in IP lysis buffer (50 mM Tris-HCl pH
7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and protease inhibitors).
Protein concentration was determined using a BCA Protein Assay Kit
(Pierce; Thermo Fisher Scientific, Inc.). Equal amounts of protein
lysate (500 µg) were incubated with 2 µg anti-CDK5 antibody (Abcam)
overnight at 4°C with gentle rotation. Protein A/G agarose beads
(Thermo Fisher Scientific, Inc.) were added and incubated for an
additional 2 h at 4°C. Input controls (30 µg of total protein
lysate without immunoprecipitation) were included for western blot
analysis to confirm the presence of target proteins. Beads were
washed three times with ice-cold washing buffer (20 mM Tris-HCl pH
7.4, 150 mM NaCl, 0.1% NP-40) and proteins were eluted with SDS
sample buffer by boiling for 5 min. Eluates were analyzed using 10%
SDS-PAGE followed by western blotting using an anti-p21 antibody
(cat. no. 2947; Cell Signaling Technology, Inc.) as
aforementioned.
Protein stability assay and MG132
treatment
Protein stability assays were performed as
previously described (49). To
assess p21 protein stability, BCPAP and TPC-1 cells were treated
with cycloheximide (CHX; MilliporeSigma) at a final concentration
of 50 µg/ml to inhibit protein synthesis. Cells were collected at
0, 2, 4 and 6 h post-CHX treatment. For proteasome inhibition,
cells were treated with 10 µM MG132 (Calbiochem; Merck KGaA) for 6
h prior to lysis. Cell lysates were prepared and protein levels of
p21 and CDK5 were determined by western blotting as aforementioned.
β-Actin (Cell Signaling Technology, Inc.) was used as the loading
control.
Immunocytochemistry
Immunocytochemistry was performed as previously
described (3). Briefly, cells were
fixed for 5 min in 4% paraformaldehyde and 2% sucrose in PBS at
room temperature. Subsequently, the cells were permeabilized and
blocked in PBS buffer containing 0.1% Triton X-100 and 5% bovine
serum albumin (Cyrus Bioscience, Lot no. 1260601) for 15 min at
room temperature. The primary antibodies CDK5 (Santa Cruz
Biotechnology, Inc.; cat. no. sc-249; dilution 1:100) and p21 (Cell
Signaling Technology, Inc.; cat. no. 2947; dilution 1:200) were
diluted in 5% BSA in PBS and added to the cells for overnight
incubation at 4°C. Then, the cells were incubated with Alexa Fluor
488-conjugated secondary antibodies (Thermo Fisher Scientific,
Inc.; cat. no. A-11008; dilution 1:1,000) and Alexa Fluor
546-conjugated secondary antibodies (Thermo Fisher Scientific,
Inc.; cat. no. A-11003; dilution 1:1,000) for 1 h at room
temperature. The cells were washed again with PBS and
counterstained with DAPI (Thermo Fisher Scientific, Inc.) for 5 min
at room temperature. Cells were subsequently washed with PBS,
mounted using Fluoromount-G (Southern Biotech; cat. no. 0100-01)
and analyzed using a fluorescence microscope (Olympus BX-51;
Olympus Corporation) and a Zeiss LSM510 confocal microscope (Carl
Zeiss AG).
Bioinformatics analysis for
protein-protein interactions
The 3D structures of proteins were visualized as
previously described (3). The
interaction between CDK5 and p21 was predicted using AlphaFold 3.0,
an AI-based protein structure prediction tool (50). The protein structures were
visualized and analyzed for interaction sites using UCSF Chimera
software (Resource for Biocomputing, Visualization, and
Informatics, University of California, San Francisco, CA, USA).
Chimera was used to map potential interaction interfaces and
validate the predicted binding sites.
TCGA data analysis
TCGA data analysis was performed as previously
reported (5). Expression data and
clinical information for patients with TC were downloaded from TCGA
using the UCSC Xena browser (https://xenabrowser.net/). Pearson correlation
analysis was performed to evaluate the relationship between CDK5
and p21 mRNA expression levels. Kaplan-Meier survival analysis was
conducted using the ‘survival’ package in R to compare overall
survival between high and low-expression groups for CDK5 and p21.
Kaplan-Meier survival analysis was performed using the ‘survival’
package in R. Differential expression analysis between tumor and
normal tissues was assessed using the Wilcoxon rank-sum test.
Violin plots were generated to visualize the stage-specific
expression of CDK5 and p21 across different stages of TC.
Differential expression analysis was conducted to compare CDK5 and
p21 expression in tumor compared with normal tissues, with
statistical significance assessed using the Wilcoxon rank-sum test.
Statistical analyses and visualizations, including Pearson
correlation, Kaplan-Meier survival analysis, violin plots, and
differential expression analysis, were conducted using R software
(R Core Team, 2023). R: A language and environment for statistical
computing. R Foundation for Statistical Computing, Vienna, Austria
(http://www.R-project.org/).
Statistical analysis
All statistical analyses were conducted using
GraphPad Prism version 8.0.1 (Dotmatics). Data are presented as the
mean ± SD of at least three independent experiments. Comparisons
between two groups were performed using an unpaired Student's
t-test. Pathological stage plots were obtained from GEPIA
(http://gepia.cancer-pku.cn/), and
differential gene expression was analyzed by one-way ANOVA followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
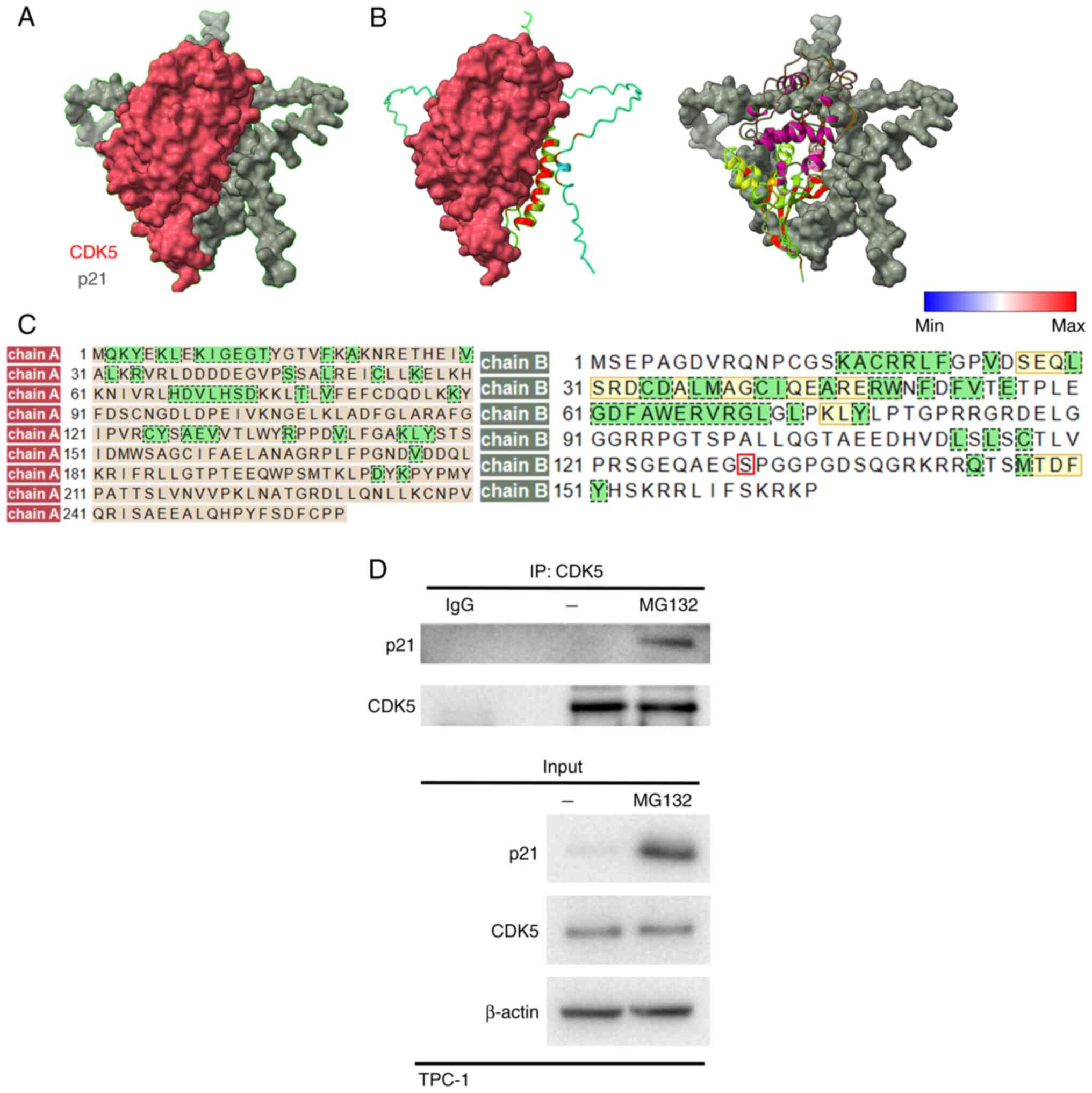
CDK5 interacts with p21
The interaction between CDK5 and p21CIP1
is pivotal for understanding the regulatory mechanisms underlying
cell cycle progression and tumor suppression in TC. To investigate
this interaction, AlphaFold 3.0, a cutting-edge AI-based protein
structure prediction tool, was used to generate a model of the
CDK5-p21 protein-protein interaction. The accuracy of AlphaFold in
predicting protein structures based on amino acid sequences
provided a reliable foundation for the analysis (50). Using data generated from AlphaFold,
Chimera was used to visualize and analyze the interaction sites
between CDK5 and p21. The analysis revealed that CDK5 directly
interacted with specific regions of the p21 protein, indicating
potential sites for regulation through phosphorylation and other
post-translational modifications (Fig.
