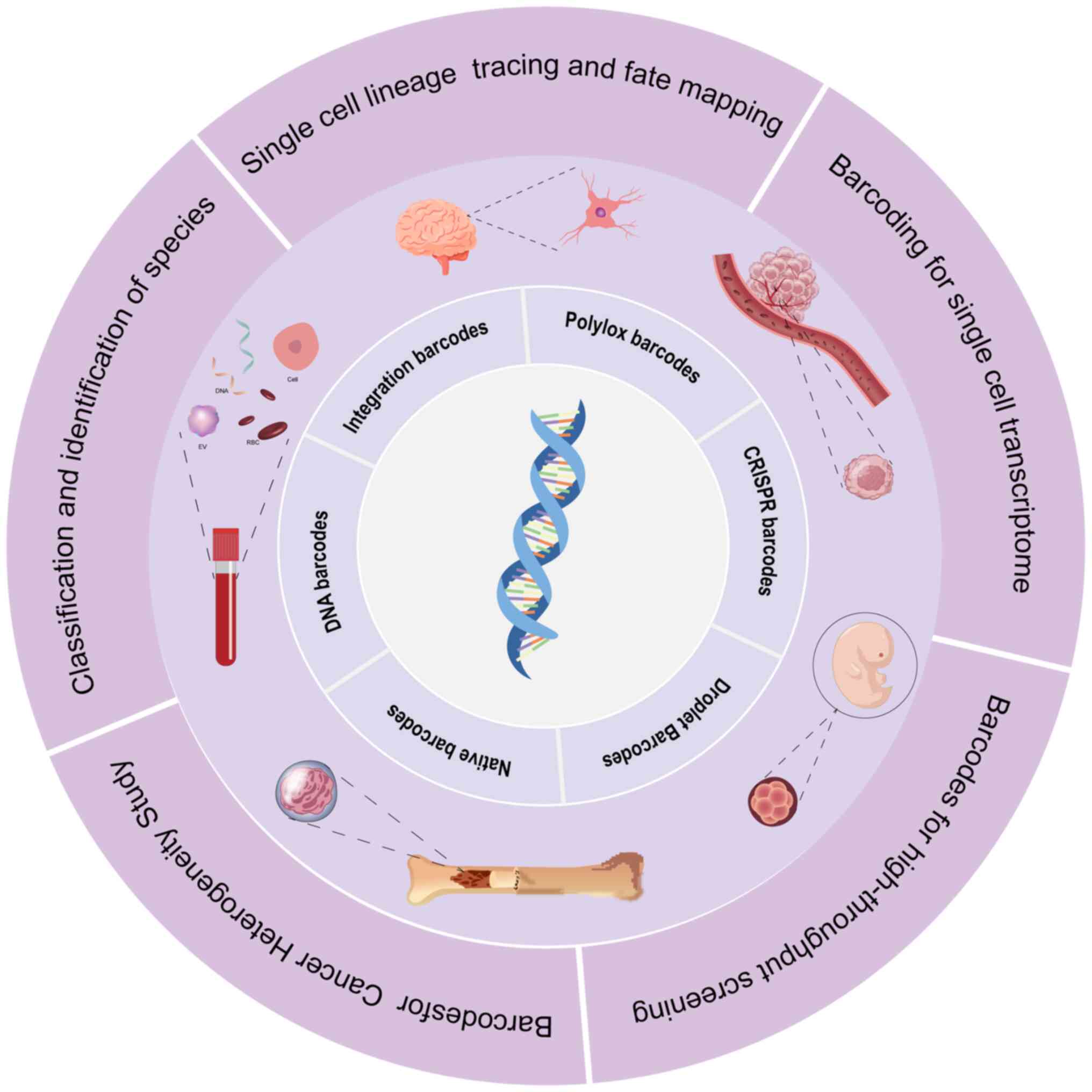
Introduction
Each cell follows a distinct developmental
trajectory and history. Mutations occur during cellular development
due to factors such as the cell cycle, fluctuations in the cellular
microenvironment, developmental processes, senescence and infection
(1). Multicellular organisms
integrate trillions of highly specialized cells derived from a
single zygote, continually developing specific genomic landscapes
during differentiation to form complex functional entities
(2). Extensive research has been
conducted on cells [including cell lineage tracing (3) clonal behavior and dynamics (4) and single-cell transcriptomics
(5). The introduction of nucleic
acid sequence barcoding technology has provided a powerful tool for
the aforementioned studies. Prior to this, various methods were
employed to label cells, such as time-lapse microscopy for direct
observation in Caenorhabditis elegans (6), dye injection, transplantation, viral
transduction (7), or genetic
recombination of fluorescent proteins to mark (8–13)
and track cells of interest. Despite the power of these methods,
they have limitations, including a limited number of generations
still marked after dye injection, non-physiological settings of
cell transplantation, low frequency of viral barcode insertion, or
a limited number of fluorescent proteins available to mark complex
tissues and these methods are labor-intensive. There is a need for
higher throughput, improved quantification and the ability to track
large numbers of single-cell systems simultaneously to further
advance the field.
Therefore, the concept of nucleic acid sequence
barcoding has been introduced to effectively address the
aforementioned issues in cell research (14,15).
Cell barcoding technology marks each cell in highly heterogeneous
cell populations with a unique barcode sequence. Nucleic acid
sequence barcodes are inherited from parent cells to progeny along
developmental trajectories, thus allowing the reconstruction of
their genetic relationships by deciphering the nucleotide sequence
information within the barcodes (16). This technology can track cells or
molecules across temporal and spatial boundaries. In theory, the
number of possible barcodes is limitless, allowing the parallel
tracking of millions of cells. These barcodes used for marking are
akin to a cell's ‘identification card’, enabling the rapid and
accurate identification of different cell types (17). In recent years, the application of
nucleic acid sequence barcoding has expanded across various fields.
For instance, deciphering clonal dynamics in disease (18), further improving the detection of
natural or synthetic barcodes and performing multimodal cell state
measurements at the single-cell resolution can improve
understanding of the origins of human diseases, explore cancer
heterogeneity (19) and
investigate cancer metastasis mechanisms (20). Using mice as a model, clustered
regularly interspaced short palindromic repeats (CRISPR) barcoding
has constructed lineage trees throughout the developmental process,
providing a viable multifunctional platform for in vivo
barcoding and lineage tracing in animal model systems (16). Additionally, in the field of cancer
liquid biopsies, cell barcoding has been applied to reveal the
immunological characteristics of cancer and the heterogeneity of
extracellular vesicles (21,22).
Furthermore, ‘RNA barcodes’ have been developed to elucidate the
combinatorial signaling pathways driving embryonic stem cell (ESC)
differentiation (23). However,
cell barcoding technology still faces challenges and risks. The
first is the issue of incorrect assignment due to barcode
similarity, especially in genetic barcoding, where highly similar
DNA tags may be erroneously allocated. Other inherent risks include
mutations in genetic markers during PCR and conventional library
sequencing steps, or partial loss of barcode reads in single-cell
data when using transcriptomic libraries (7). Moreover, the dynamic changes of
barcodes present limitations in tracking clonal evolution,
especially in scenarios requiring the tracking of clones over time
(18). Lastly, the data analysis
and computational complexity of cell barcoding are high and the
development and application of these methods remain a
challenge.
With the rapid development of microfluidic
technology and advancements in RNA sequencing, precise analysis at
the single-cell level has been achieved (24). By integrating cellular barcoding
techniques, high-throughput transcriptomic analyses can be
conducted on a vast number of cells within heterogeneous
populations, thereby elucidating population structures, the
interplay of gene expression and the heterogeneity during
differentiation processes (25).
Previously, DNA barcodes for spatial omics have been described and
the integration of these barcodes with fluorescence microscopy has
been explored, with a focus on cellular localization and tissue
structure studies (26). Although
the present review advanced the application of spatial omics, it
did not delve into the challenges and diverse applications of
nucleic acid barcodes in lineage tracing and disease modeling. By
contrast, the present review focused on the complex challenges and
broad applications of nucleic acid barcodes in these fields.
Additionally, previous studies have mainly concentrated on the
application of barcodes in lineage tracing, particularly in stem
cell biology and cancer research (17,27).
However, these studies did not comprehensively address the emerging
complexities of barcode technology, such as high-throughput
sequencing and the integration of multidimensional data. More
importantly, while the literature reviews the historical
development of lineage tracing methods and their applications in
developmental biology and cancer research (3), it does not cover the latest advances
and breakthroughs in barcode technology. The present review
examined nucleic acid sequence-based barcoding strategies, which
are primarily categorized into two main types: Natural barcodes and
synthetic barcodes. It further discussed the advantages, challenges
and potential solutions of nucleic acid sequence barcoding in
practical applications; despite significant progress in nucleic
acid sequence barcoding technology, there is still room for
improvement in sensitivity, specificity, cost-effectiveness and
multiplexing capabilities. Lastly, the present review projected the
future development of nucleic acid sequence barcoding technology.
With the progress in synthetic biology and gene editing
technologies, this field is expected to witness further innovations
and breakthroughs. In the future, we may see the emergence of more
efficient and cost-effective barcoding systems, which will markedly
advance biomedical research and clinical applications. It is
expected that nucleic acid sequence barcoding technology will make
a more substantial contribution to understanding complex biological
processes and improving human health (Fig. 1).
Principles and types
Since the inception of nucleic acid sequence
barcodes, a variety of distinct barcode types have been developed,
including Polylox barcodes, CRISPR barcodes, integration barcodes,
droplet barcodes and native barcodes (3). Nucleic acid sequence barcodes
distinctly label each cell within a highly heterogeneous cell
population through unique barcode sequences. As the developmental
trajectory unfolds, barcode sequence information is inherited from
progenitor cells to their progeny, thereby elucidating lineage
history (28). The principles,
advantages, limitations, applications and challenges associated
with various barcode strategies is comprehensively discussed later
and summarized in Table I.
 | Table I.Barcoding technology. |
Table I.
Barcoding technology.
| Barcode type | Principle | Advantages | Disadvantages | Applications | Challenges |
|---|
| CRISPR-Cas9 | Uses the
CRISPR-Cas9 system to insert barcode | 1. High specificity
and accuracy | 1. Barcode
insertion may interfere with Gene expression | 1. Used for cell
tracking | 1. Optimizing
editing efficiency and accuracy |
| barcodes | sequences at
specific genomic locations guided by gRNA. Each | 2. Enables
quantification and tracking of individual cells or EVs | 2. Requires precise
gene editing techniques | 2. Post-genome
editing tracking | 2. Challenges in
cross-species applications |
|
| barcode corresponds
to a different cell or EV | 3. Strong
scalability | 3. Low editing
efficiency possible | 3. Single-cell
analysis | 3. Biosafety
concerns |
| Polylox
barcodes | Based on the
Polylox system, which generates | 1. Multiple barcode
system | 1. System
complexity may lead to instability in barcode insertion | 1. Used for cell
population tracking | 1. Barcode
variability |
|
| multiple unique
barcode genes randomly inserted into the | 2. High-throughput
tagging and tracking in cell populations | 2. Barcode
mutations may reduce tracking accuracy | 2. Cancer
research | 2. Long-term
stability of the system |
|
| genome for
high-throughput analysis in cell populations | 3. Suitable for
long-term cell fate tracking |
| 3. Long-term cell
fate analysis, etc. | 3. Challenges in
multi-cell applications |
| Integration
barcodes | Uses insertion
barcode systems where | 1. Long-term
stability | 1. Barcode
insertion may be biased by genomic location | 1. Used for
long-term cell line tracking | 1. Selection bias
in insertion sites |
|
| viral or other
vectors directly insert barcode sequences into the target cell
genome, | 2. No external
intervention required; barcodes are stably passed on during cell
division | 2. Potential
interference with target cell function | 2. Stem cell
research | 2. Potential
conflict with cell function |
|
| which are inherited
during cell division | 3. Precise
single-cell tracking |
| 3. Genome editing
tracking | 3. Requires
efficient viral or vector systems |
| Droplet
barcodes | Microfluidic
droplets are used to partition and | 1. High throughput
and sensitivity | 1. Droplet
stability can affect analysis | 1. Used for
single-cell analysis | 1. Stability and
consistency of microfluidic technology |
|
| tag individual
cells or EVs, enabling high- | 2. Precise
single-cell or single-EV analysis | 2. Requires complex
microfluidic devices | 2. High-throughput
single-molecule sequencing | 2. Equipment and
data analysis costs |
|
| throughput parallel
analysis | 3. Can perform
multiparametric analysis for large-scale data generation | 3. High complexity
in data analysis | 3. EV analysis | 3. Droplet marking
errors |
| Native
barcodes | Relies on natural
barcodes, such as RNA or DNA | 1. No external
insertion required, naturally occurring system | 1. Dependent on the
presence and stability of natural markers | 1. Used for cell
tracking, RNA studies | 1. Limitations of
natural markers |
|
| barcodes, which are
directly recognized by | 2. Suitable for
molecular-level tagging | 2. Potential
competition with target molecules or systems for binding | 2. Species
separation based on natural markers | 2. Stability of
natural markers in the system |
|
| biological markers
without external insertion | 3. Relatively
stable markers for long-term monitoring |
|
| 3. Sensitivity
issues in localization and recognition |
Principles of cellular barcoding
DNA cellular barcoding is a high-throughput method
widely used to track cell lineages across various fields (18,29),
including hematopoiesis (7,30,31),
development, cancer (20,32–34)
and infectious disease dynamics (35). It employs a unique, inheritable DNA
sequence integrated into the genome of an ancestral cell, with
descendants detected through sequencing. In theory, the number of
cellular barcodes is limitless, with the number of possible
barcodes being 4n, where n is the length of the sequence
(since each position can encode for one of the four bases). Thus, a
random 10-base pair (bp) barcode can assume any of 410
(~106) different sequences, while a random 30-bp barcode
can assume any of 430 (~1018) different
sequences, each serving as a unique identifier. Regarding the
design principles, barcodes are typically categorized into random
and semi-random types. Random barcodes, such as those used by
Macosko et al in 2015 (36)
who combined cellular barcoding with microfluidic technology for
high-throughput cell labeling, consist of completely randomized
nucleotide sequences. Semi-random barcodes, on the other hand,
leverage recombinase-based or gene-editing technologies such as
CRISPR/Cas9 and are integrated into cellular DNA via viral vectors
or other methods for cell marking (37,38).
As cellular barcoding technology continues to evolve, researchers
have designed various types of DNA cellular barcodes to suit
different research needs and experimental designs. The design and
application of these barcodes provide a powerful tool for gaining
deep insights into cellular behaviors and biological processes
(17).