1A-D). This interaction is significant as it implies modulation
of the stability of p21 and activity in TC cells, potentially
contributing to enhanced cell proliferation and malignancy. To
experimentally validate the CDK5-p21 interaction, IP assays were
performed using the TC cell line TPC-1. Additionally, cells were
treated with MG132, a proteasome inhibitor, to prevent proteasomal
degradation and ensure the detection of any ubiquitin-dependent
degradation products. The IP analysis confirmed that CDK5
biochemically interacted with p21 protein in TPC-1 cells,
reinforcing the computational predictions and suggesting a direct
regulatory relationship between CDK5 and p21 in TC (Fig. 1D). These findings provide a
comprehensive understanding of the CDK5-p21 interaction,
demonstrating its potential role in modulating cell cycle
regulation and promoting the malignancy of TC.
CDK5 reduces p21 protein stability and
induces TC cell proliferation
To investigate the role of CDK5 in the regulation of
p21CIP1 in TC cells, several experiments were performed
to analyze protein interactions, degradation and cellular
localization. TC cells transfected with either an empty vector (EV)
or CDK5 were treated with CHX to inhibit protein synthesis. Western
blot analysis showed that p21 levels decreased more rapidly in
cells overexpressing CDK5 compared with the EV control. This
suggested that CDK5 accelerated p21 degradation (Fig. 2A and B). Next, cells were treated
with MG132, a proteasome inhibitor, to block protein degradation.
In mock-treated cells, overexpression of CDK5 resulted in lower p21
levels compared with the EV control. In MG132-treated cells, p21
levels did not differ markedly between CDK5-overexpressing cells
and EV controls. In addition, IHC was used to further confirm the
results. In control cells (vector), p21 was normally expressed,
while in CDK5-overexpressing cells, p21 levels were visibly
reduced. Upon MG132 treatment, p21 levels were restored in
CDK5-overexpressing cells, confirming the role of
proteasome-mediated degradation (Fig.
2C-E). The data collectively demonstrate that CDK5
downregulates p21 in TC cells by promoting its ubiquitin-dependent
degradation via the proteasome pathway. This regulatory mechanism
suggests a significant role for CDK5 in modulating cell cycle
progression and tumor development in TC by targeting p21 for
degradation. To further elucidate the mechanism by which CDK5
regulated p21, Co-IP experiments were performed to analyze the
interaction between CDK5 and p21. Wild-type (WT) p21 and its
phosphorylation-deficient mutant (MT; S130A) were compared. Western
blot analysis revealed that CDK5 effectively interacted with WT, as
shown by the co-precipitation of CDK5 and p21 proteins (Fig. 2F). However, the intensity of the
interaction signal was lower for MT p21, suggesting that the S130
site influenced the strength of the CDK5-p21 interaction (Fig. 2F). To assess the functional impact
of CDK5-mediated p21 regulation on cell proliferation, TC cells
were analyzed for their proliferation rates in the presence of WT
and MT p21. The results showed that CDK5 overexpression markedly
enhanced cell proliferation when WT p21 was present. By contrast,
cells expressing MT p21 (S130A) displayed suppressed CDK5-mediated
proliferation (Fig. 2G),
suggesting that the phosphorylation of p21 at the S130 site is
essential for CDK5 to promote cell proliferation. These findings
indicated that the phosphorylation-deficient mutant p21 (S130A)
blocks the ability of CDK5 to enhance cell proliferation, thereby
acting as a dominant-negative form to counteract CDK5-mediated
oncogenic effects. The data collectively suggested that CDK5
promoted cell proliferation in TC by targeting p21 for
phosphorylation-dependent degradation and that MT p21 can serve as
a potential therapeutic tool to inhibit CDK5-driven tumor
progression.
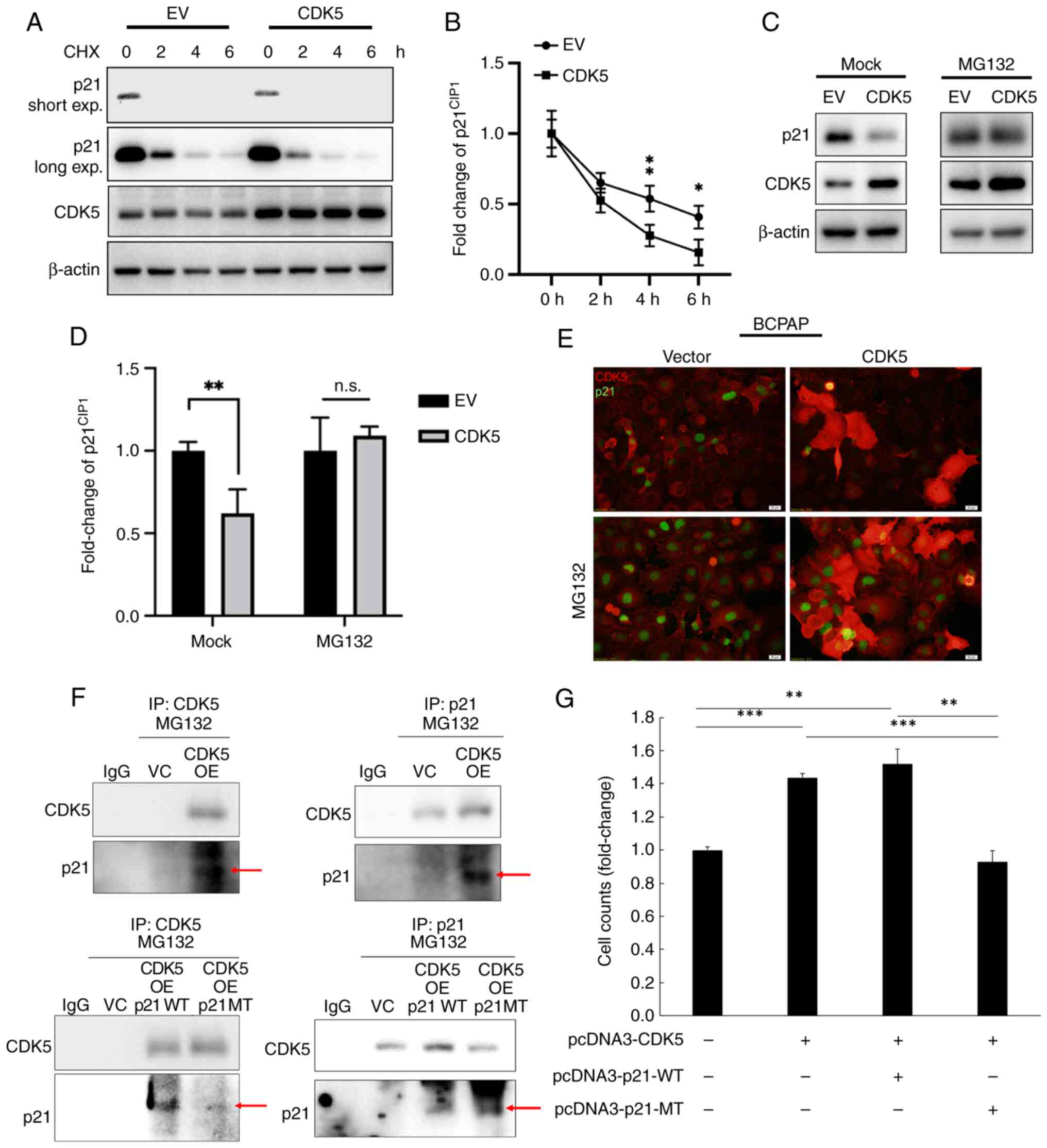 | Figure 2.CDK5 downregulates p21 in TC cells.