Types of barcode design and
challenges
Polylox barcodes
Design
The Polylox system facilitates high-resolution
lineage tracing and multiplexing, enabling concurrent tracking of
multiple cell populations. This system offers valuable insights
into tissue regeneration, developmental biology and cancer
progression. Understanding the cellular origin of various cell
types or tumors can uncover critical biological mechanisms
(39). In the seminal work by Cai
et al a recombinase-based nucleic acid sequence barcode
technology was first introduced. This technology employs Cre
recombinase to excise or invert specific DNA sequences, which
interact with a series of open reading frames of fluorescent
proteins, followed by analysis through fluorescence imaging
(40). However, the repetitive
action of Cre may result in random recombination and collapse of
the target array. In 2017, Pei et al (41,42)
used the Polylox barcode system to elucidate the in vivo
fate of hematopoietic stem cells and it was further applied it in
2019 for lineage tracing in mice (41,42).
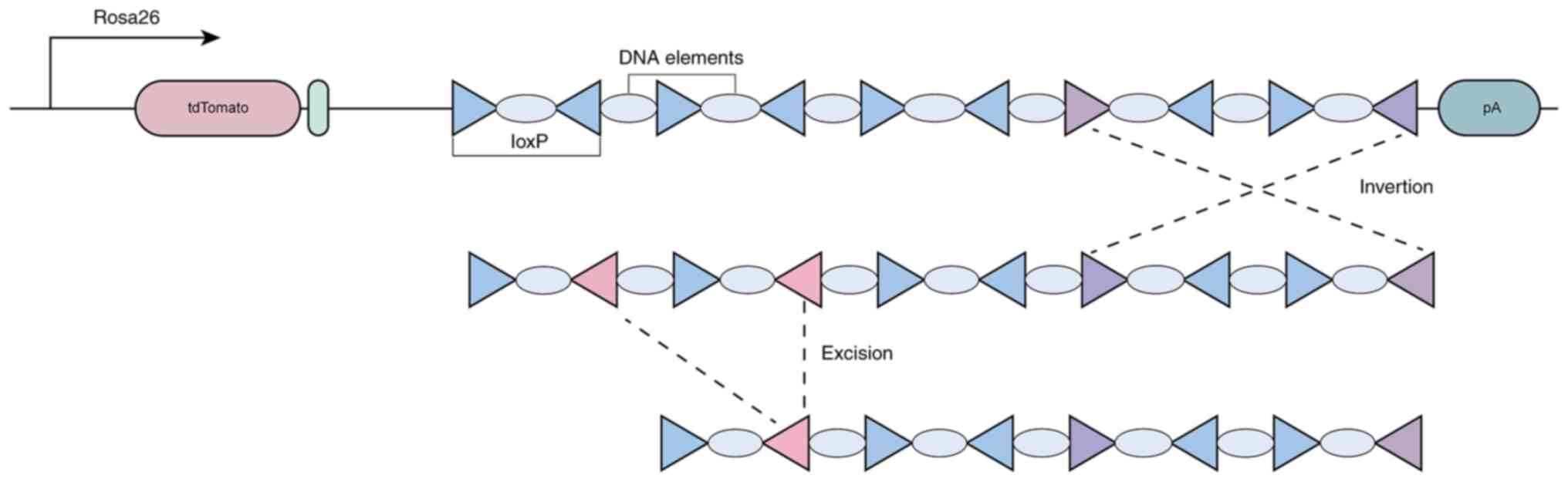
The Polylox system is an advanced in vivo cellular lineage
tracing technology that introduces artificially synthesized
recombination substrates into specific loci in mice (such as the
Rosa26 locus) and employs the transient activity of Cre recombinase
to induce random DNA recombination, generating unique DNA barcodes
(Fig. 2). The Polylox system
comprises 10 loxP sites, spaced 11 base pairs apart, with the nine
intermediate DNA fragments carrying unique sequences from the
Arabidopsis AT2G21770 gene, thereby forming the ‘alphabet’
of the barcode (3,43,44).
This innovation confers the Polylox barcode with enhanced
functionality, offering a more robust and scalable fate mapping
method compared to traditional techniques, with high precision, as
well as superior multiplexing capabilities and long-term tracking
potential. The versatile application of the system renders it
suitable for a broad range of experimental models. It can be
employed in studies on stem cell behavior, tumor progression, or
immune cell tracking, providing a versatile approach for diverse
biological investigations, all without the necessity of exogenous
markers. The Polylox system employs an endogenous genetic barcode
to track cells, offering a more natural and generalizable method of
tracking (41). However, the
complexity of system setup, such as the insertion of multiple
recombinase target sites (loxP or other variants), may present
technical challenges and restrict its use in certain laboratories
(41). Additionally, the system
may result in mosaic expression in certain cells and not all cells
may undergo recombination events, potentially introducing bias when
analyzing cellular fate. Another consideration is tissue-specific
recombination efficiency, as different tissues and developmental
stages exhibit variable recombination efficiencies. This could lead
to discrepancies between barcode outcomes and expected results.
During this process, barcode drift or loss may occur, resulting in
insufficient representation of certain barcodes over time,
particularly in rapidly dividing or regenerating tissues (45). It is also important to note that,
during the tracking of large cell populations, barcode overlap may
occur, complicating the differentiation of closely related cells or
their progeny (44).
Challenge
The Polylox barcode system generates barcodes by
chemically inducing alterations to specific sequences. However, the
insertion of these barcodes is inherently stochastic, resulting in
uneven distribution and expression across cell populations
(46,47). This issue can be addressed by
optimizing the chemical induction conditions and reaction system to
enhance the uniformity and stability of barcode insertion. Barcodes
are frequently employed in cellular contexts; however, their
introduction may alter cellular states, thereby confounding the
interpretation of biological outcomes. Moreover, the recovery rates
of both cells and barcodes pose a potential risk. Given the
stochastic nature of barcode insertion, some cells may fail to
successfully generate a barcode, resulting in low recovery rates.
Employing multiple rounds of selection can ensure that a sufficient
number of cells receive functional barcodes, while the development
of optimized capture techniques can further improve recovery rates.
It is well-established that the diversity of barcodes facilitates
the generation of millions, or even tens of millions, of distinct
barcodes. However, the intrinsic randomness of the Polylox barcode
generation process further complicates data analysis. This
challenge is particularly pronounced in large-scale cell
populations, where the accurate separation and identification of
barcode sequences constitutes a significant obstacle. To address
this challenge, statistical model algorithms, such as Bayesian
analysis, have been developed to handle the diverse barcode data,
thereby enhancing the accuracy of data analysis (47–49).
CRISPR Cas9-based barcodes
Design
The CRISPR-Cas9-based barcoding technology exploits
the potential of the CRISPR-Cas9 system to introduce specific DNA
modifications (that is, barcodes) into the genomes of individual
cells (50). These barcodes,
comprising unique synthetic DNA sequences, function as molecular
tags, facilitating the tracing of cell lineages and associating
genetic information with various cellular attributes, including
gene expression and therapeutic responses. A variety of CRISPR-Cas9
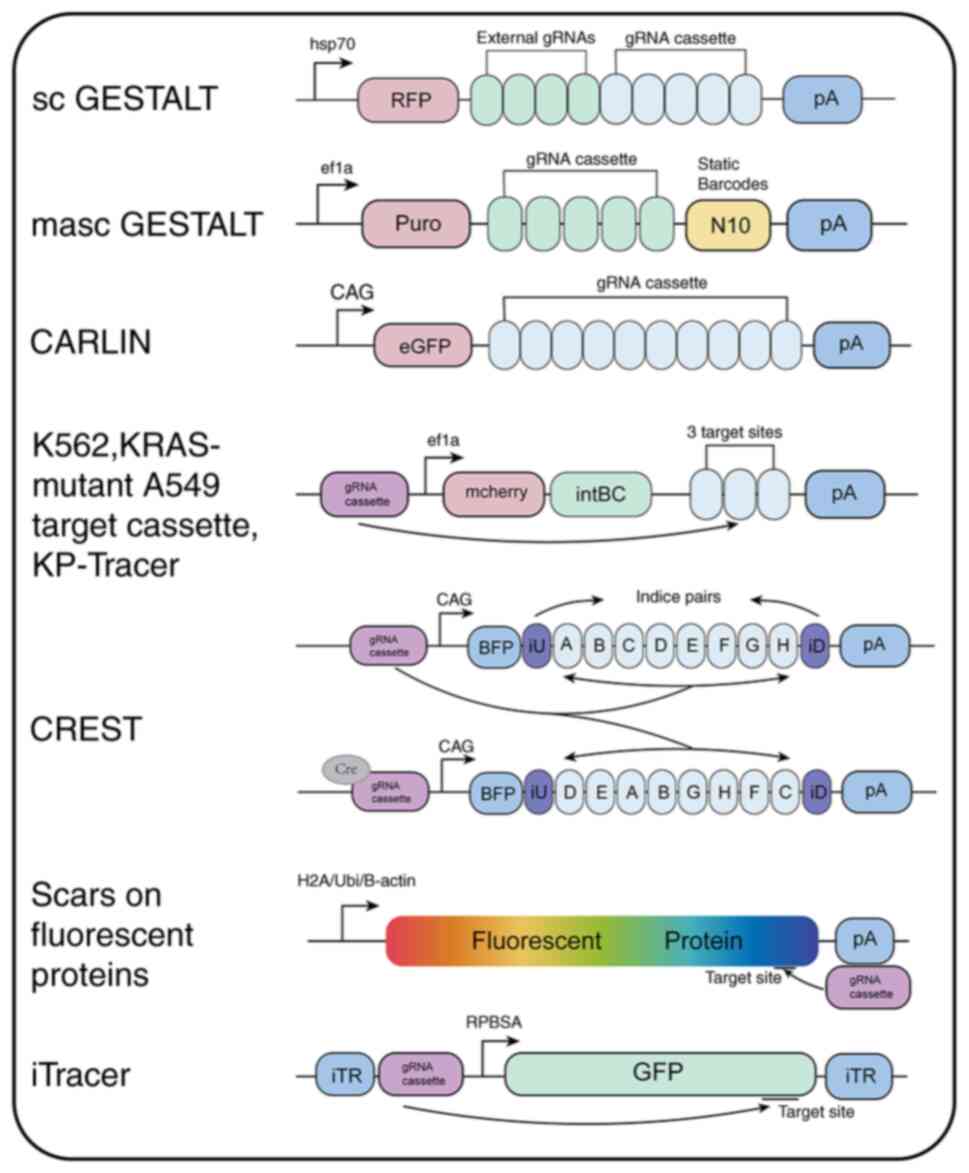
barcode systems have been developed, including GESTALT, scGESTALT,
macsGESTALT, synthetic barcodes delivered via piggyBac transposase,
CREST and ScarTrace (51–56) (Fig.
3). In zebrafish models, a CRISPR/Cas9-based synthetic barcode
system was evaluated, capable of accumulating informational
mutations during cellular division events and throughout the
organism's developmental process (47). This system is applicable to a wide
range of organisms. However, due to saturation issues, the GESTALT
system is restricted to early embryonic stages and exhibits reduced
precision in reconstructing complex lineage trees. To overcome this
limitation, single-cell RNA sequencing (scRNA-seq) technology is
incorporated, as demonstrated by scGESTALT and its applications in
zebrafish and macsGESTALT mouse models (51,53,54).
This technology facilitates barcode editing at multiple time
points, capturing lineage information from later developmental
stages. The scGESTALT method is applicable to various multicellular
organisms, enabling the characterization of molecular identities
and lineage histories of thousands of cells throughout development
and disease progression (53). As
an inducible CRISPR-Cas9-based lineage recorder, macsGESTALT
efficiently captures single-cell transcription and phylogenetic
data (54). When applied to an
in vivo model of pancreatic cancer metastasis, macsGESTALT
revealed that metastatic activity peaked at a specific late-stage
hybrid epithelial-mesenchymal transition (EMT) state (51,53,54).
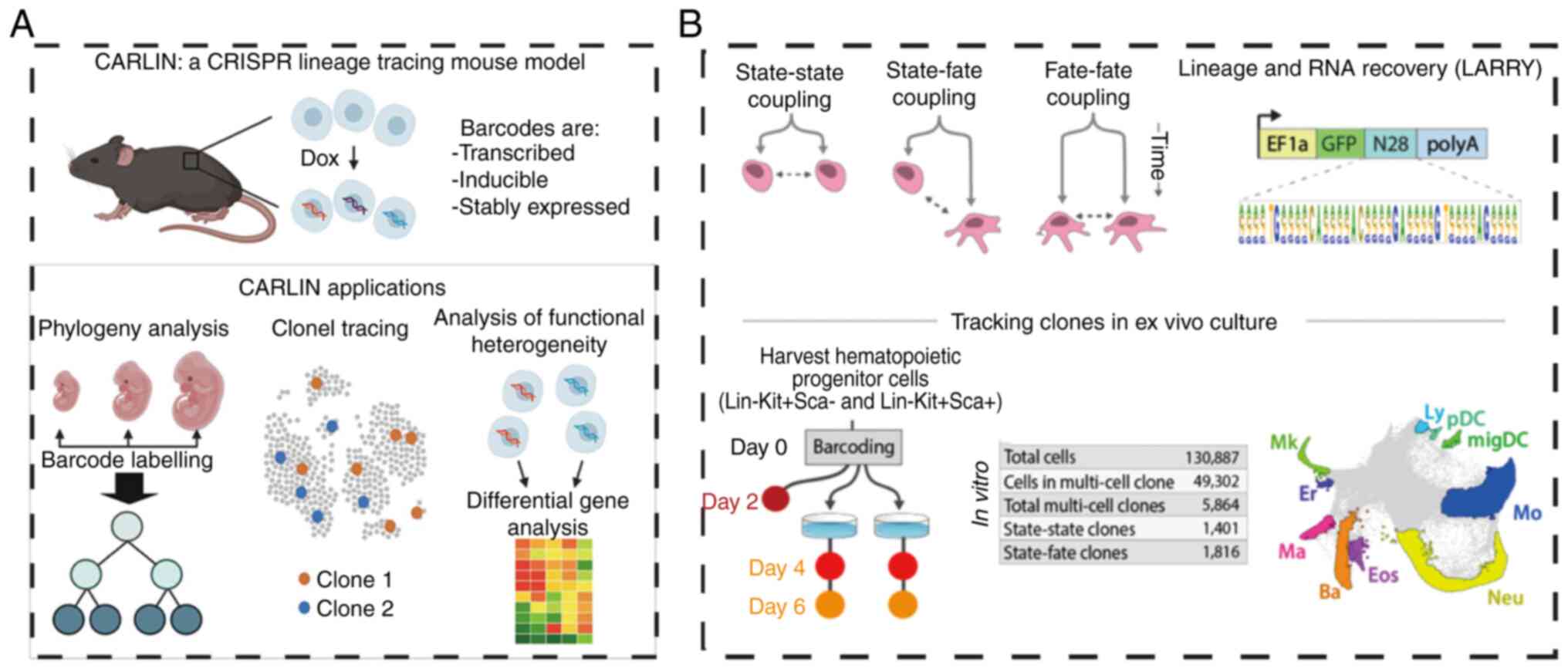
Additionally, CARLIN technology has been employed to identify
inherent biases in the clonal activity of fetal liver hematopoietic
stem cells (HSCs), enabling an unbiased, global analysis of both
lineage history and gene expression in single mouse cells (55). Another variant of the
CRISPR/Cas9-based synthetic barcode approach uses the piggyBac
transposon, which efficiently integrates synthetic barcodes and
active specific guide RNAs (sgRNAs) into target cells. In an
initial model, engineered K562 cells containing a piggyBac library
with sgRNAs and their respective target site boxes (including an
inducible Cas9 system) were introduced into fertilized eggs to
reconstruct lineage development during mouse embryogenesis
(56). This model was subsequently
optimized to enable the direct implantation of Kras-mutant A549
cells into mouse lung tissue or the generation of a genetically
engineered mouse model of induced lung adenocarcinoma (KP-Tracer),
facilitating the tracking of tumor evolution via barcode
information (57,58). Moreover, a CRISPR-based
lineage-specific tracing method, CREST, was developed for clonal
tracing in Cre mice. Using two complementary strategies based on
CREST, the single-cell lineage of the developing mouse ventral
midbrain was mapped. The CREST method and its strategies allow for
comprehensive single-cell lineage analysis, offering new insights
into the molecular programming of neurodevelopment (59). Fluorescent proteins, in addition to
serving as standalone color barcodes, were integrated with
CRISPR-Cas9-mediated nucleic acid sequence barcodes to create
fluorescent protein genetic markers. Most CRISPR/Cas9-based lineage
tracing methods employ an inducible editing system, enabling
temporal and/or spatial regulation within specific developmental
timeframes to track diverse cell lineages. These methods have also
been widely used to study lineage relationships across various cell
types (38,60). However, CRISPR/Cas9 editing may
induce off-target effects, necessitating highly specific sgRNA
sequences to precisely target synthetic barcodes and avoid
unintended mutations (61,62). Furthermore, non-homologous end
joining repair mechanisms inevitably generate insertion-deletion
mutations of varying lengths, presenting challenges for clonal
analysis. Lastly, multiple double-strand breaks in synthetic
barcodes can lead to complete loss of barcode information, thus
reducing the accuracy of lineage tracing analyses.