(A) Western blot analysis of p21 protein expression levels in BCPAP
cells transfected with EV or a CDK5 overexpression plasmid. Cells
were treated with 50 µg/ml CHX for 0, 2, 4 or 6 h to inhibit
protein synthesis. Short and long exposures of p21 are shown. (B)
Quantification of p21 protein levels. Data are presented as the
fold change relative to time 0 for EV and CDK5 transfected cells.
(C) Western blot analysis of p21 and CDK5 protein expression levels
in BCPAP cells transfected with EV or CDK5, with or without
treatment with 10 µM MG132 for 6 h. (D) Quantification of p21
protein expression. Data are presented as fold change relative to
EV-transfected mock-treated cells. (E) Immunofluorescence staining
of p21 (green) in BCPAP cells transfected with vector or CDK5, with
or without MG132 treatment. The results indicate that CDK5
overexpression reduces p21 levels in TC cells and this effect was
reversed following proteasome inhibition. Scale bar, 20 µm. (F)
Co-immunoprecipitation analysis of CDK5 and p21 was performed to
investigate the interaction between CDK5 and p21 in thyroid cancer
cells. Panels show the interaction of CDK5 with WT p21 and MT p21
following proteasome inhibition using MG132. Red arrows indicate
the presence of p21 in the immunoprecipitated complex. (G) Cell
counting assay was performed to evaluate p21 WT and MT p21 on cell
proliferation. For all blots, β-actin was used as the loading
control. Values are presented as the mean ± SD from three
independent experiments. Data were compared using an unpaired
Student's t-test. *P<0.05, **P<0.01, ***P<0.001. CDK5,
cyclin-dependent kinase; 5TC, thyroid cancer; EV, empty vector;
HCX, cycloheximide; n.s., not significant; WT, wild-type; MT, S130A
mutant. |
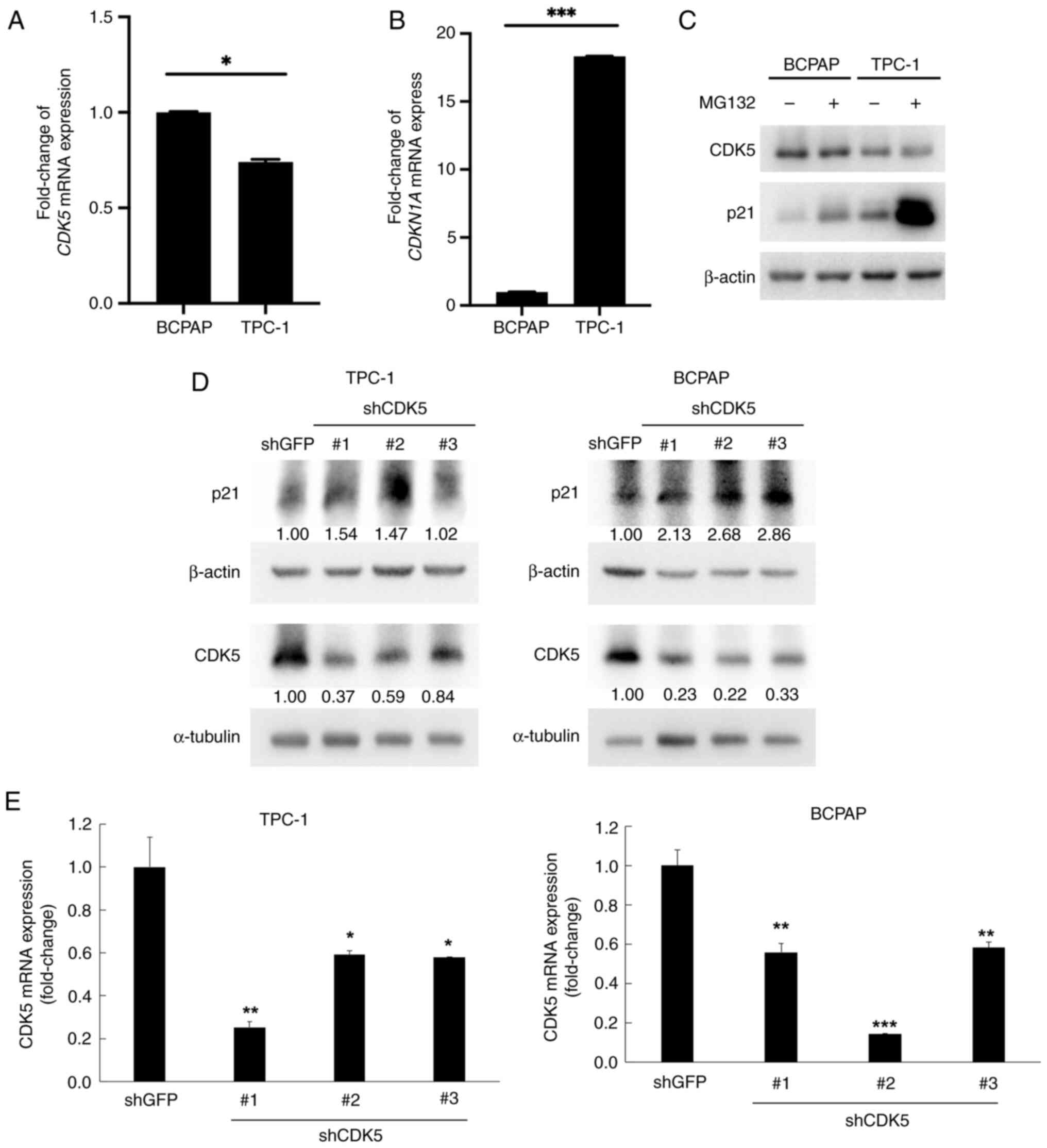
CDK5 negatively regulates p21
expression in TC cells
To further elucidate the regulatory relationship
between CDK5 and p21CIP1 in TC cells, several
experiments examining mRNA and protein expression levels, as well
as the effects of CDK5 knockdown were performed. RT-qPCR was used
to measure the mRNA expression levels of CDK5 in BCPAP and TPC-1
cells. The results showed markedly higher CDK5 mRNA expression in
BCPAP cells compared with TPC-1 cells (Fig. 3A). In addition, TPC-1 cells had
markedly higher p21 mRNA expression levels than BCPAP cells
(Fig. 3B). Western blot analysis
was performed to examine the protein expression levels of CDK5 and
p21 in BCPAP and TPC-1 cells, both with and without MG132
treatment. In the absence of MG132, TPC-1 cells showed higher
levels of CDK5 and lower levels of p21 expression compared with
BCPAP cells. MG132 treatment increased p21 levels in both cell
lines, but the increase was more pronounced in TPC-1 cells,
indicating that CDK5 promoted p21 degradation through the
proteasome pathway (Fig. 3C). In
addition, TPC-1 and BCPAP cells were transfected with shRNAs
targeting CDK5 (shCDK5 #1, shCDK5 #2 and shCDK5 #3) or a control
shRNA (shGFP). Following CDK5 knockdown, p21 protein expression
levels were measured. Cells with CDK5 expression knocked down
exhibited a significant increase in p21 levels compared with the
shGFP control (Fig. 3D and E).
This indicated that CDK5 negatively regulates p21 expression, as
knockdown of CDK5 led to an increase in p21 protein expression
levels.