Challenge
The CRISPR-Cas9 barcode technology typically inserts
barcode sequences into the target genome through precise gene
editing. However, despite the high targeting fidelity of the CRISPR
system, off-target effects remain a concern, potentially leading to
barcode insertion into non-target genomes and thereby increasing
the data complexity (63). To
minimize off-target effects, researchers have developed improved
Cas9 variants, such as high-fidelity Cas9, to enhance specificity
and have designed sgRNAs to further reduce off-target activity
(64). Several barcode
technologies utilizing CRISPR-Cas9 gene editing have been
developed, addressing various challenges. However, issues such as
insufficient recovery rates of both cells and barcodes persist. In
certain cells, CRISPR-mediated barcode insertion may be incomplete
or may fail to occur successfully due to variations in DNA repair
mechanisms, thereby affecting subsequent lineage tracing and
sequencing data (65). The use of
more robust viral vectors or optimized CRISPR-Cas9 systems, in
conjunction with multiple rounds of selection, can help ensure a
higher barcode insertion efficiency. Furthermore, the insertion of
barcodes into cells may interfere with the original gene
expression, potentially leading to erroneous data in clonal and
genetic analyses (65,66). Finally, the integration of barcode
technologies with sequencing methods enables the reading of barcode
sequences; however, due to off-target effects, precise localization
of barcode insertion sites and robust computational support are
essential for identifying and quantifying barcode data in complex
cell populations (67).
Researchers have exploited high-throughput sequencing and
specialized bioinformatics tools, such as CRISPResso, to align and
correct potential off-target insertion points, optimizing barcode
recovery and analysis (68–71).
Integration barcodes
Design
Integration barcodes, also referred to as genetic
barcodes, are short sequences positioned at the ends of transposons
or viral integration sites, typically at the 3′ end of a reporter
gene (such as GFP) and the 5′ end of the adjacent 30-LTR. These
barcodes facilitate the unique labeling of individual cells by
integrating into the host genome, thereby enabling their
differentiation and tracking in subsequent experimental analyses
(72). Due to the high
transduction efficiency of retroviral and lentiviral vectors, along
with the availability of standardized experimental protocols,
transposons or lentiviruses are commonly employed to integrate
barcodes into the host genome. During barcode generation, the
original marked cells pass genetic markers to progeny, leading to
the formation of unique barcode sequences that are randomly
generated (19). Although unique
labeling of cells has been accomplished, current methodologies
encounter challenges in efficiently reading barcode sequences
during single-cell RNA sequencing (scRNA-seq) (3). This limitation poses challenges for
the precise tracking and analysis of cells. In response to this
challenge, integration barcode technology has undergone several
improvements. Currently, barcodes are classified into two primary
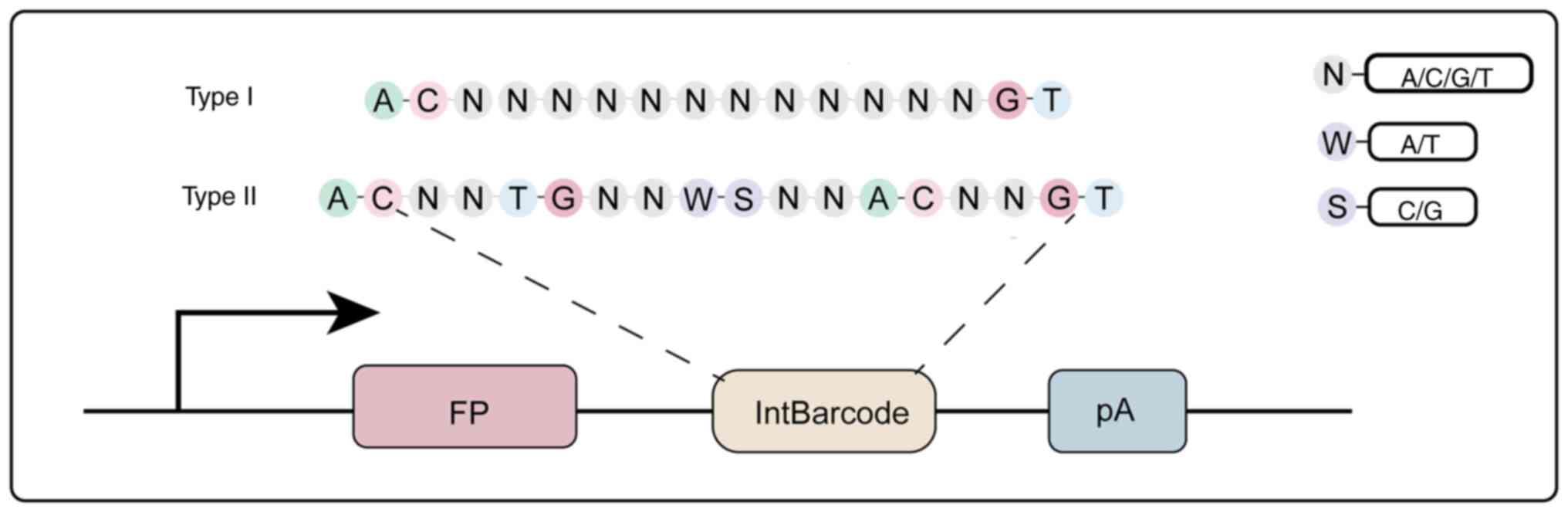
types based on their design: Random barcodes and semi-random
barcodes. Random barcodes typically consist of four nucleotides (A,
G, C, T) randomly arranged and denoted as (n) (72,73),
as illustrated in Fig. 4. While
these barcodes exhibit considerable diversity, theoretically
accommodating a vast number of unique sequences, they also present
several drawbacks. First, the amplification of these barcodes
necessitates the PCR process, which may induce sequence biases or
small insertions and deletions. Second, the complete randomness of
the sequences, devoid of structural information, complicates
subsequent decoding efforts. In contrast, semi-random barcodes
feature a more structured design, often incorporating a combination
of strong and weak base sequences. For example, a previous design
used strong bases (such as S-C, G) and weak bases (such as w-A, T)
(72,74). This approach presents significant
advantages over fully random barcodes. Notably, semi-random
barcodes exhibit a discernible sequence pattern, facilitating
easier identification and retrieval during sequencing. Moreover,
these barcodes can be directly integrated into vectors without the
need for PCR amplification, thus circumventing the biases and
insertions associated with PCR (7). The inherent structural regularity of
semi-random barcodes not only maintains their efficiency but also
enhances their stability and reliability. The primary advantage of
integration barcode technology lies in its capacity to efficiently
label and track cells, enabling researchers to precisely identify
the genetic characteristics of individual cells within complex
cellular populations (75). With
ongoing advances in barcode design (from random to semi-random
barcodes and even the combination of distinct barcode strategies)
this technology holds considerable promise across various
applications in genomics, drug screening and gene editing. However,
challenges persist, including amplification errors and issues
related to sequence read accuracy, which necessitate further
optimization in future research endeavors.
Challenge
Integration barcodes are typically introduced into
the genomes of target cells through viral vectors or other delivery
systems. However, due to the inherent randomness of insertion, the
distribution of barcodes within cell populations is often uneven,
which may affect the reliability of experimental results (76). The integration of barcodes is
influenced by DNA repair mechanisms, potentially leading to the
insertion of barcodes at random genomic loci. In single-cell and
lineage tracking applications, this randomness may introduce biases
or inaccuracies, as barcodes may integrate into non-representative
genomic regions. For instance, insertions into non-coding regions
or functionally irrelevant sites could affect gene expression or
cellular pathways, thereby confounding the analysis (77,78).
Furthermore, the insertion of barcodes may present a potential
conflict with cellular function. Insertion at critical genomic loci
could disrupt essential genes or regulatory elements, potentially
interfering with cell survival, division, or differentiation,
leading to unintended cellular changes (79,80).
In cancer research, such insertions could alter tumor biology,
obscure tumor heterogeneity and hinder accurate understanding of
distinct tumor subtypes. Finally, the integration of barcodes
relies on viral vectors to deliver the barcode sequences. However,
the efficiency of these vectors can be unstable, leading to low
integration rates or ineffective barcode delivery, thereby
compromising experimental outcomes. To address this challenge, it
is crucial to develop efficient and stable viral vector systems
that ensure the effective and reproducible integration of barcodes
into target cells (81,82). Additionally, the cost and
complexity associated with the development and optimization of such
systems may pose significant challenges, particularly in
high-throughput experiments.
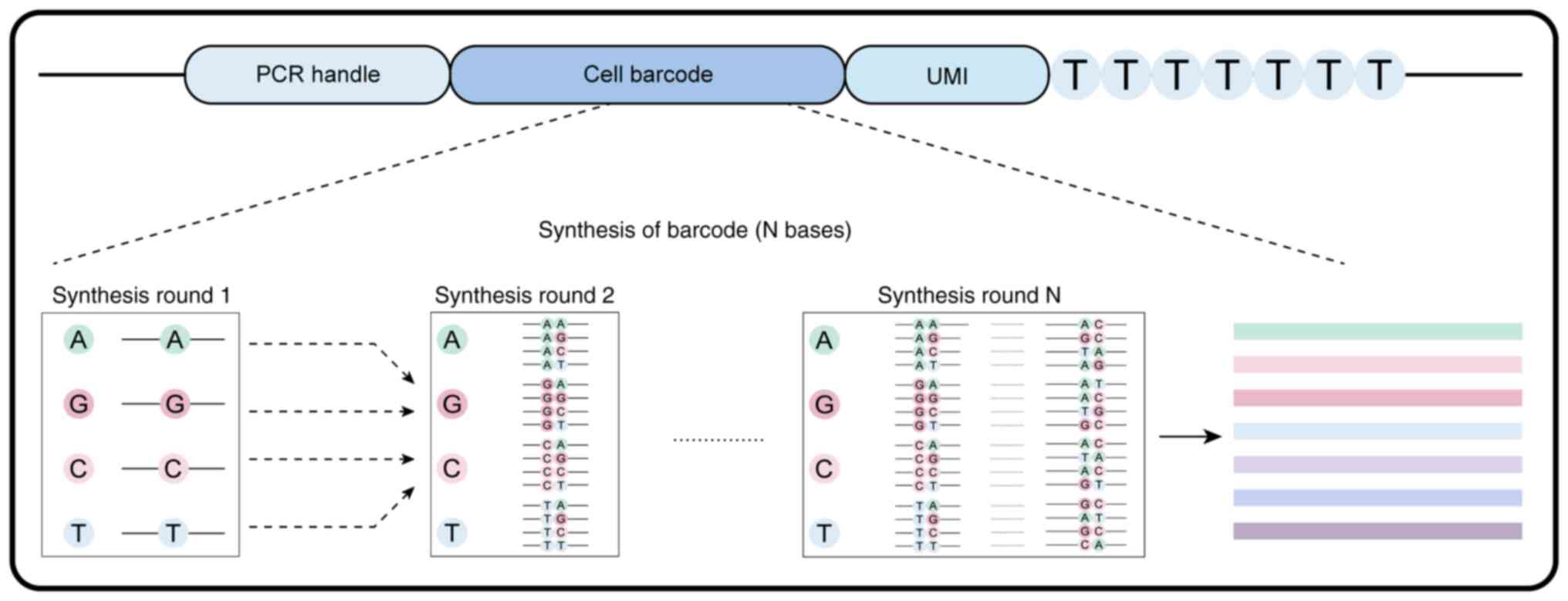
Droplet barcoding
Design
Nucleic acid sequence barcodes are comprised of
random, semi-random and non-random components. Random barcodes are
generated by permuting various nucleotide elements and adjusting
base positions and sequence lengths to achieve both diversity and
uniqueness. The design of these random barcodes includes: i) PCR
handles for reverse transcription and amplification; ii) cellular
barcodes; iii) unique molecular identifiers (UMIs) for
distinguishing PCR duplicates; and iv) a 30 bp oligo-dT sequence
for capturing polyadenylated mRNA and initiating reverse
transcription (36,83)(Fig.
5). Several barcode platforms, including Drop-Seq, InDrop and
10X Genomics, have been developed based on the principle of random
barcodes (5,24,36,84).
Prior to these, similar approaches, such as CEL-Seq and Smart-seq2
(85,86) employed random sequence barcodes for
cell labeling. While CEL-Seq is highly effective in gene expression
analysis, it has limited resolution when detecting complex
alternative splicing events or analyzing full-length transcript
isoforms (85). By contrast,
methods such as Smart-seq offer enhanced efficiency, resulting in
the development of two versions of SMART-seq. These versions
address some of the limitations of other single-cell RNA sequencing
technologies by capturing and analyzing complete transcript
sequences, including both coding and non-coding regions. Despite
the high sensitivity of SMART-Seq2, the method is still susceptible
to amplification biases, which may compromise the accuracy of gene
expression quantification, particularly for low-abundance genes
(86). The random barcode design
used by the Drop-Seq, InDrop and 10X Genomics platforms is largely
similar, incorporating PCR handles, cellular barcodes, UMIs, poly-T
cleavage regions and T7 promoters (24,36,87).