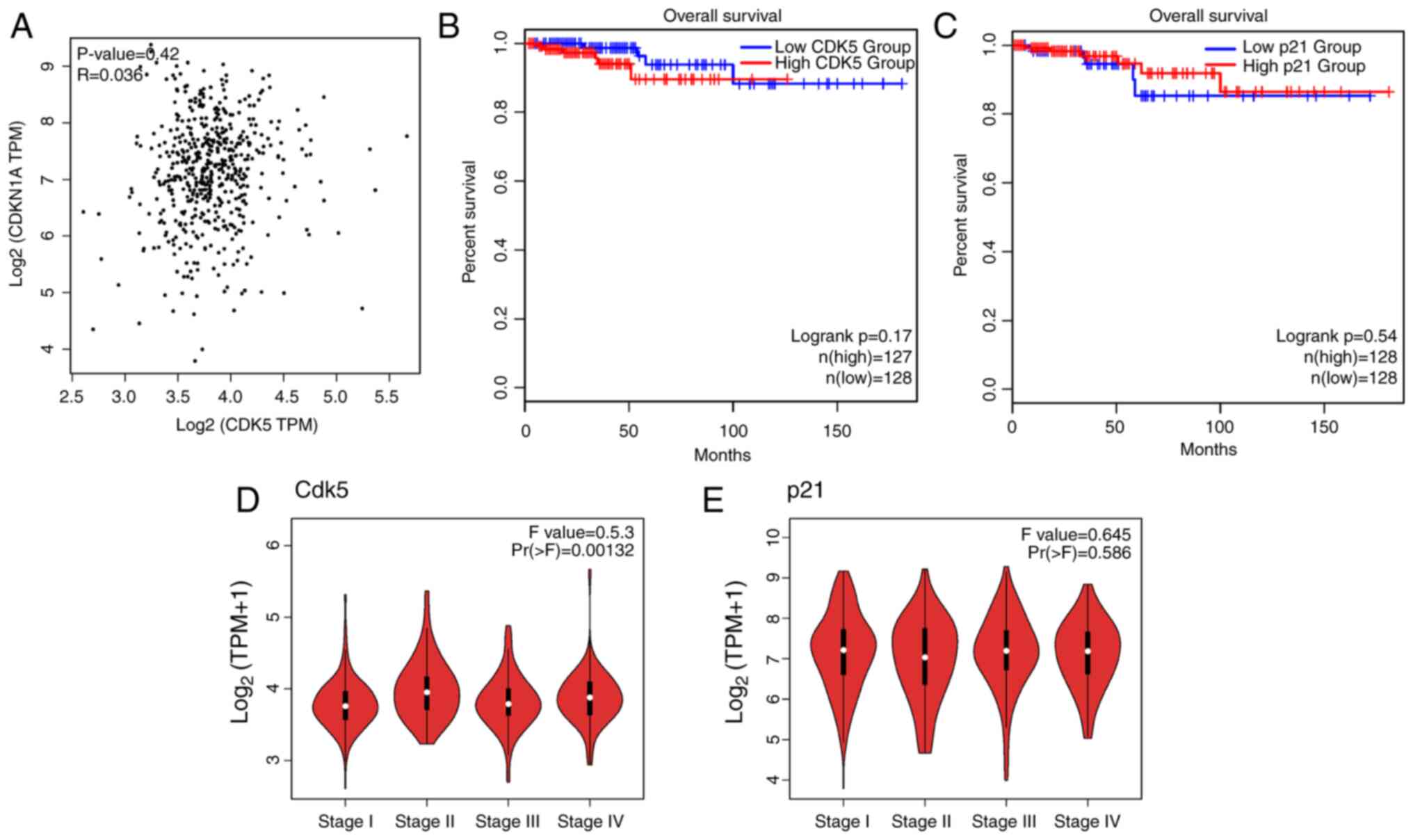
CDK5 contributes to tumor malignancy
in patients with TC
To understand the clinical relevance of CDK5 and
p21CIP1 expression in TC, data from TCGA were analyzed.
The analysis included correlation, survival, stage-specific
expression and differential expression between tumor and normal
tissues. A scatter plot showing the correlation between CDK5 and
p21 mRNA expression levels in TC samples is shown in Fig. 4. The correlation coefficient (R)
was 0.42, with a P-value <0.03, indicating a significant
correlation between CDK5 and p21 expression in TC tissues (Fig. 4A). In addition, patients with high
CDK5 expression had worse overall survival compared with those with
low CDK5 expression (Fig. 4B).
Conversely, patients with high p21 expression tended to have
improved overall survival (Fig.
4C), indicating a negative correlation between CDK5 and p21
expression. Violin plots were used to show the expression levels of
CDK5 and p21 across different stages of TC. CDK5 expression
increases with advancing cancer stage, showing a difference between
early-stage (Stage I/II) and late-stage cancer (Stage III/IV)
(Fig. 4D). However, p21 expression
decreased with advancing cancer stage, with a difference between
early and late stages (Fig. 4E).
Analysis of data from TCGA revealed a significant negative
correlation between CDK5 and p21 expression in TC, supporting the
experimental findings that CDK5 negatively regulated p21. Survival
analysis indicated that high CDK5 expression was associated with
poorer overall survival, while high p21 expression tended to be
linked with improved survival outcomes. Additionally, CDK5
expression increased and p21 expression decreased with advancing
cancer stage, suggesting their roles in TC progression.
Differential expression analysis confirmed that CDK5 was
up-regulated and p21 was down-regulated in TC tissues compared with
normal tissues. These findings underscore the clinical relevance of
targeting the CDK5-p21 axis in TC treatment.
CDK5 and p21 expression in TC
specimens are correlated with tumor aggressiveness
IHC staining was performed to assess CDK5 and p21
expression in TC specimens from 11 patients (Fig. 5A). The expression profiles were
analyzed in relation to tumor stage, sex and malignancy features
(TNM staging; Table I and Data S1). In all cases, CDK5 and p21 were
localized to the nuclei and cytoplasm of tumor cells. A distinct
pattern emerged where patients with more advanced TNM stages, lymph
node involvement, or recurrent tumors exhibited higher CDK5 and
lower p21 expression (Fig. 5 and
Table I). Notably, high CDK5
expression, particularly in patients 2, 3 and 9, was associated
with more advanced-stage cancer (II, III and IVA) and malignancy
features such as recurrence and invasion into surrounding
structures. By contrast, moderate CDK5 and p21 expression were
predominantly observed in early-stage cancers (Stage I), such as in
patients 1, 6, 7, 8 and 11, where tumors remained smaller and
confined to the thyroid. Notably, high p21 expression, as seen in
patient 5, was rare and may suggest the presence of a unique tumor
suppression pathway (Fig. 5A,
Table I and Data S1). A comparative analysis of
protein expression revealed that patients with more advanced cancer
had a predominance of high CDK5 and low p21 expression,
particularly in those with nodal involvement or invasive features,
while early-stage patients had higher expression levels of p21
relative to CDK5 (Fig. 5B and C).
These observations suggest a potential role of CDK5 in promoting
tumor progression and a tumor-suppressive function for p21, with
differential CDK5 and p21 expression profiles (Fig. 5), serving as valuable indicators
for assessing tumor progression and identifying high-risk
patients.
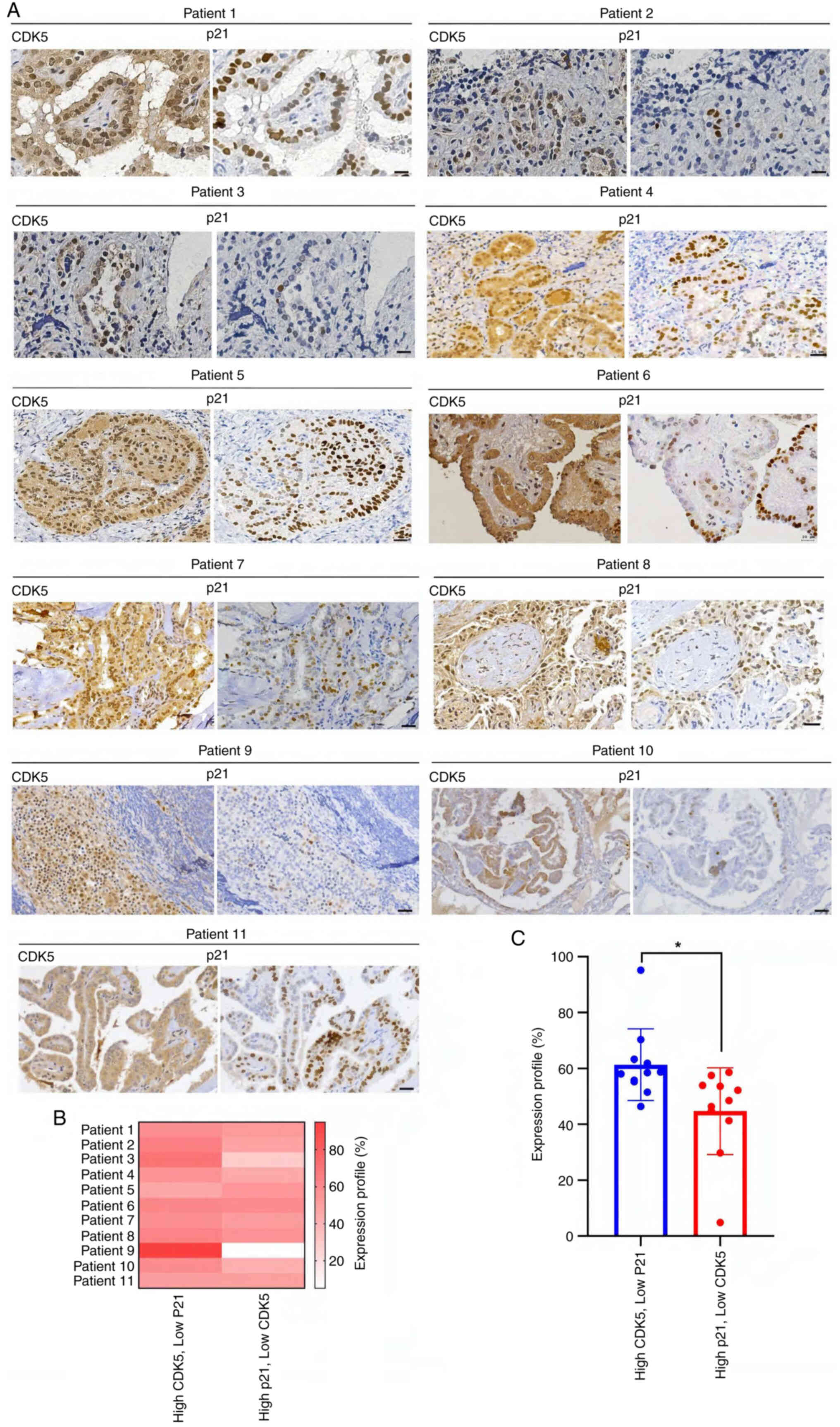 | Figure 5.IHC analysis of CDK5 and p21 nuclear
expression in papillary thyroid cancer patient specimens. (A) IHC
staining for CDK5 and p21 in patients with thyroid cancer
demonstrating the relationship between CDK5 and p21 nuclear
expression. Left panels, CDK5 staining showed strong nuclear
localization in several patient samples, particularly in tumor
cells. The nuclear expression of CDK5 was more prominent in
aggressive regions of the thyroid cancer samples, suggesting a role
in tumor proliferation. Right panels, p21 staining was inversely
correlated with CDK5. In samples where CDK5 was strongly expressed
in the nucleus, p21 nuclear staining was weak or absent.
Conversely, in areas were CDK5 expression was low, p21 nuclear
localization was more evident. This expression pattern supported
the hypothesis that CDK5 negatively regulated p21 expression at the
nuclear level, contributing to cancer cell cycle dysregulation and
malignancy. Scale bar, 50 µm. (B) The heatmap illustrates the
correlation between nuclear CDK5 and p21 expression across multiple
samples, with darker red indicating higher CDK5 expression and
lower p21 levels. (C) Significant differences in expression
patterns between groups with high CDK5/low p21 and low CDK5/high
p21 expression (*P<0.05). This suggested that elevated nuclear
CDK5 was associated with decreased p21 expression, supporting the
hypothesis that CDK5 promoted thyroid cancer progression. IHC,
immunohistochemistry; CDK5, cyclin-dependent kinase. |
Discussion
The results of the present study provided novel
insights into the role of CDK5 and p21 in the progression and
malignancy of human TC. The interaction between CDK5 and p21 was
shown to play a critical role in cell cycle regulation, impacting
the proliferation of cancer cells and consequently, the progression
of TC.
TC is the most prevalent type of thyroid malignancy,
representing ~80% of all TC cases (51,52).
Despite its generally favorable prognosis, characterized by slow
growth and a high survival rate, a significant subset of patients
presents with aggressive disease (53). These aggressive forms are marked by
recurrence, resistance to treatment and distant metastases, which
substantially worsen patient outcomes (54–56).
Understanding the molecular mechanisms driving TC progression is
vital for developing more effective therapies.
CDK5, a serine/threonine kinase, is known for its
role in neuronal development and function (2–4).
However, its aberrant activity has been increasingly linked to
various types of cancer, where it influences critical processes
such as cell migration, invasion and metastasis (57–59).
CDK5 exerts these effects by interacting with various cellular
proteins, including those involved in cell cycle regulation
(22,60,61).
p21, a well-established cyclin-dependent kinase inhibitor, serves
as a crucial tumor suppressor by inhibiting cell cycle progression
and thereby preventing uncontrolled cell proliferation (22). The degradation of p21 can lead to
the loss of cell cycle control, fostering an environment conducive
to tumor growth and malignancy (22,62).
In the context of TC, the interaction between CDK5 and p21 has not
been explored, to the best of the authors' knowledge. Considering
the role of CDK5 in other types of cancer and the findings of the
present study, which show its interaction with p21 across various
cancer types (22), it was
hypothesized that CDK5 may interact with p21 to regulate TC cell
proliferation, potentially contributing to the malignancy of
TC.
The results of the present study demonstrated that
CDK5 interacted with p21, specifically targeting the
phosphorylation site at S130 and facilitated its
ubiquitin-dependent degradation in TC cells. The regulation of p21
degradation is a highly orchestrated process involving specific
ubiquitin chain types and recognition domains, which are critical
for its proteasomal targeting (63). The K48-linked polyubiquitin chains
are the primary signals for p21 degradation, facilitated by domains
such as the PIP box and KRR motif that enable interactions with key
E3 ubiquitin ligases (63). In TC,
dysregulation of these mechanisms may enhance p21 degradation,
contributing to unchecked proliferation and tumor progression.
Upstream regulatory factors, particularly those influencing CDK5
activation, such as its co-activators p35 and p39 and oncogenic
signaling pathways such as PI3K/AKT and MAPK, probably modulate the
interaction between CDK5 and p21 (64). These pathways may influence CDK5
activity through post-translational modifications, including
phosphorylation of CDK5 or p21, altering their binding affinity and
promoting p21 destabilization (22,63,65).