However, there are subtle differences in materials, barcode
capacity and post-demulsification reactions. Drop-Seq, developed by
Macosko et al, enables the tagging of hundreds of cells for
single-cell genomic analysis. Nucleic acid barcodes are integrated
with polyacrylamide microspheres or particles, which are then
linked to target cells and encapsulated into nanoliter-sized
droplets via a microfluidic platform (36). Nevertheless, limitations remain,
such as issues with cell capture efficiency, dropout events and
limited resolution for rare cell types. Like Drop-Seq, InDrop
employs microfluidic droplets to isolate single cells, but with
distinct bead chemistry and barcode designs that provide higher
sensitivity (24). InDrop can
capture full-length transcripts, offering more detailed insights
into gene expression, alternative splicing and transcript isoforms,
making it particularly suitable for studying gene isoform diversity
at the single-cell level (24).
10X Genomics offers high sensitivity, particularly in detecting
low-abundance transcripts, thus enabling researchers to gain deeper
insights into the gene expression dynamics of rare or low-expressed
genes. This system uses unique molecular identifiers (UMIs) to
improve quantification accuracy and minimize amplification biases.
However, it captures only a portion of each transcript (typically
the 3′ end), which may limit its ability to assess full-length
transcript diversity or alternative splicing events comprehensively
(88).
Challenge
In droplet-based barcode systems, individual cells
are encapsulated within microdroplets and tagged with unique
barcode sequences. However, in complex systems, cross-droplet
contamination and barcode swapping are common issues, which may
result in cross-contamination of barcodes between cells (89,90).
To mitigate such concerns, single-cell separation technologies,
such as microfluidic chips, should be employed during droplet
generation to reduce barcode cross-contamination (91,92).
Currently, the mRNA capture efficiency within droplets is low, at
only 7% (93). Despite robust
quantification through UMI filtering, this low efficiency hampers
the reliable detection of genes with fewer than 20–50 transcripts
per cell (90). This issue, which
affects single-cell RNA sequencing, necessitates the implementation
of improved cell lysis methods or optimized enzymatic reactions
during library preparation (94).
Additionally, as the barcodes in this system are random, they do
not permit the association of a given barcode with a specific cell
identification. Background noise, a critical factor contributing to
the low signal-to-noise ratio in this technology, originates
primarily from non-specific binding molecules, residual substances
in solutions and cross-contamination between cells (95). This noise compromises data
accuracy, particularly in high-throughput screening and cancer
research, where it may obscure critical biological signals.
Therefore, reducing non-specific binding agents and enhancing the
purity of single-cell analysis is essential. The use of refined
reagents, optimized culture conditions and more sensitive
sequencing platforms can help mitigate background noise (95,96).
To extract barcode sequence information, sequencing technologies
are indispensable; however, current sequencing methods face
limitations in detecting genes of extremely low abundance,
especially at the single-cell level, where sequencing depth is
often insufficient. Moreover, high-throughput sequencing
technologies themselves are prone to errors. Consequently,
sequencing workflows must be optimized, increased sequencing depth
must be implemented and multiplex amplification strategies employed
to improve the detection of low-abundance genes, reduce sequencing
error rates and integrate error-correction algorithms to enhance
data quality (69). The vast
volume of data generated by barcodes necessitates the use of
efficient algorithms and computational platforms. The integration
of various computational models is essential for data
consolidation, enabling a deeper understanding of the relationship
between barcodes and gene expression, thereby enhancing data
interpretation capabilities.
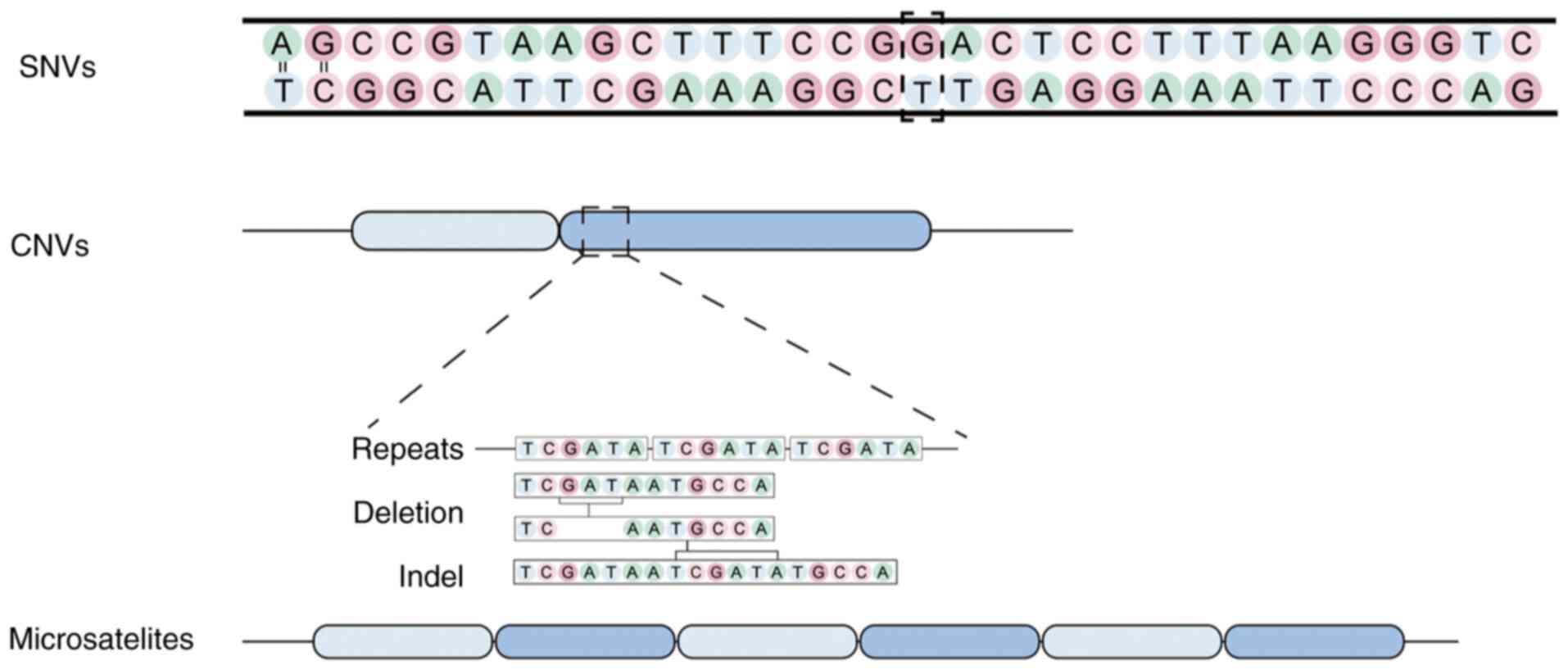
Native barcodes
Design
Somatic mutation barcoding technology represents an
advanced and increasingly indispensable method for tracking and
analyzing somatic mutations at the single-cell level. This method
is key for investigating genetic alterations linked to disease
progression, aging and cellular evolution. The primary types of
mutations detectable through somatic mutation barcoding include
microsatellite instability (MSI), copy number variations (CNVs)
(97), single nucleotide variants
(SNVs) (98) and mitochondrial DNA
mutations (Fig. 6). MSI involves
alterations in the length of short, repetitive DNA sequences,
typically indicative of defective DNA repair mechanisms and is
commonly associated with cancer and various genetic disorders
(99,100). CNVs refer to changes in the copy
number of specific genomic regions, frequently linked to
neurological disease, cancer and developmental disorders. SNVs
represent single base-pair changes in the DNA sequence, which are
pivotal for understanding disease susceptibility and genetic
diversity. Although mitochondrial DNA mutations remain less well
understood, they play a crucial role in various metabolic and
neurodegenerative diseases, influencing cellular energy production
and function. The application of somatic mutation barcoding
technology in biomedical research and clinical diagnostics holds
revolutionary potential (101–103). It enables researchers to capture
the full complexity of somatic mutations at the single-cell level,
offering an unprecedented approach for analyzing genetic diversity
within tissue. In oncology, this technology is particularly
valuable, as clonal evolution within tumors frequently dictates
therapeutic resistance and metastasis. It also provides new
opportunities for studying neurological diseases, as analyzing
somatic mutations in neuronal populations may elucidate the
mechanisms underlying diseases such as Alzheimer's disease, autism
and schizophrenia. Furthermore, as somatic mutations accumulate in
various tissues throughout aging, this technology can deepen our
understanding of the aging process and age-related diseases
(104,105). The advantages of somatic mutation
barcoding technology are clear. Its high sensitivity facilitates
the detection of rare mutations in heterogeneous cell populations
and allows for longitudinal monitoring of mutations in response to
treatment or environmental factors. By using unique molecular
barcodes, researchers can distinguish mutations that arise in
different cells, thereby facilitating the analysis of clonal
evolution and cellular lineage (106). Additionally, the application of
this technology provides novel insights into the relationship
between somatic mutations and disease mechanisms, potentially
leading to the development of personalized therapeutic strategies.
Despite its numerous advantages, somatic mutation barcoding
technology faces several challenges (107). Sequencing errors, amplification
biases and cross-contamination can result in inaccuracies in
mutation identification and quantification, particularly when
addressing low-abundance mutations, which are prevalent in complex
diseases such as cancer and neurological disorders. Moreover,
distinguishing pathogenic mutations from benign ones remains a
significant challenge, as not all mutations contribute to disease
development. To overcome these limitations, advances in sequencing
technologies, error-correction algorithms and bioinformatics tools
are essential. Innovations, such as enhanced error-correction
methods, longer read lengths and more sensitive sequencing
platforms, are anticipated to improve (108–110).
Challenge
Native barcodes are derived from natural variations
or mutations, rather than exogenous sequences, which inherently
restrict their insertion sites and diversity. This limitation may
lead to an insufficient number and diversity of barcodes, rendering
them inadequate for large-scale tracking purposes. To mitigate this
limitation, a combination of diverse natural variations and
mutations, coupled with specific cell-labeling techniques, may be
employed to augment barcode diversity and resolution. These
barcodes generally rely on inherent cellular markers, such as
specific gene expressions or mutations, which often exhibit uneven
expression levels across different cell types and lack universal
applicability. Moreover, certain cell types may lack sufficient
natural markers, or the markers may be difficult to detect with
adequate sensitivity. The range of natural markers is restricted
and their levels are susceptible to fluctuations in cell state and
environmental factors, resulting in inconsistent marker expression.
To overcome this limitation, a combination of various natural
markers or alternative exogenous labels is required to increase
marker diversity, while high-sensitivity techniques, such as
single-molecule RNA sequencing, must be employed to enhance
detection sensitivity. Stability is also a critical factor to
consider when utilizing natural barcodes, as they are often
influenced by external factors such as cell division, environmental
changes and epigenetic modifications. Throughout research, barcodes
may undergo alterations during cell development, compromising their
stability and impeding accurate lineage tracing. Furthermore,
long-term stability may deteriorate over extended periods,
rendering sustained tracking difficult. Barcode stability can be
enhanced through the optimization of probe design and the use of
advanced imaging techniques, such as fluorescence confocal or
super-resolution microscopy, to improve spatial localization.
Additionally, sensitivity remains a critical concern, especially in
low-abundance or rare cell populations, where natural barcode
concentrations may be too low for effective signal detection.
Finally, the intrinsic nature of native barcodes leads to
heightened background noise interference, complicating data
interpretation. The application of data-cleaning techniques to
reduce background noise and multi-channel data fusion methods can
improve the accuracy of barcode detection.
Applications of barcodes
Single cell lineage tracing and fate
mapping
Lineage tracing has emerged as a powerful tool for
studying tissue development, homeostasis and disease, particularly
when combined with experimental manipulation of signaling pathways
that regulate cell fate decisions. The evolution of lineage tracing
methods has advanced from traditional techniques, such as optical
microscopy and dye-based cell labeling, to more sophisticated
approaches, including recombinase-based systems, barcode
technologies and natural gene mutations (111–113). This evolution has enabled the
tracking of thousands of cells simultaneously, facilitated by
advances in barcode analysis technologies, from PCR (114) to sequencing methods, microarrays
(15) Sanger sequencing (115,116) high-throughput sequencing
(7,31) and single-cell RNA sequencing
(scRNA-seq) (117). Since these
foundational studies, barcoding has been widely adopted for fate
mapping in both multicellular organisms and single-cell microbial
communities. In cell biology, this includes investigating clonal
relationships among cell types (18), analyzing differentiation
trajectories (118), identifying
molecular signatures tied to clonal identity, resolving lineage
relationships and dissecting cellular dynamics (119). At present, fate mapping extends
to cancer research, including studies on cancer heterogeneity,
clonal evolution, drug resistance and metastasis mechanisms.
Barcodes in this context serve as both static and cumulative
markers (118). Polylox barcodes
(44), transposon-based barcodes
(120) and CRISPR-based barcodes
(38) are commonly used in lineage
tracing, with applications across various systems. Integrated
barcodes are often employed to mark progenitor cells, with this
approach being widely used in hematopoietic stem cell research
(44,121). By tracking individual cells and
their progeny, these studies have linked specific transcriptional
programs to various differentiation states, highlighting the
dynamic relationship between transcriptional regulation and lineage
commitment. Tracking the lineage history of cells is essential for
addressing fundamental biological questions. For instance, in 2020,
Bowling et al (55) used a
CRISPR-based array system to track lineage in mice, uncovering
intrinsic biases in fetal liver hematopoietic stem cell clonal
activity and providing insights into stem cell responses to injury
(Fig. 7A). In the same year,
Weinreb et al (118)
employed expressed DNA barcodes to perform clonal tracing of
transcriptomes over time, applying this approach to investigate
fate determination during hematopoiesis. Unlike CRISPR-based
methods, this technique does not require lineage tree inference to
establish sister-cell relationships; it exhibits remarkably low
single-cell barcode dropout rates and eliminates the need for
delivering multiple components. The study identified states that
initiate fate potential and mapped them onto a continuous
transcriptional landscape. (Fig.
7B). Lineage tracing has broader applications, including fate
mapping in entire organisms (52),
vertebrate brains (51), EMT in
cancer and cancer xenograft metastasis (122). The application of barcode
technologies in lineage tracing has also addressed several key
challenges. For instance, barcodes in dynamic systems, such as
cancer stem cells or regenerative tissues, offer valuable insights
into cellular identity plasticity and lineage decisions. These
methods enable the exploration of tissue heterogeneity, revealing
how different cell populations within the same tissue can behave
based on their lineage and gene expression profiles. Despite the
advances, several challenges remain. High-throughput barcode
generation and integration of multi-dimensional data (e.g.,
genomic, transcriptomic and epigenomic profiles) from
lineage-traced cells still pose technical difficulties (7,123,124). Moreover, distinguishing between
genetic mutations and transcriptional shifts in lineage tracing
experiments requires careful control of experimental conditions and
sophisticated computational methods. These challenges underscore
the need for ongoing refinement of lineage tracing techniques and
the development of more scalable and precise methods for
high-dimensional data integration. lineage tracing, particularly
when integrated with barcode technology, has transformed our
understanding of cellular fate, lineage commitment and plasticity.
Its applications in basic research and disease modeling, especially
in cancer, provide a valuable framework for investigating the
complexities of cellular behavior during development and disease
(123,125).
Barcoding for single cell
transcriptome
Single-cell transcriptomics is used to investigate
the gene expression profiles of individual cells. This technology
is pivotal in uncovering cellular heterogeneity, identifying novel
cellular subpopulations and is indispensable for understanding the
functional roles of diverse cell types within tissues. Among the
primary methodologies used in single-cell transcriptomics is the
droplet barcode technology (24).
Prior to this, several methodologies, including SMART-seq, CEL-seq
and Cyt-seq, were developed to study embryonic samples (126,127), the early developmental stages of
the nematode Caenorhabditis elegans and the transcriptomes
of rare cells within the hematopoietic system (128). Nevertheless, these approaches
remain constrained by limitations such as low throughput (129), reduced specificity and
sensitivity and PCR bias (130).
The emergence of microfluidic technologies has substantially
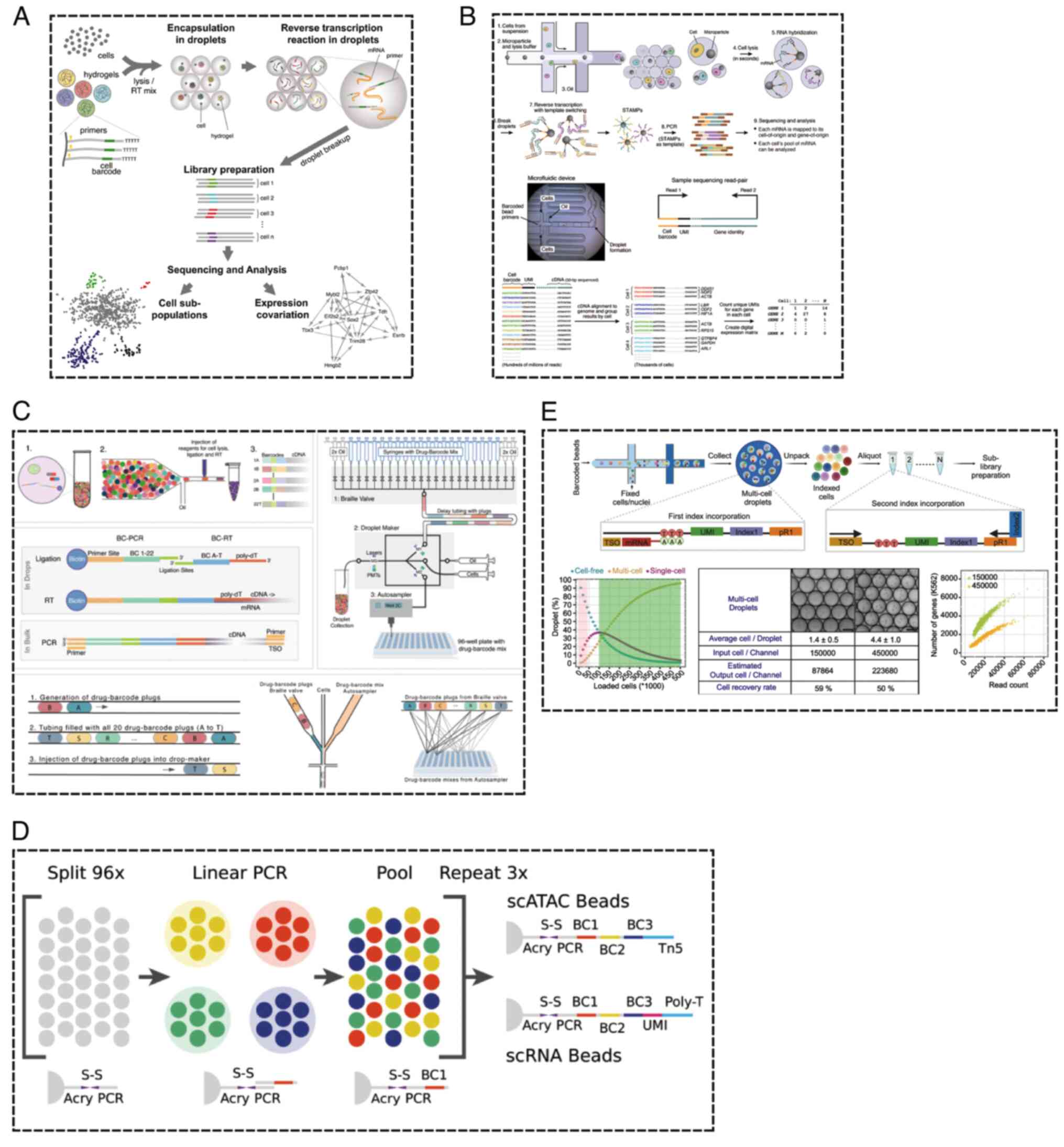
mitigated these challenges. Klein et al (24) integrated microfluidic technology
with barcode beads to devise a calibration technique suitable for
RNA sequencing of thousands of individual cells, widely applied in
sequencing analyses of embryonic stem cells and their
differentiated forms. This technology offers boundless scalability
in cell processing, facilitating highly efficient cell capture,
rapid sample collection and minimal technical noise (Fig. 8A) (24).
In the same year, the development of the Drop-seq
platform introduced a rapid strategy for analyzing thousands of
individual cells. This platform analyzed the transcriptomes of
44,808 retinal cells from mice, constructing a molecular gene
expression map encompassing both established retinal cell types and
novel candidate subtypes (Fig. 8B)
(36). Single-cell sequencing
offers several advantages, including the precise identification of
genetic differences between healthy and aberrant cells, as well as
the construction of cellular maps for entire organisms, tissues and
microbiomes. Nevertheless, existing chromatin extraction methods
remain prohibitively expensive and there are currently no publicly
available tools for the efficient processing of tens of thousands
of cells. Moreover, although certain single-cell RNA sequencing
tools are available at no cost, they often exhibit limitations in
sensitivity and practicality. To address these issues, Florian
et al developed HyDrop, an innovative open-source platform
that surmounts these obstacles (5). This platform employs a novel type of
barcode bead and refines existing microfluidic protocols using
open-source reagents. These advancements offer users a more
intuitive workflow while enhancing sequencing sensitivity without
incurring additional costs. Leveraging this platform, thousands of
single-cell RNA and open chromatin profiles were successfully
extracted from the brains of mice and fruit flies (Fig. 8C) (5).
Traditional droplet-based microfluidic methods often
generate a significant number of droplet-only cells, failing to
fully exploit barcode beads and reagents. Although combinatorial
indexing on microplates proves more effective for barcode usage, it
is labor-intensive (84,131,132). To address this, researchers have
developed a combinatorial indexing system based on overloading and
dissociation (OAK) for ultra-high-throughput single-cell
multi-omics analysis. Initial partitioning is performed using a
droplet barcode system, followed by a second round of
identity-tagged references to achieve combinatorial indexing. The
OAK system boasts ultra-high throughput, broad compatibility, high
sensitivity and a simplified workflow, making it a powerful tool
for large-scale molecular analysis, particularly for rare cell
populations (Fig. 8E) (133). In recent years, this technology
has also been applied to drug screening. Mathur et al
(134) proposed an expandable
microfluidic workflow, named Combi-Seq, which uses transcriptomic
changes as a readout for drug effects, screening hundreds of drug
combinations within microliter-sized droplets (Fig. 8D). Applying Combi-Seq, they
screened 420 drug combinations for their effects on the K562 cell
transcriptome, using ~250 single-cell droplets per condition,
successfully predicting synergistic and antagonistic drug
interactions and their pathway activities.
Barcodes for high-throughput
screening
High-throughput screening technology has become a
crucial tool in the study of genetic variations and perturbations,
driving advancements in our understanding of biological systems and
enhancing the application of biotechnologies. The development of
large-scale screenings and genetically diverse variant libraries
has played a central role in advancing this field. At the genomic
scale, RNA interference (135,136) and CRISPR-based technologies have
been widely employed to screen pooled libraries (137), investigating cellular phenotypes
such as cell viability and transcriptomes at single-cell
resolution. Traditional screening methods are often constrained by
low throughput, extended durations and high costs. Although
progress in genome-scale screening has enabled the assessment of
mammalian gene functions, these methods still rely on simple
phenotype readouts that cannot differentiate between similar
responses induced by distinct mechanisms. The advent of
droplet-based single-cell RNA sequencing technology has introduced
a powerful platform for high-throughput screening (138). When paired with CRISPR-based
transcriptional interference platforms, this has facilitated the
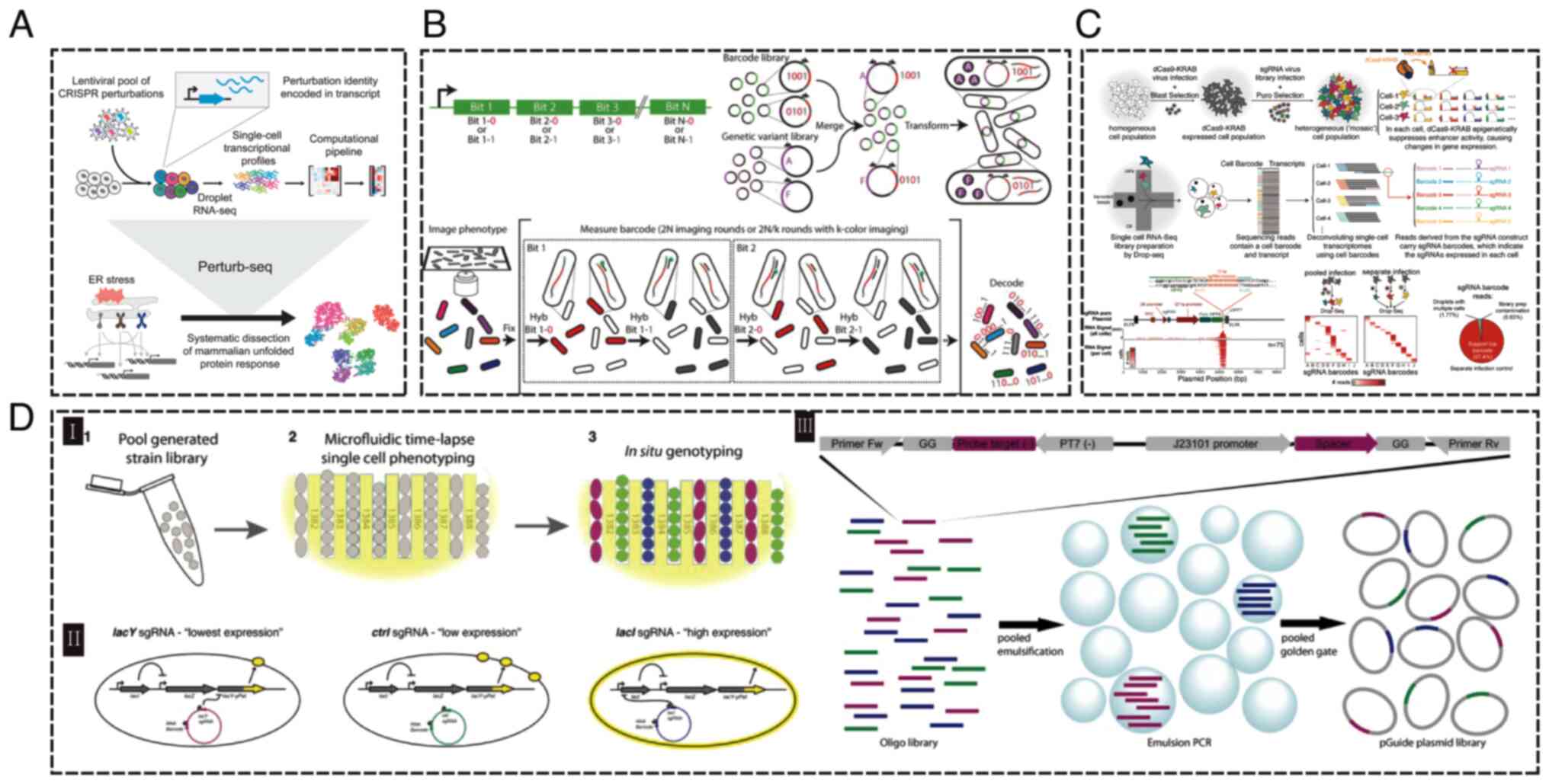
development of the ‘perturb-seq’ method. (Fig. 9A) (139). This approach is not only easily
implementable but also highly scalable, allowing parallel screening
at the single-cell level to generate rich phenotype data. Using
this technology, studies on the unfolded protein response in
mammals have elucidated how three sensors within the endoplasmic
reticulum monitor various types of stress. Furthermore,
CRISPR-based transcriptional interference has been combined with
scRNA-seq to dissect immune circuits. Nucleic acid barcodes also
play a pivotal role in phenotype analysis (140), particularly in situations
requiring high-resolution imaging to localize cellular morphology,
proteins and RNA. Emanuel et al (141) introduced an innovative method
that combines genetic variant barcodes with multiplexed
fluorescence in situ hybridization (FISH) technology
(141). Each genetic variant is
associated with a unique nucleic acid barcode, enabling
identification via multiplexed FISH imaging. This method identifies
brighter and more photostable YFAST fluorescent protein variants
from a pool of 60,000 variants (141) (Fig.
9B). By integrating genome engineering, nucleic acid barcode
technologies, high-resolution microscopic imaging and
microfluidics, researchers can now conduct high temporal and
spatial resolution studies of complex phenotypes in live cells,
enabling precise control of single strains through single-cell
observation. However, effective studies on the localization of
phenotypes related to intracellular features or their correlation
with specific genotypes are still limited (142). Xie et al (143) developed the Mosaic-seq platform,
which integrates CRISPR barcodes with transcriptomic data and sgRNA
regulators, enabling the engineering and analysis of multiple
enhancer activities at the single-cell level (Fig. 9C). Lawson et al (144) expanded advanced live-cell
microscopy techniques to bacterial strain libraries generated
through pooling (Fig. 9D). They
successfully confirmed the presence of plasmid-encoded strains
using single-molecule fluorescence lifetime imaging, where the
plasmid expressed sgRNA and employed dCas9 interference to suppress
distinct genes within the Escherichia coli genome. This
method effectively addresses the challenge of characterizing
complex dynamic phenotypes within genetic libraries of various
bacterial strains. For instance, it enables the screening of how
changes in regulatory or coding sequences affect the timing,
localization, or functionality of gene products, or how shifts in
the expression of a set of genes influence the intracellular
dynamics of a reporter gene.