Furthermore, additional molecular partners may facilitate p21
ubiquitination in TC; for example, PCNA acts as a scaffold for
CRL4Cdt2-mediated p21 ubiquitination during replication
stress, while E3 ligases such as MDM2 play similar roles in other
cellular contexts (66). The
involvement of deubiquitinates such as USP7 or USP10 further
complicates the regulation by counteracting ubiquitin-dependent
degradation, stabilizing p21 under certain conditions. These
molecular interactions create a finely tuned balance of p21 levels,
where aberrant regulation can favor tumor progression. Exploring
these pathways in TC provides critical insights into the mechanisms
underlying p21 degradation and CDK5 activity, offering potential
therapeutic opportunities to stabilize p21, restore cell cycle
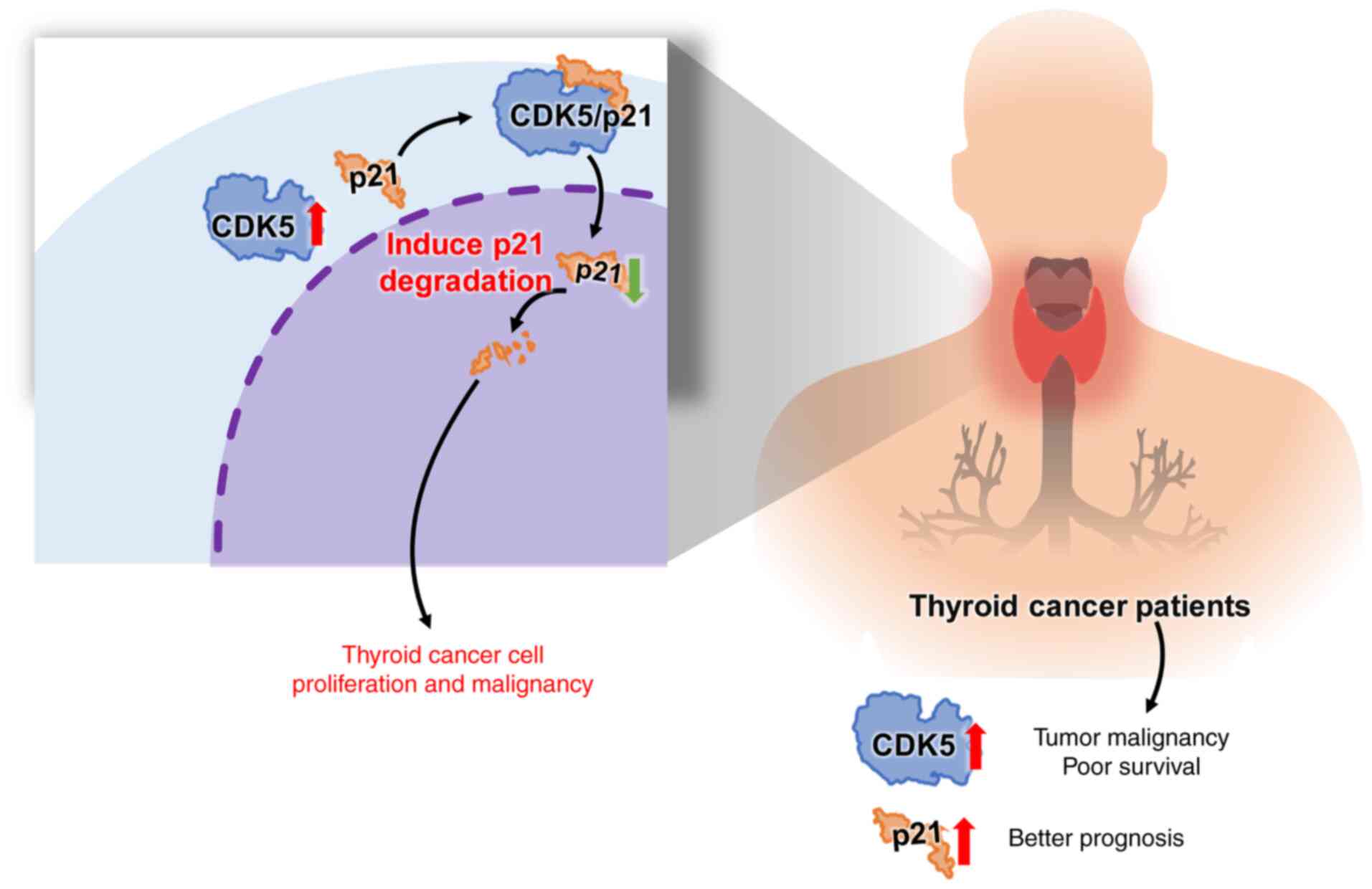
regulation and inhibit CDK5-driven oncogenic processes (Fig. 6). Therefore, advanced bioinformatic
tools, such as AlphaFold 3.0 (50,67)
were used to predict and analyze the protein-protein interaction
between CDK5 and p21. These predictions were experimentally
validated using IP assays, which confirmed the biochemical
interaction between CDK5 and p21 in TC cells. Additionally,
analysis of clinical data from TCGA revealed a significant negative
correlation between CDK5 and p21 expression levels in TC tissues.
High CDK5 expression was associated with poor overall survival,
while high p21 expression was correlated with improved survival
outcomes. This suggested that CDK5 contributes to TC progression by
potentially promoting the degradation of p21, thereby enabling
uncontrolled cell proliferation and disrupting cell cycle
regulation (Fig. 6).
In addition, the present study presented evidence
linking the expression of CDK5 and p21 to TC progression, with
specific correlations to tumor aggressiveness, TNM stage, sex and
age from clinical specimens. Based on the data presented in
Fig. 5, several hypotheses can be
discussed regarding these findings. The results of the
immunohistochemistry analysis suggested a clear association between
the expression of CDK5/p21 and tumor stage. Patients with higher
TNM stages, lymph node metastasis, or recurrence (such as Patients
2 and 3) demonstrated high CDK5 and low p21 expression. In these
patients, CDK5 may be contributing to the enhanced tumor
invasiveness and metastatic potential, while the reduced expression
of p21, an inhibitor of cell cycle progression, could be
facilitating unchecked cellular proliferation. This observation
supported the hypothesis that CDK5 functions as an oncogene in TC,
promoting malignancy, while p21 acts as a tumor suppressor.
Patients with higher CDK5 expression, particularly those with
advanced TNM staging, lymph node involvement, or recurrence,
exhibited more aggressive tumor characteristics. These findings
suggested that CDK5 promoted tumor progression, invasion and
metastasis, making it a potential therapeutic target for aggressive
TCs. Reduced p21 expression in more advanced cases of cancer (such
as Patients 2, 3, 9 and 10) may lead to uncontrolled cell growth,
contributing to tumor invasion and metastasis. These results
highlighted the importance of p21 in maintaining tumor suppressive
activity. This supported the hypothesis that an imbalance between
CDK5 and p21 may be linked to cancer recurrence and the development
of secondary malignancies. In summary, the findings suggested that
CDK5 and p21 played crucial roles in TC progression. High CDK5
expression correlated with aggressive tumor features, while high
p21 expression was associated with early-stage, less invasive
cancers. These observations provide a basis for further
investigation into the roles of CDK5 and p21 as potential
therapeutic targets or prognostic biomarkers in TC, with
implications for patient stratification and treatment.
In the present study, how CDK5 could serve as a
novel therapeutic target in TC by targeting p21 through degradation
and altering tumor progression was shown. The observed variability
in p21 expression levels across different stages of TC suggested
its potential as a biomarker along with CDK5 expression status as
another biomarker for disease progression and treatment response,
enabling improved stratification of patients based on their tumor
biology. Furthermore, the interplay between CDK5
expression/activation and p21 stability highlighted the need for
future studies to evaluate small molecule inhibitors that
specifically target CDK5 or its upstream regulators to inhibit CDK5
activation or prevent CDK5-p21 interaction (22,65,68,69).
This approach could be particularly beneficial in the subset of
patients with TC with aggressive disease, where current treatments
are less effective. Future research should focus on developing
small molecule inhibitors of CDK5 or exploring gene therapy
approaches to downregulate CDK5 expression in TC cells (70). These strategies could be tested in
preclinical models to evaluate their efficacy in halting TC
progression. Additionally, combining CDK5 inhibitors with existing
therapies could potentially enhance treatment outcomes (71–74),
offering a novel avenue for improving the prognosis of patients
with advanced TC. However, while the present study provided
valuable insights into the role of the CDK5-p21 axis in TC, there
are certain limitations to consider. First, the findings were based
on in vitro experiments, which, although useful for
understanding molecular mechanisms, might not fully reflect the
complexity of the tumor microenvironment or systemic factors
observed in clinical settings. Second, the study lacked animal
experiments, which are crucial for validating the in vitro
findings and understanding the physiological and pathological
relevance of the CDK5-p21 axis in vivo. Third, while the
analysis included 11 patient cases, the sample size was relatively
small, which may limit the broader applicability of the findings.
Finally, the detailed molecular mechanisms underlying CDK5-mediated
p21 ubiquitination, including the involvement of specific E3
ubiquitin ligases and regulatory pathways, remain to be fully
clarified and warrant further investigation, although the CDK5-p21
interaction was identified in the present study. Despite these
limitations, the results laid a strong foundation for future
studies to further validate these findings in more complex models
and larger patient cohorts.
In conclusion, this study distinguished itself by
examining the role of CDK5 in regulating p21 via phosphorylation at
the S130 site, a relatively unexplored mechanism in TC. This
specific phosphorylation event introduces an additional regulatory
layer that markedly affects the stability and function of p21,
which has not been adequately addressed in TC. The results of the
present study highlighted the critical role of the CDK5-p21
interaction in TC and highlighted novel diagnostic and therapeutic
targets that could be exploited to improve patient outcomes.
Further exploration of CDK5 inhibitors in clinical settings may
pave the way for more effective treatments of this common yet often
aggressive cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Science and
Technology Council, Taiwan (grant nos. 109-2320-B-005-004-MY3,
109-2911-I-005-003 and 112-2320-B-005-008 to H. Lin); joint grant
of Taichung Veterans General Hospital and National Chung Hsing
University (grant no. TCVGH-NCHU1117614 to H. Lin). Tungs' Taichung
MetroHarbor Hospital Grant (grant nos. TTMHH-R1120062 and
TTMHH-R1130044) and Tungs' Taichung MetroHarbor Hospital and
National Chung Hsing University Grant (grant no.