Exploring cancer heterogeneity
Human cell lines have been pivotal in cancer
research, driving discoveries of oncogenic mechanisms and
therapeutic targets. Tumor heterogeneity, characterized by
differences in tumor cell growth rate, invasive ability and
regulation by drug-sensitive genes or molecules, results from
genetic, epigenetic and environmental influences (145). Additionally, cancer heterogeneity
depends on extrinsic factors such as time, the tumor
microenvironment and interventions. Linking the cellular behavior
of cancer cells with their molecular profiles can aid in proposing
more effective methods to predict clinical outcomes and treat
cancer patients. Cancer cells originate from individual malignant
cells in the body and accumulate mutations in their DNA as they
proliferate (146). Thus, tumors
evolve into highly distinct cancer clones over time. With the
advent of next-generation sequencing technologies (such as
sc-RNA-seq), tumor heterogeneity and clonal frequency can be
further investigated before and after treatment. However,
carcinogenesis involves millions of cells and current technologies
cannot fully cover such a large, heterogeneous cell population. The
introduction of nucleic acid sequence barcoding provides a solution
by labeling individual cells with a ‘unique’ barcode. This barcode
can be used alongside sequencing to track progenitor cells, as it
is heritably replicated and inherited by daughter cells during cell
division (17,37,72,147) In recent years, The development of
cancer is primarily driven by the processes of clonal selection and
clonal expansion. However, the non-genetic mechanisms underlying
intratumoral heterogeneity and malignant clonal heterogeneity
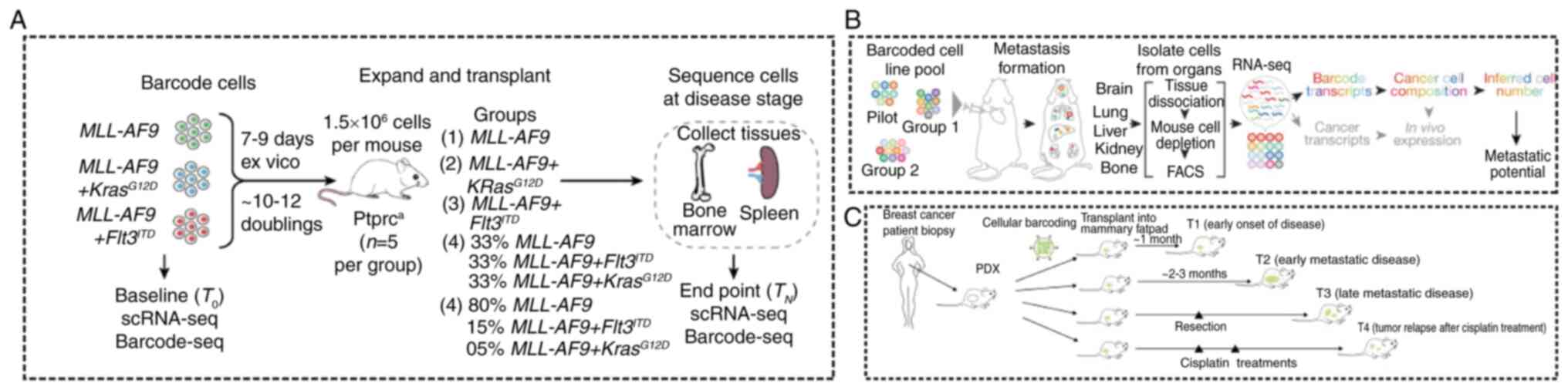
remain largely poorly understood. Fennell et al (148) developed an innovative expression
barcoding strategy, termed Single-cell Profiling and Lineage
Tracing (SPLINTR). Using a mouse model of acute myeloid leukemia,
the study employed isogenic clonal tracking to demonstrate, for the
first time, that the clonal output characteristics of leukemia stem
cells are critical determinants of their sensitivity to
chemotherapy. By using SPLINTR technology, the team was able to
simultaneously track the dynamic changes of thousands of malignant
clones in a time-resolved manner, thereby uncovering principles of
clonal adaptation that extend beyond the cancer genome. Notably,
the study identified Slpi, a pivotal mediator of malignant clonal
dominance that is exclusively detectable in vivo. This
discovery offers new theoretical insights and research directions
toward a deeper understanding of the mechanisms underlying cancer
clonal evolution (Fig. 10A). In
addition, Merino et al (20) aimed to uncover the subclonal
relationship between primary triple-negative breast cancer and its
metastasis. They employed cellular barcoding of two untreated
triple-negative breast cancer patient-derived xenografts to track
thousands of barcoded clones across primary tumors and metastases.
Their findings suggest that cancer dissemination might relate to
persistent shedding of material (Fig.
10C). Furthermore, Jin et al (149) employed an in vivo
barcoding strategy to assess the metastatic potential of human
cancer cell lines in mouse xenografts. They developed a
first-generation metastasis map (MetMap) that elucidated
organ-specific metastasis patterns (Fig. 10B). Further research into
barcoding technology enables precise mapping of individual cell
behavior at the cellular level and comprehensive analysis of
intra-tumor heterogeneity mechanisms. Additionally, it facilitates
molecular characterization of cancer clones, enhancing
understanding of their characteristics. Barcoding technology holds
substantial potential in advancing cancer research across cloning,
metastasis, origin, development, treatment and drug selection.
Extracellular vesicle analysis
Extracellular vehicles (EVs), membrane-enclosed
structures released by cells, are typically classified into several
subgroups based on size or origin (150,151). Rich in molecular cargo, EVs play
a pivotal role in cellular signaling and communication through the
transport of bioactive substances and are involved in the signaling
pathways associated with numerous diseases, including cancer
(152–154). In recent years, EVs have emerged
as valuable biomarkers for disease diagnosis and therapeutic
monitoring (155). Small EVs
(sEVs), derived from multicellular organisms, display a highly
heterogeneous vesicular repertoire. Since the surface protein
composition of EVs markedly influences their biological behavior
(156), there is a growing need
for high-throughput, single-EV analytical methods to more precisely
characterize EV subpopulations (157). In 2021, Ko et al (158) introduced an antibody-based
immunosequencing method that combines droplet microfluidics with
nucleic acid sequence barcodes. This technique uses droplet
microfluidics to partition EVs and barcodes, followed by sequencing
of the barcode-antibody-DNA constructs, enabling multiplexed
protein analysis from individual nanosized EVs to delineate their
protein composition (Fig. 11A).
This method offers the advantages of high sensitivity and
throughput, with potential for further refinement to detect
low-abundance proteins, such as enhancing the signal-to-noise
ratio, reducing nonspecific binding and improving sequencing depth.
In 2022, a novel alternative method based on droplet barcode
sequencing (DBS) technology was proposed for the detection of
individual sEVs (22) (Fig. 11B). This scalable method requires
no specialized equipment or barcode gel beads and demonstrated that
DBS-Pro facilitates the analysis of individual sEVs with a mixing
rate of <2%. The approach was applied to analyze surface
proteins of over 120,000 sEVs derived from non-small-cell lung
cancer (NSCLC) cell lines and malignant pleural effusion samples
from patients with NSCLC. This method enables the surface protein
analysis of single EVs and the characterization of expanded sEV
subtypes. However, due to the limited quantity of material
available, this strategy may be impractical for clinical
applications. Furthermore, a multiplexed approach based on droplet
barcode technology has been developed, in which lipid nanoparticles
carrying single-stranded DNA barcodes are fused with control
particles, enabling the simultaneous tracking of EVs from different
cell lines within a single in vivo experiment (159). In addition to barcode-based
analysis of EV subpopulations, CRISPR-assisted single-barcode-based
screening of regulators of sEV release has been employed (Fig. 11C) to identify key players in sEV
release (160). This screening
uses the interaction between gRNA and a deathCas9 fusion protein
linked to sEV markers, combined with CRISPR-gRNA barcode encoding
of sEVs (Fig. 11D) (159). Barcode quantification enables
large-scale, parallel estimations of sEV release from individual
cells. The barcode-sEV and CRISPR-based screening approach allows
for genome-wide exploration of sEV release regulators, facilitating
the identification of previously unrecognized regulators and the
discovery of exosome/exosome-like properties, such as CD63+/CD9+
sEVs and the synchronous release of CD9+ sEVs during the cell
cycle. In comparison with traditional one-to-one analyses, this
multiplexed approach offers markedly higher throughput for
identifying sEV release regulators, independent of the effects of
sEV abundance in culture media. Despite significant advances in
sequencing-based single-EV protein analysis, several challenges
persist that hinder broader clinical application. These challenges
include the isolation of individual EVs, efficient capture of
low-abundance proteins, clinical feasibility and the need for
comprehensive and accurate insights into the biological functions
of EVs. The integration of nucleic acid sequence barcodes with
sequencing-based single-EV protein analysis has revolutionized the
study of extracellular vesicle biology. With its high sensitivity,
scalability and ability to provide multiparametric analyses, this
method holds substantial potential for advancing our understanding
of EVs as biomarkers for disease diagnosis, prognosis and
treatment. However, technical obstacles remain, particularly in the
isolation of single EVs, optimization of sequencing technologies
and the interpretation of complex datasets. Continued innovation in
these areas will pave the way for the routine clinical application
of single-EV analysis, opening new frontiers for precision
medicine.
 | Figure 11.EV profiling. (A) An antibody-based
immunosequencing method that enables multiplexed measurement of
protein molecules from single nanometer-sized electric vehicles.
(B) The principle of droplet barcode sequencing analysis for
protein analysis, used for the quantification of surface proteins
on individual vesicles. (C) CIBER screening utilizes the
interaction between gRNA and death Cas9 fused with sEV markers,
along with barcode-encoded sEVs via CRISPR-gRNA. Barcode
quantification allows for the large-scale, parallel estimation of
the amount of sEVs released from each individual cell. (D) EVs from
16 cell lines were used to generate a unique barcode-hEV library,
through fusion with LNPs carrying single-stranded DNA barcodes as
controls. Reprinted with permission from references (22,157–159,) gRNA, guide RNA; sEV, small
extracellular vehicles; CRISPR, clustered regularly interspaced
short palindromic repeats; hEV, hybridized EV; LNPs, lipid
nanoparticles. |
Conclusions and perspectives
In the 1950s, the elucidation of the double helix
structure of DNA opened a new chapter in life science. The
introduction of nucleic acid barcoding has had a similarly profound
effect on the biomedical field. Given the role of DNA in genetic
replication, nucleic acid barcoding serves as a robust tool for
cell labeling and tracking. The diversity and uniqueness of nucleic
acid barcodes imbue them with high throughput, resolution and
accuracy. Recent technological advances have further enabled
barcoding to offer a comprehensive, multi-modal understanding of
tissue cell composition across temporal and spatial dimensions
(161). Looking ahead, nucleic
acid barcoding will extend beyond high-throughput screening,
single-cell lineage tracking and the characterization of tumor
heterogeneity. It holds promise for clinical cancer diagnosis,
adjuvant therapy, exosome detection and subpopulation
classification based on surface markers (18,19).
Additionally, nucleic acid sequence barcoding is increasingly
applied in areas such as active protease sensing (162), multiplex nucleic acid detection
platforms (163), bacterial
single-cell RNA sequencing for toxin regulation prediction
(164) and DNA sequence-based
optical-free single-cell spatial proteomics (165) to infer the spatial distribution
of cell surface proteins. After nearly two decades of development,
nucleic acid sequence barcoding offers a comprehensive, multimodal
understanding of tissues and whole organisms across temporal and
spatial dimensions. However, several challenges continue to impede
its broader application. First, the diversity of barcodes does not
yet comprehensively cover all cell types in a number of organs and
biases introduced during barcoding remain unaddressed. Second,
obtaining barcode sequence readouts requires library creation and
target sequencing, which do not provide direct results. Lastly,
despite advances in sequencing technology, inaccuracies,
information loss and high error rates persist, limiting the
reliability of results.
New nucleic acid sequence barcoding libraries and
applications are reported annually, facilitating increasingly
detailed and comprehensive analyses of cell and cancer
heterogeneity. This technology has filled gaps left by other
sequencing methods, offering broader insights into biological
development, cloning and disease research. As the technology
continues to evolve, the integration of optical barcoding with
nucleic acid sequence barcoding holds promise for spatially
localizing cells and analyzing gene expression profiles in
unprecedented detail (115,166–168). Addressing the limitations of
barcode diversity, sequencing accuracy and data integration will
require continued innovation. The development of barcode libraries
that can comprehensively cover a broader spectrum of cell types,
coupled with improvements in sequencing technology and error
correction, will markedly enhance the power of nucleic acid
barcoding. In addition, expanding the application of multi-modal
data, including genomic, transcriptomic, proteomic and spatial
information, will provide a deeper understanding of the mechanisms
driving disease. Machine learning and AI-driven approaches will
also play a key role in making sense of this complex data and
enabling more accurate predictions about disease progression and
treatment response. The integration of optical barcoding techniques
into nucleic acid sequence barcoding holds significant potential
for spatially resolving cellular dynamics within tissues, opening
new avenues for research and clinical applications.
Interdisciplinary collaborations are essential to
address these challenges. Bioinformaticians, computational
biologists, molecular biologists and clinicians must work together
to refine the technology, improve error correction and optimize
data analysis tools. Engineers and physicists can contribute by
developing next-generation sequencing technologies and advancing
optical barcoding methods. Collaborations with clinicians and
oncologists are crucial for translating these advances into
clinical settings, particularly for the detection of tumor
heterogeneity and monitoring therapeutic responses. Such
collaborations will undoubtedly accelerate the translation of
nucleic acid barcoding technology into practical, clinically
relevant applications, transforming our approach to disease
diagnosis, treatment and prevention.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific and
Technological Innovation Major Base of Guangxi (grant no.
2022-36-Z05) and the Guangxi Science and Technology Major Program
(grant no. AA24263028).
Availability of data and materials
Not applicable.
Authors' contributions
YW was responsible for conceptualization,
investigation, visualization and writing the original draft. FL was
responsible for conceptualization, funding acquisition, validation,
visualization, writing, reviewing and editing. Data authentication
is not applicable. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Loewer A and Lahav G: We are all
individuals: Causes and consequences of non-genetic heterogeneity
in mammalian cells. Curr Opin Genet Dev. 21:753–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Y, Zhang X and Wang Z: Cellular
barcoding: From developmental tracing to anti-tumor drug discovery.
Cancer Lett. 567:2162812023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen C, Liao Y and Peng G: Connecting past
and present: Single-cell lineage tracing. Protein Cell. 13:790–807.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weinreb C and Klein AM: Lineage
reconstruction from clonal correlations. Proc Natl Acad Sci USA.
117:17041–17048. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Rop FV, Ismail JN, Bravo González-Blas
C, Hulselmans GJ, Flerin CC, Janssens J, Theunis K, Christiaens VM,
Wouters J, Marcassa G, et al: HyDrop enables droplet based
single-cell ATAC-seq and single-cell RNA-seq using dissolvable
hydrogel beads. Elife. 11:e739712022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sulston JE, Schierenberg E, White JG and
Thomson JN: The embryonic cell lineage of the nematode
Caenorhabditis elegans. Dev Biol. 100:64–119. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu R, Neff NF, Quake SR and Weissman IL:
Tracking single hematopoietic stem cells in vivo using
high-throughput sequencing in conjunction with viral genetic
barcoding. Nat Biotechnol. 29:928–933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weber K, Thomaschewski M, Warlich M, Volz
T, Cornils K, Niebuhr B, Täger M, Lütgehetmann M, Pollok JM,
Stocking C, et al: RGB marking facilitates multicolor clonal cell
tracking. Nat Med. 17:504–509. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weber K, Bartsch U, Stocking C and Fehse
B: A multicolor panel of novel lentiviral ‘Gene Ontology’ (LeGO)
vectors for functional gene analysis. Mol Ther. 16:698–706. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gomez-Nicola D, Riecken K, Fehse B and
Perry VH: In-vivo RGB marking and multicolour single-cell tracking
in the adult brain. Sci Rep. 4:75202014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weber K, Mock U, Petrowitz B, Bartsch U
and Fehse B: Lentiviral gene ontology (LeGO) vectors equipped with
novel drug-selectable fluorescent proteins: New building blocks for
cell marking and multi-gene analysis. Gene Ther. 17:511–520. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mohme M, Maire CL, Riecken K, Zapf S,
Aranyossy T, Westphal M, Lamszus K and Fehse B: Optical barcoding
for single-clone tracking to study tumor heterogeneity. Mol Ther.
25:621–633. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shembrey C, Smith J, Grandin M, Williams
N, Cho HJ, Mølck C, Behrenbruch C, Thomson BN, Heriot AG, Merino D
and Hollande F: Longitudinal monitoring of intra-tumoural
heterogeneity using optical barcoding of patient-derived colorectal
tumour models. Cancers (Basel). 14:5812022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schepers K, Swart E, van Heijst JW,
Gerlach C, Castrucci M, Sie D, Heimerikx M, Velds A, Kerkhoven RM,
Arens R and Schumacher TN: Dissecting T cell lineage relationships
by cellular barcoding. J Exp Med. 205:2309–2318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Heijst JW, Gerlach C, Swart E, Sie D,
Nunes-Alves C, Kerkhoven RM, Arens R, Correia-Neves M, Schepers K
and Schumacher TN: Recruitment of antigen-specific CD8+
T cells in response to infection is markedly efficient. Science.
325:1265–1269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalhor R, Kalhor K, Mejia L, Leeper K,
Graveline A, Mali P and Church GM: Developmental barcoding of whole
mouse via homing CRISPR. Science. 361:eaat98042018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kebschull JM and Zador AM: Cellular
barcoding: Lineage tracing, screening and beyond. Nat Methods.
15:871–879. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sankaran VG, Weissman JS and Zon LI:
Cellular barcoding to decipher clonal dynamics in disease. Science.
378:eabm58742022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Serrano A, Berthelet J, Naik SH and Merino
D: Mastering the use of cellular barcoding to explore cancer
heterogeneity. Nat Rev Cancer. 22:609–624. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Merino D, Weber TS, Serrano A, Vaillant F,
Liu K, Pal B, Di Stefano L, Schreuder J, Lin D, Chen Y, et al:
Barcoding reveals complex clonal behavior in patient-derived
xenografts of metastatic triple negative breast cancer. Nat Commun.
10:7662019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ko J, Wang Y, Carlson JCT, Marquard A,
Gungabeesoon J, Charest A, Weitz D, Pittet MJ and Weissleder R:
Single extracellular vesicle protein analysis using immuno-droplet
digital polymerase chain reaction amplification. Adv Biosyst.
4:e19003072020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Banijamali M, Höjer P, Nagy A, Hååg P,
Gomero EP, Stiller C, Kaminskyy VO, Ekman S, Lewensohn R, Karlström
AE, et al: Characterizing single extracellular vesicles by droplet
barcode sequencing for protein analysis. J Extracell Vesicles.
11:e122772022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yeo GHT, Lin L, Qi CY, Cha M, Gifford DK
and Sherwood RI: A multiplexed barcodelet single-cell RNA-Seq
approach elucidates combinatorial signaling pathways that drive ESC
differentiation. Cell Stem Cell. 26:938–950.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klein AM, Mazutis L, Akartuna I,
Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA and Kirschner
MW: Droplet barcoding for single-cell transcriptomics applied to
embryonic stem cells. Cell. 161:1187–1201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perié L, Duffy KR, Kok L, de Boer RJ and
Schumacher TN: The branching point in erythro-myeloid
differentiation. Cell. 163:1655–1662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schueder F, Unterauer EM, Ganji M and
Jungmann R: DNA-Barcoded fluorescence microscopy for spatial omics.
Proteomics. 20:e19003682020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Howland KK and Brock A: Cellular barcoding
tracks heterogeneous clones through selective pressures and
phenotypic transitions. Trends Cancer. 9:591–601. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kretzschmar K and Watt FM: Lineage
Tracing. Cell. 148:33–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Perié L and Duffy KR: Retracing the in
vivo haematopoietic tree using single-cell methods. FEBS Lett.
590:4068–4083. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kok L, Masopust D and Schumacher TN: The
precursors of CD8+ tissue resident memory T cells: From lymphoid
organs to infected tissues. Nat Rev Immunol. 22:283–293. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Naik SH, Perié L, Swart E, Gerlach C, van
Rooij N, de Boer RJ and Schumacher TN: Diverse and heritable
lineage imprinting of early haematopoietic progenitors. Nature.
496:229–232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dhimolea E, De Matos Simoes R, Kansara D,
Al'Khafaji A, Bouyssou J, Weng X, Sharma S, Raja J, Awate P,
Shirasaki R, et al: An embryonic diapause-like adaptation with
suppressed myc activity enables tumor treatment persistence. Cancer
Cell. 39:240–256.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Echeverria GV, Ge Z, Seth S, Zhang X,
Jeter-Jones S, Zhou X, Cai S, Tu Y, McCoy A, Peoples M, et al:
Resistance to neoadjuvant chemotherapy in triple-negative breast
cancer mediated by a reversible drug-tolerant state. Sci Transl
Med. 11:eaav09362019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Echeverria GV, Powell E, Seth S, Ge Z,
Carugo A, Bristow C, Peoples M, Robinson F, Qiu H, Shao J, et al:
High-resolution clonal mapping of multi-organ metastasis in triple
negative breast cancer. Nat Commun. 9:50792018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blundell JR and Levy SF: Beyond genome
sequencing: Lineage tracking with barcodes to study the dynamics of
evolution, infection, and cancer. Genomics. 104((6 Pt A)): 417–430.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Macosko EZ, Basu A, Satija R, Nemesh J,
Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck
EM, et al: Highly parallel genome-wide expression profiling of
individual cells using nanoliter droplets. Cell. 161:1202–1214.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Naik SH, Schumacher TN and Perié L:
Cellular barcoding: A technical appraisal. Exp Hematol. 42:598–608.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Spanjaard B, Hu B, Mitic N,
Olivares-Chauvet P, Janjuha S, Ninov N and Junker JP: Simultaneous
lineage tracing and cell-type identification using
CRISPR-Cas9-induced genetic scars. Nat Biotechnol. 36:469–473.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wagner DE and Klein AM: Lineage tracing
meets single-cell omics: opportunities and challenges. Nat Rev
Genet. 21:410–427. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai D, Cohen KB, Luo T, Lichtman JW and
Sanes JR: Improved tools for the Brainbow toolbox. Nat Methods.
10:540–547. 2013. View Article : Google Scholar
|
|
41
|
Pei W, Feyerabend TB, Rössler J, Wang X,
Postrach D, Busch K, Rode I, Klapproth K, Dietlein N, Quedenau C,
et al: Polylox barcoding reveals haematopoietic stem cell fates
realized in vivo. Nature. 548:456–460. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pei W, Wang X, Rössler J, Feyerabend TB,
Höfer T and Rodewald HR: Using Cre-recombinase-driven Polylox
barcoding for in vivo fate mapping in mice. Nat Protoc.
14:1820–1840. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
McLellan MA, Rosenthal NA and Pinto AR:
Cre- loxP-mediated recombination: General principles and
experimental considerations. Curr Protoc Mouse Biol. 7:1–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pei W, Shang F, Wang X, Fanti AK, Greco A,
Busch K, Klapproth K, Zhang Q, Quedenau C, Sauer S, et al:
Resolving fates and single-cell transcriptomes of hematopoietic
stem cell clones by polyloxexpress barcoding. Cell Stem Cell.
27:383–395.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Becher B, Waisman A and Lu LF: Conditional
gene-targeting in mice: Problems and solutions. Immunity.
48:835–836. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McDavid A, Finak G, Chattopadyay PK,
Dominguez M, Lamoreaux L, Ma SS, Roederer M and Gottardo R: Data
exploration, quality control and testing in single-cell qPCR-based
gene expression experiments. Bioinformatics. 29:461–467. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kharchenko PV, Silberstein L and Scadden
DT: Bayesian approach to single-cell differential expression
analysis. Nat Methods. 11:740–742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fan J, Slowikowski K and Zhang F:
Single-cell transcriptomics in cancer: Computational challenges and
opportunities. Exp Mol Med. 52:1452–1465. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu T, Park S, Zhu J, Wang Y, Zhan X, Wang
X, Wang L, Zhu H and Wang T: Overcoming expressional drop-outs in
lineage reconstruction from single-cell RNA-sequencing data. Cell
Rep. 34:1085892021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang JY and Doudna JA: CRISPR technology:
A decade of genome editing is only the beginning. Science.
379:eadd86432023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Raj B, Wagner DE, McKenna A, Pandey S,
Klein AM, Shendure J, Gagnon JA and Schier AF: Simultaneous
single-cell profiling of lineages and cell types in the vertebrate
brain. Nat Biotechnol. 36:442–450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
McKenna A, Findlay GM, Gagnon JA, Horwitz
MS, Schier AF and Shendure J: Whole-organism lineage tracing by
combinatorial and cumulative genome editing. Science.
353:aaf79072016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Raj B, Gagnon JA and Schier AF:
Large-scale reconstruction of cell lineages using single-cell
readout of transcriptomes and CRISPR-Cas9 barcodes by scGESTALT.
Nat Protoc. 13:2685–2713. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Simeonov KP, Byrns CN, Clark ML, Norgard
RJ, Martin B, Stanger BZ, Shendure J, McKenna A and Lengner CJ:
Single-cell lineage tracing of metastatic cancer reveals selection
of hybrid EMT states. Cancer Cell. 39:1150–1162.e9. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bowling S, Sritharan D, Osorio FG, Nguyen
M, Cheung P, Rodriguez-Fraticelli A, Patel S, Yuan WC, Fujiwara Y,
Li BE, et al: An engineered CRISPR-Cas9 mouse line for simultaneous
readout of lineage histories and gene expression profiles in single
cells. Cell. 181:1410–1422.e27. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chan MM, Smith ZD, Grosswendt S, Kretzmer
H, Norman TM, Adamson B, Jost M, Quinn JJ, Yang D, Jones MG, et al:
Molecular recording of mammalian embryogenesis. Nature. 570:77–82.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Quinn JJ, Jones MG, Okimoto RA, Nanjo S,
Chan MM, Yosef N, Bivona TG and Weissman JS: Single-cell lineages
reveal the rates, routes, and drivers of metastasis in cancer
xenografts. Science (New York, NY). 371:eabc19442021. View Article : Google Scholar
|
|
58
|
Yang D, Jones MG, Naranjo S, Rideout WM
III, Min KHJ, Ho R, Wu W, Replogle JM, Page JL, Quinn JJ, et al:
Lineage tracing reveals the phylodynamics, plasticity, and paths of
tumor evolution. Cell. 185:1905–1923.e25. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie L, Liu H, You Z, Wang L, Li Y, Zhang
X, Ji X, He H, Yuan T, Zheng W, et al: Comprehensive spatiotemporal
mapping of single-cell lineages in developing mouse brain by
CRISPR-based barcoding. Nat Methods. 20:1244–1255. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Alemany A, Florescu M, Baron CS,
Peterson-Maduro J and Van Oudenaarden A: Whole-organism clone
tracing using single-cell sequencing. Nature. 556:108–112. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Allen F, Crepaldi L, Alsinet C, Strong AJ,
Kleshchevnikov V, De Angeli P, Páleníková P, Khodak A, Kiselev V,
Kosicki M, et al: Predicting the mutations generated by repair of
Cas9-induced double-strand breaks. Nat Biotechnol. Nov
27–2018.(Epub ahead of print).
|
|
62
|
Schmidt ST, Zimmerman SM, Wang J, Kim SK
and Quake SR: Quantitative analysis of synthetic cell lineage
tracing using nuclease barcoding. ACS Synth Biol. 6:936–942. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Varanasi A and Wilson E: The Applications
of the CRISPR/Cas9 Gene-Editing System in Treating Human Diseases.
J Stud Res. 11:1–17. 2022. View Article : Google Scholar
|
|
64
|
Choubisa V and Sharma V: Unveiling neural
network potential in forecasting CRISPR effects and off-target
prophecies for gene editing. Int J Sci Res Arch. 10:252–259. 2023.
View Article : Google Scholar
|
|
65
|
Moreno-Ayala R and Junker JP: Single-cell
genomics to study developmental cell fate decisions in zebrafish.
Brief Funct Genomics. Mar 30–2021.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yabe IM, Truitt LL, Espinoza DA, Wu C,
Koelle S, Panch S, Corat MAF, Winkler T, Yu KR, Hong SG, et al:
Barcoding of macaque hematopoietic stem and progenitor cells: A
robust platform to assess vector genotoxicity. Mol Ther Methods
Clin Dev. 11:143–154. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Amrani N, Gao XD, Liu P, Edraki A, Mir A,
Ibraheim R, Gupta A, Sasaki KE, Wu T, Donohoue PD, et al: NmeCas9
is an intrinsically high-fidelity genome-editing platform. Genome
Biol. 19:2142018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao L, Liu Z, Levy SF and Wu S:
Bartender: A fast and accurate clustering algorithm to count
barcode reads. Bioinformatics. 34:739–747. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Johnson MS, Venkataram S and Kryazhimskiy
S: Best practices in designing, sequencing, and identifying random
DNA barcodes. J Mol Evol. 91:263–280. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Störtz F and Minary P: crisprSQL: A novel
database platform for CRISPR/Cas off-target cleavage assays.