TTMHH-NCHULS112004) in Taiwan.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding authors.
Authors' contributions
MO, ST and HL contributed to the conception and
design of the study. MT, MO, SS and PC were responsible for data
acquisition. MO, SS, PC, YL, CC, HK, FL and MC analyzed and
interpreted the data. MT, MO, CC, HK, FL, ST, and HL drafted the
manuscript and revised it critically for important intellectual
content. ST and HL provided supervision and confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was conducted with the approval of
the Institutional Review Board (IRB) under protocol number 113003.
All experimental procedures followed ethical guidelines, and
informed consent was obtained from all TC patients involved in this
study. Thyroid tissue samples were collected from a cohort of TC
patients. The samples were preserved and processed for IHC analysis
to evaluate the expression of specific proteins. Clinical data on
patients' age, sex, and cancer staging were obtained from the
hospital's electronic medical records. All samples were anonymized
to ensure patient confidentiality in accordance with IRB
regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Oner M, Lin E, Chen MC, Hsu FN, Shazzad
Hossain Prince GM, Chiu KY, Teng CJ, Yang TY, Wang HY, Yue CH, et
al: Future aspects of CDK5 in prostate cancer: From pathogenesis to
therapeutic implications. Int J Mol Sci. 20:38812019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oner M, Chen MC, Cheng PT, Li YH, Cheng
YC, Celik A, Soong SW, Hsu LW, Lin DY, Hossain Prince GMS, et al:
Impact of metformin on neocortical development during pregnancy:
Involvement of ERK and p35/CDK5 pathways. Chemosphere.
358:1421242024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oner M, Chen MC, Cheng PT and Lin H:
Metformin inhibits nerve growth factor-induced sympathetic neuron
differentiation through p35/CDK5 inhibition. Am J Physiol Cell
Physiol. 326:C1648–C1658. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oner M, Cheng PT, Wang HY, Chen MC and Lin
H: Metformin alters dendrite development and synaptic plasticity in
rat cortical neurons. Biochem Biophys Res Commun. 710:1498742024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oner M, Lin E, Chiu KY, Chen MC, Prince
GMSH, Lai CH, Hsieh JT, Wang HY and Lin HO: p35/CDK5 regulates
bladder cancer proliferation and migration and promotes higher
tumor grade and poor survival rate in patients with bladder cancer.
Anticancer Res. 44:543–553. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue CH, Oner M, Chiu CY, Chen MC, Teng CL,
Wang HY, Hsieh JT, Lai CH and Lin H: RET Regulates human medullary
thyroid cancer cell proliferation through CDK5 and STAT3
activation. Biomolecules. 11:8602021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen MC, Chen KC, Chang GC, Lin H, Wu CC,
Kao WH, Teng CJ, Hsu SL and Yang TY: RAGE acts as an oncogenic role
and promotes the metastasis of human lung cancer. Cell Death Dis.
11:2652020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen MC, Huang CY, Hsu SL, Lin E, Ku CT,
Lin H and Chen CM: Retinoic acid induces apoptosis of prostate
cancer DU145 cells through cdk5 overactivation. Evid Based
Complement Alternat Med. 2012:5807362012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu FN, Chen MC, Lin KC, Peng YT, Li PC,
Lin E, Chiang MC, Hsieh JT and Lin H: Cyclin-dependent kinase 5
modulates STAT3 and androgen receptor activation through
phosphorylation of Ser727 on STAT3 in prostate cancer
cells. Am J Physiol Endocrinol Metab. 305:E975–E986. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuo HS, Hsu FN, Chiang MC, You SC, Chen
MC, Lo MJ and Lin H: The role of Cdk5 in retinoic acid-induced
apoptosis of cervical cancer cell line. Chin J Physiol. 52:23–30.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin E, Chen MC, Huang CY, Hsu SL, Huang
WJ, Lin MS, Wu JC and Lin H: All-trans retinoic acid induces DU145
cell cycle arrest through Cdk5 activation. Cell Physiol Biochem.
33:1620–1630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin H, Chen MC, Chiu CY, Song YM and Lin
SY: Cdk5 regulates STAT3 activation and cell proliferation in
medullary thyroid carcinoma cells. J Biol Chem. 282:2776–2784.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin H, Chen MC and Ku CT: Cyclin-dependent
kinase 5 regulates steroidogenic acute regulatory protein and
androgen production in mouse Leydig cells. Endocrinology.
150:396–403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Prince G, Yang TY, Lin H and Chen MC:
Mechanistic insight of cyclin-dependent kinase 5 in modulating lung
cancer growth. Chin J Physiol. 62:231–240. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Teng CJ, Cheng PT, Cheng YC, Tsai JR, Chen
MC and Lin H: Dinaciclib inhibits the growth of acute myeloid
leukemia cells through either cell cycle-related or ERK1/STAT3/MYC
pathways. Toxicol In Vitro. 96:1057682024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Foy R, Crozier L, Pareri AU, Valverde JM,
Park BH, Ly T and Saurin AT: Oncogenic signals prime cancer cells
for toxic cell overgrowth during a G1 cell cycle arrest. Mol Cell.
83:4047–4061.e6. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dimri GP, Nakanishi M, Desprez PY, Smith
JR and Campisi J: Inhibition of E2F activity by the
cyclin-dependent protein kinase inhibitor p21 in cells expressing
or lacking a functional retinoblastoma protein. Mol Cell Biol.
16:2987–2997. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakanishi M, Kaneko Y, Matsushime H and
Ikeda K: Direct interaction of p21 cyclin-dependent kinase
inhibitor with the retinoblastoma tumor suppressor protein. Biochem
Biophys Res Commun. 263:35–40. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hauge S, Macurek L and Syljuåsen RG: p21
limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell
Cycle. 18:834–847. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pisonero-Vaquero S, Soldati C, Cesana M,
Ballabio A and Medina DL: TFEB modulates p21/WAF1/CIP1 during the
DNA damage response. Cells. 9:11862020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang PH, Chen MC, Peng YT, Kao WH, Chang
CH, Wang YC, Lai CH, Hsieh JT, Wang JH, Lee YT, et al: Cdk5
directly targets nuclear p21CIP1 and promotes cancer cell growth.
Cancer Res. 76:6888–6900. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Zhang YJ, Zhao HY, Zhai QL, Zhang
Y and Shen YF: The impact of R213 mutation on p53-mediated p21
activity. Biochimie. 99:215–218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen DW, Lang BHH, McLeod DSA, Newbold K
and Haymart MR: Thyroid cancer. Lancet. 401:1531–1544. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sinha RA and Yen PM: Metabolic messengers:
Thyroid hormones. Nat Metab. 6:639–650. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Basolo F, Macerola E, Poma AM and
Torregrossa L: The 5th edition of WHO classification of tumors of
endocrine organs: changes in the diagnosis of follicular-derived
thyroid carcinoma. Endocrine. 80:470–476. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Modica R, Benevento E and Colao A:
Endocrine-disrupting chemicals (EDCs) and cancer: New perspectives
on an old relationship. J Endocrinol Invest. 46:667–677. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ijaz K and Yin F: Papillary thyroid
carcinoma with squamous dedifferentiation: A potential diagnostic
pitfall. Anticancer Res. 43:255–258. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ohashi R: Solid variant of papillary
thyroid carcinoma: An under-recognized entity. Endocr J.
67:241–248. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang SY, Ahn HR, Youn HJ and Jung SH:
Prognosis of papillary thyroid carcinoma in relation to
preoperative subclinical hypothyroidism. Ann R Coll Surg Engl.
103:367–373. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee JS, Lee JS, Yun HJ, Kim SM, Chang H,
Lee YS, Chang HS and Park CS: Aggressive subtypes of papillary
thyroid carcinoma smaller than 1 cm. J Clin Endocrinol Metab.
108:1370–1375. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mao J, Zhang Q, Zhang H, Zheng K, Wang R
and Wang G: Risk factors for lymph node metastasis in papillary
thyroid carcinoma: A systematic review and meta-analysis. Front
Endocrinol (Lausanne). 11:2652020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu J, Deng Y, Liu T, Zhou J, Jia X, Xiao
T, Zhou S, Li J, Guo Y, Wang Y, et al: Lymph node metastasis
prediction of papillary thyroid carcinoma based on transfer
learning radiomics. Nat Commun. 11:48072020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang D, Zhu XL and Jiang J: Papillary
thyroid carcinoma with breast and bone metastasis. Ear Nose Throat
J. 102:259–262. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Daniels GH: Follicular thyroid carcinoma:
A perspective. Thyroid. 28:1229–1242. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pelizzo MR, Mazza EI, Mian C and Merante
Boschin I: Medullary thyroid carcinoma. Expert Rev Anticancer Ther.
23:943–957. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang J and Barletta JA: Anaplastic thyroid
carcinoma. Semin Diagn Pathol. 37:248–256. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tjokorda Gde Dalem Pemayun, . Current
diagnosis and management of thyroid nodules. Acta Med Indones.
48:247–257. 2016.PubMed/NCBI
|
|
39
|
Roman BR, Randolph GW and Kamani D:
Conventional thyroidectomy in the treatment of primary thyroid
cancer. Endocrinol Metab Clin North Am. 48:125–141. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fullmer T, Cabanillas ME and Zafereo M:
Novel therapeutics in radioactive iodine-resistant thyroid cancer.
Front Endocrinol (Lausanne). 12:7207232021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brierley JD: Update on external beam
radiation therapy in thyroid cancer. J Clin Endocrinol Metab.
96:2289–2295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Salvatore D, Santoro M and Schlumberger M:
The importance of the RET gene in thyroid cancer and therapeutic
implications. Nat Rev Endocrinol. 17:296–306. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dong X, Akuetteh PDP, Song J, Ni C, Jin C,
Li H, Jiang W, Si Y, Zhang X, Zhang Q and Huang G: Major vault
protein (MVP) associated with BRAF V600E mutation is an
immune microenvironment-related biomarker promoting the progression
of papillary thyroid cancer via MAPK/ERK and PI3K/AKT pathways.
Front Cell Dev Biol. 9:6883702022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nikiforov YE and Nikiforova MN: Molecular
genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol.
7:569–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nozhat Z and Hedayati M: PI3K/AKT Pathway
and its mediators in thyroid carcinomas. Mol Diagn Ther. 20:13–26.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chou CK, Chi SY, Hung YY, Yang YC, Fu HC,
Wang JH, Chen CC and Kang HY: Clinical impact of androgen
receptor-suppressing miR-146b expression in papillary thyroid
cancer aggressiveness. J Clin Endocrinol Metab. 108:2852–2861.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng PT, Cheng YC, Oner M, Li YH, Chen
MC, Wu JH, Chang TC, Celik A, Liu FL, Wang HY, et al: Antrodia
salmonea extract inhibits cell proliferation through regulating
cell cycle arrest and apoptosis in prostate cancer cell lines. Chin
J Physiol. 65:209–214. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen CY, Li YH, Liao WL, Oner M, Cheng YC,
Liu FL, Cheng PT, Celik A, Wu JH, Lai CH, et al: Antrodia salmonea
extracts regulate p53-AR signaling and apoptosis in human prostate
cancer LNCaP cells. Evid Based Complement Alternat Med.
2022:70331272022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Abramson J, Adler J, Dunger J, Evans R,
Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick
J, et al: Accurate structure prediction of biomolecular
interactions with AlphaFold 3. Nature. 630:493–500. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lee YK, Rovira A, Carroll PV and Simo R:
Management of aggressive variants of papillary thyroid cancer. Curr
Opin Otolaryngol Head Neck Surg. 32:125–133. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Qu N, Chen D, Ma B, Zhang L, Wang Q, Wang
Y, Wang H, Ni Z, Wang W, Liao T, et al: Integrated proteogenomic
and metabolomic characterization of papillary thyroid cancer with
different recurrence risks. Nat Commun. 15:31752024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Coca-Pelaz A, Shah JP, Hernandez-Prera JC,
Ghossein RA, Rodrigo JP, Hartl DM, Olsen KD, Shaha AR, Zafereo M,
Suarez C, et al: Papillary thyroid cancer-aggressive variants and
impact on management: A narrative review. Adv Ther. 37:3112–3128.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Filetti S, Durante C, Hartl D, Leboulleux
S, Locati LD, Newbold K, Papotti MG and Berruti A; ESMO Guidelines
Committee. Electronic address, : simpleclinicalguidelines@esmo.org:
Thyroid cancer: ESMO clinical practice guidelines for diagnosis,
treatment and follow-up†. Ann Oncol. 30:1856–1883. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gunabushanam G: Perfluorobutane-enhanced
US helps differentiate benign lymph nodes from papillary thyroid
cancer metastases. Radiology. 307:e2305812023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schonfeld SJ, Morton LM, Berrington de
Gonzalez A, Curtis RE and Kitahara CM: Risk of second primary
papillary thyroid cancer among adult cancer survivors in the United
States, 2000–2015. Cancer Epidemiol. 64:1016642020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao GB, Sun Y, Fang RD, Wang Y, Wang Y and
He QY: Post-translational modifications of CDK5 and their
biological roles in cancer. Mol Biomed. 2:222021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jin X, Yang C, Fan P, Xiao J, Zhang W,
Zhan S, Liu T, Wang D and Wu H: CDK5/FBW7-dependent ubiquitination
and degradation of EZH2 inhibits pancreatic cancer cell migration
and invasion. J Biol Chem. 292:6269–6280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mandl MM, Zhang S, Ulrich M, Schmoeckel E,
Mayr D, Vollmar AM and Liebl J: Inhibition of Cdk5 induces cell
death of tumor-initiating cells. Br J Cancer. 116:912–922. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu C, Zhai X, Zhao B, Wang Y and Xu Z:
Cyclin I-like (CCNI2) is a cyclin-dependent kinase 5 (CDK5)
activator and is involved in cell cycle regulation. Sci Rep.
7:409792017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang J, Li H and Herrup K: Cdk5 nuclear
localization is p27-dependent in nerve cells: Implications for cell
cycle suppression and caspase-3 activation. J Biol Chem.
285:14052–14061. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bautista L, Knippler CM and Ringel MD:
p21-activated kinases in thyroid cancer. Endocrinology.
161:bqaa1052020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu Z and Hunter T: Ubiquitylation and
proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2)
CDK inhibitors. Cell Cycle. 9:2342–2352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Takasugi T, Minegishi S, Asada A, Saito T,
Kawahara H and Hisanaga S: Two degradation pathways of the p35 Cdk5
(cyclin-dependent kinase) activation subunit, dependent and
independent of ubiquitination. J Biol Chem. 291:4649–4657. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang S, Lu Z, Mao W, Ahmed AA, Yang H,
Zhou J, Jennings N, Rodriguez-Aguayo C, Lopez-Berestein G, Miranda
R, et al: CDK5 regulates paclitaxel sensitivity in ovarian cancer
cells by modulating AKT activation, p21Cip1- and p27Kip1-mediated
G1 cell cycle arrest and apoptosis. PLoS One. 10:e01318332015.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Havens CG and Walter JC: Mechanism of
CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev.
25:1568–1582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jumper J, Evans R, Pritzel A, Green T,
Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A,
Potapenko A, et al: Highly accurate protein structure prediction
with AlphaFold. Nature. 596:583–589. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Harper JW, Elledge SJ, Keyomarsi K,
Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley
L, Swindell E, et al: Inhibition of cyclin-dependent kinases by
p21. Mol Biol Cell. 6:387–400. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Malumbres M: Cyclin-dependent kinases.
Genome Biol. 15:1222014. View
Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hirai H, Kawanishi N and Iwasawa Y: Recent
advances in the development of selective small molecule inhibitors
for cyclin-dependent kinases. Curr Top Med Chem. 5:167–179. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ardelt MA, Fröhlich T, Martini E, Müller
M, Kanitz V, Atzberger C, Cantonati P, Meßner M, Posselt L, Lehr T,
et al: Inhibition of cyclin-dependent kinase 5: A strategy to
improve sorafenib response in hepatocellular carcinoma therapy.
Hepatology. 69:376–393. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lenjisa JL, Tadesse S, Khair NZ,
Kumarasiri M, Yu M, Albrecht H, Milne R and Wang S: CDK5 in
oncology: Recent advances and future prospects. Future Med Chem.
9:1939–1962. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pozo K and Bibb JA: The emerging role of
Cdk5 in cancer. Trends Cancer. 2:606–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang M, Zhang L, Hei R, Li X, Cai H, Wu
X, Zheng Q and Cai C: CDK inhibitors in cancer therapy, an overview
of recent development. Am J Cancer Res. 11:1913–1935.
2021.PubMed/NCBI
|