Nucleic Acids Res. 49((D1)): D855–D861. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Cradick TJ, Qiu P, Lee CM, Fine EJ and Bao
G: COSMID: A web-based tool for identifying and validating
CRISPR/Cas off-target sites. Mol Ther Nucleic Acids. 3:e2142014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bystrykh LV and Belderbos ME: Clonal
analysis of cells with cellular barcoding: When numbers and sizes
matter. Methods Mol Biol. 1516:57–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rodriguez-Fraticelli AE, Wolock SL,
Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA,
Klein AM and Camargo FD: Clonal analysis of lineage fate in native
haematopoiesis. Nature. 553:212–216. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bramlett C, Jiang D, Nogalska A, Eerdeng
J, Contreras J and Lu R: Clonal tracking using embedded viral
barcoding and high-throughput sequencing. Nat Protoc. 15:1436–1458.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ordon J, Thouin J, Nakano RT, et al:
Simultaneous tracking of near-isogenic bacterial strains in
synthetic Arabidopsis microbiota by chromosomally-integrated
barcodes. 2023.
|
|
76
|
Leibovich N and Goyal S: Limitations and
Optimizations of Cellular Lineages Tracking. biorxiv. doi:.
https://doi.org/10.1101/2023.03.15.532767
|
|
77
|
Frenkel M, Hujoel MLA, Morris Z and Raman
S: Discovering chromatin dysregulation induced by protein-coding
perturbations at scale. bioRxiv. doi:. https://doi.org/10.1101/2023.09.20.555752
|
|
78
|
Blois S, Goetz BM, Bull JJ and Sullivan
CS: Interpreting and de-noising genetically engineered barcodes in
a DNA virus. PLoS Comput Biol. 18:e10101312022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Anwar SL, Wulaningsih W and Lehmann U:
Transposable elements in human cancer: Causes and consequences of
deregulation. Int J Mol Sci. 18:9742017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ohigashi I, Yamasaki Y, Hirashima T and
Takahama Y: Identification of the transgenic integration site in
immunodeficient tgε26 human CD3ε transgenic mice. PLoS One.
5:e143912010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wolff JH and Mikkelsen JG: Delivering
genes with human immunodeficiency virus-derived vehicles: Still
state-of-the-art after 25 years. J Biomed Sci. 29:792022.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Porter SN, Baker LC, Mittelman D and
Porteus MH: Lentiviral and targeted cellular barcoding reveals
ongoing clonal dynamics of cell lines in vitro and in vivo. Genome
Biol. 15:R752014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kivioja T, Vähärautio A, Karlsson K, Bonke
M, Enge M, Linnarsson S and Taipale J: Counting absolute numbers of
molecules using unique molecular identifiers. Nat Methods. 9:72–74.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Fan HC, Fu GK and Fodor SP: Combinatorial
labeling of single cells for gene expression cytometry. Science.
347:12583672015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hashimshony T, Wagner F, Sher N and Yanai
I: CEL-Seq: Single-cell RNA-Seq by multiplexed linear
amplification. Cell Rep. 2:666–673. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Picelli S, Björklund ÅK, Faridani OR,
Sagasser S, Winberg G and Sandberg R: Smart-seq2 for sensitive
full-length transcriptome profiling in single cells. Nat Methods.
10:1096–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z,
Huang Y and Wang J: Comparative analysis of droplet-based
ultra-high-throughput single-cell RNA-Seq systems. Mol Cell.
73:130–142.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zheng GX, Terry JM, Belgrader P, Ryvkin P,
Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et
al: Massively parallel digital transcriptional profiling of single
cells. Nat Commun. 8:140492017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Rotem A, Ram O, Shoresh N, Sperling RA,
Schnall-Levin M, Zhang H, Basu A, Bernstein BE and Weitz DA:
High-throughput single-cell labeling (Hi-SCL) for RNA-Seq using
drop-based microfluidics. PLoS One. 10:e01163282015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Pinglay S, Lalanne JB, Daza RM, Koeppel J,
Li X, Lee DS and Shendure J: Multiplex generation and single-cell
analysis of structural variants in mammalian genomes. Science.
387:eado59782025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lan F, Demaree B, Ahmed N and Abate AR:
Single-cell genome sequencing at ultra-high-throughput with
microfluidic droplet barcoding. Nat Biotechnol. 35:640–646. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li J, Zhang R, Li T, et al:
Ultra-high-throughput microbial single-cell whole genome sequencing
for genome-resolved metagenomics. 2022. View Article : Google Scholar
|
|
93
|
Shembekar N, Chaipan C, Utharala R and
Merten CA: Droplet-based microfluidics in drug discovery,
transcriptomics and high-throughput molecular genetics. Lab Chip.
16:1314–1331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Suea-Ngam A, Howes PD, Srisa-Art M and
deMello AJ: Droplet microfluidics: From proof-of-concept to
real-world utility? Chem Commun (Camb). 55:9895–9903. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sheng C, Lopes R, Li G, et al:
Probabilistic machine learning ensures accurate ambient denoising
in droplet-based single-cell omics. 2022. View Article : Google Scholar
|
|
96
|
Imoto Y: Comprehensive Noise Reduction in
Single-Cell Data with the RECODE Platform. biorxiv. doi:.
https://doi.org/10.1101/2024.04.18.590054
|
|
97
|
Sun C, Kathuria K, Emery SB, Kim B,
Burbulis IE, Shin JH; Brain Somatic Mosaicism Network, ; Weinberger
DR, Moran JV, Kidd JM, et al: Mapping recurrent mosaic copy number
variation in human neurons. Nat Commun. 15:42202024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bo L, Liang Y and Lin G: Cancer
classification based on multiple dimensions: SNV patterns. Comput
Biol Med. 151((Pt A)): 1062702022.PubMed/NCBI
|
|
99
|
McConnell MJ, Lindberg MR, Brennand KJ,
Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS,
Vermeesch JR, Hall IM and Gage FH: Mosaic copy number variation in
human neurons. Science. 342:632–637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Addis L, Ahn JW, Dobson R, Dixit A,
Ogilvie CM, Pinto D, Vaags AK, Coon H, Chaste P, Wilson S, et al:
Microdeletions of ELP4 are associated with language impairment,
autism spectrum disorder, and mental retardation. Hum Mutat.
36:842–850. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xiong E, Liu P, Deng R, Zhang K, Yang R
and Li J: Recent advances in enzyme-free and enzyme-mediated
single-nucleotide variation assay in vitro. Natl Sci Rev.
11:nwae1182024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kadam PS, Yang Z, Lu Y, Zhu H, Atiyas Y,
Shah N, Fisher S, Nordgren E, Kim J, Issadore D and Eberwine J:
Single-mitochondrion sequencing uncovers distinct mutational
patterns and heteroplasmy landscape in mouse astrocytes and
neurons. BMC Biol. 22:1622024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Dondi A, Borgsmüller N, Ferreira PF, Haas
BJ, Jacob F and Heinzelmann-Schwarz V; Tumor Profiler Consortium;
Beerenwinkel N, : De novo detection of somatic variants in
high-quality long-read single-cell RNA sequencing data. bioRxiv
[Preprint]. 2024.03.06.583775. 2024.
|
|
104
|
Ganz J, Luquette LJ, Bizzotto S, Miller
MB, Zhou Z, Bohrson CL, Jin H, Tran AV, Viswanadham VV, McDonough
G, et al: Contrasting somatic mutation patterns in aging human
neurons and oligodendrocytes. Cell. 187:1955–1970.e23. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Csordas A, Sipos B, Kurucova T, Volfova A,
Zamola F, Tichy B and Hicks DG: Cell Tree Rings: The structure of
somatic evolution as a human aging timer. Geroscience.
46:3005–3019. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
You X, Cao Y, Suzuki T, Shao J, Zhu B,
Masumura K, Xi J, Liu W, Zhang X and Luan Y: Genome-wide direct
quantification of in vivo mutagenesis using high-accuracy
paired-end and complementary consensus sequencing. Nucleic Acids
Res. 51:e1092023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Weng C, Weissman JS and Sankaran VG:
Robustness and reliability of single-cell regulatory multi-omics
with deep mitochondrial mutation profiling. bioRxiv [Preprint].
2024.08.23.609473. 2024.
|
|
108
|
Evrony GD, Lee E, Mehta BK, Benjamini Y,
Johnson RM, Cai X, Yang L, Haseley P, Lehmann HS, Park PJ and Walsh
CA: Cell lineage analysis in human brain using endogenous
retroelements. Neuron. 85:49–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Redon R, Ishikawa S, Fitch KR, Feuk L,
Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et
al: Global variation in copy number in the human genome. Nature.
444:444–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wei CJ and Zhang K: RETrace: Simultaneous
retrospective lineage tracing and methylation profiling of single
cells. Genome Res. 30:602–610. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fink J, Andersson-Rolf A and Koo BK: Adult
stem cell lineage tracing and deep tissue imaging. BMB Rep.
48:655–667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Woodworth MB, Girskis KM and Walsh CA:
Building a lineage from single cells: Genetic techniques for cell
lineage tracking. Nat Rev Genet. 18:230–244. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Li L, Bowling S, McGeary SE, Yu Q, Lemke
B, Alcedo K, Jia Y, Liu X, Ferreira M, Klein AM, et al: A mouse
model with high clonal barcode diversity for joint lineage,
transcriptomic, and epigenomic profiling in single cells. Cell.
186:5183–5199.e22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Walsh C and Cepko CL: Widespread
dispersion of neuronal clones across functional regions of the
cerebral cortex. Science. 255:434–440. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gerrits A, Dykstra B, Kalmykowa OJ, Klauke
K, Verovskaya E, Broekhuis MJ, de Haan G and Bystrykh LV: Cellular
barcoding tool for clonal analysis in the hematopoietic system.
Blood. 115:2610–2618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Golden JA, Fields-Berry SC and Cepko CL:
Construction and characterization of a highly complex retroviral
library for lineage analysis. Proc Natl Acad Sci USA. 92:5704–5708.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Khozyainova AA, Valyaeva AA, Arbatsky MS,
Isaev SV, Iamshchikov PS, Volchkov EV, Sabirov MS, Zainullina VR,
Chechekhin VI, Vorobev RS, et al: Complex analysis of single-cell
RNA sequencing data. Biochemistry (Mosc). 88:231–252. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Weinreb C, Rodriguez-Fraticelli A, Camargo
FD and Klein AM: Lineage tracing on transcriptional landscapes
links state to fate during differentiation. Science.
367:eaaw33812020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bizzotto S, Dou Y, Ganz J, Doan RN, Kwon
M, Bohrson CL, Kim SN, Bae T, Abyzov A; NIMH Brain Somatic
Mosaicism Network, ; et al: Landmarks of human embryonic
development inscribed in somatic mutations. Science. 371:1249–1253.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sun J, Ramos A, Chapman B, Johnnidis JB,
Le L, Ho YJ, Klein A, Hofmann O and Camargo FD: Clonal dynamics of
native haematopoiesis. Nature. 514:322–327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kim IS, Wu J, Rahme GJ, Battaglia S, Dixit
A, Gaskell E, Chen H, Pinello L and Bernstein BE: Parallel
single-cell RNA-Seq and genetic recording reveals lineage decisions
in developing embryoid bodies. Cell Rep. 33:1082222020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wagenblast E, Soto M, Gutiérrez-Ángel S,
Hartl CA, Gable AL, Maceli AR, Erard N, Williams AM, Kim SY,
Dickopf S, et al: A model of breast cancer heterogeneity reveals
vascular mimicry as a driver of metastasis. Nature. 520:358–362.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Akimov Y, Bulanova D, Timonen S,
Wennerberg K and Aittokallio T: Improved detection of
differentially represented DNA barcodes for high-throughput clonal
phenomics. Mol Syst Biol. 16:e91952020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
McKenna A and Gagnon JA: Recording
development with single cell dynamic lineage tracing. Development.
146:dev1697302019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang MY, Zhou Y, Lai GS, Huang Q, Cai WQ,
Han ZW, Wang Y, Ma Z, Wang XW, Xiang Y, et al: DNA barcode to trace
the development and differentiation of cord blood stem cells
(Review). Mol Med Rep. 24:8492021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Petropoulos S, Edsgärd D, Reinius B, Deng
Q, Panula SP, Codeluppi S, Plaza Reyes A, Linnarsson S, Sandberg R
and Lanner F: Single-cell RNA-Seq reveals lineage and X chromosome
dynamics in human preimplantation embryos. Cell. 165:1012–1026.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Farrell JA, Wang Y, Riesenfeld SJ, Shekhar
K, Regev A and Schier AF: Single-cell reconstruction of
developmental trajectories during zebrafish embryogenesis. Science.
360:eaar31312018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
McFarland JM, Paolella BR, Warren A,
Geiger-Schuller K, Shibue T, Rothberg M, Kuksenko O, Colgan WN,
Jones A, Chambers E, et al: Multiplexed single-cell transcriptional
response profiling to define cancer vulnerabilities and therapeutic
mechanism of action. Nat Commun. 11:42962020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Birnbaum KD: Power in numbers: Single-cell
RNA-Seq strategies to dissect complex tissues. Annu Rev Genet.
52:203–221. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lan F, Saba J, Ross TD, Zhou Z, Krauska K,
Anantharaman K, Landick R and Venturelli OS: Massively parallel
single-cell sequencing of diverse microbial populations. Nat
Methods. 21:228–235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Cusanovich DA, Daza R, Adey A, Pliner HA,
Christiansen L, Gunderson KL, Steemers FJ, Trapnell C and Shendure
J: Multiplex single-cell profiling of chromatin accessibility by
combinatorial cellular indexing. Science. 348:910–914. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhu C, Yu M, Huang H, Juric I, Abnousi A,
Hu R, Lucero J, Behrens MM, Hu M and Ren B: An ultra
high-throughput method for single-cell joint analysis of open
chromatin and transcriptome. Nat Struct Mol Biol. 26:1063–1070.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Wu B, Bennett HM, Ye X, Sridhar A,
Eidenschenk C, Everett C, Nazarova EV, Chen HH, Kim IK, Deangelis
M, et al: Overloading And unpacKing (OAK)-droplet-based
combinatorial indexing for ultra-high throughput single-cell
multiomic profiling. Nat Commun. 15:91462024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Mathur L, Szalai B, Du NH, Utharala R,
Ballinger M, Landry JJM, Ryckelynck M, Benes V, Saez-Rodriguez J
and Merten CA: Combi-seq for multiplexed transcriptome-based
profiling of drug combinations using deterministic barcoding in
single-cell droplets. Nat Commun. 13:44502022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Paddison PJ, Silva JM, Conklin DS,
Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L,
Scobie K, et al: A resource for large-scale RNA-interference-based
screens in mammals. Nature. 428:427–431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Silva JM, Marran K, Parker JS, Silva J,
Golding M, Schlabach MR, Elledge SJ, Hannon GJ and Chang K:
Profiling essential genes in human mammary cells by multiplex RNAi
screening. Science. 319:617–620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Datlinger P, Rendeiro AF, Schmidl C,
Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A,
Alpar D and Bock C: Pooled CRISPR screening with single-cell
transcriptome readout. Nat Methods. 14:297–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Duncombe TA and Dittrich PS: Droplet
barcoding: Tracking mobile micro-reactors for high-throughput
biology. Curr Opin Biotechnol. 60:205–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Adamson B, Norman TM, Jost M, Cho MY,
Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et
al: A multiplexed single-cell CRISPR screening platform enables
systematic dissection of the unfolded protein response. Cell.
167:1867–1882.e21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Jaitin DA, Weiner A, Yofe I, Lara-Astiaso
D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A
and Amit I: Dissecting immune circuits by linking CRISPR-pooled
screens with single-cell RNA-Seq. Cell. 167:1883–1896.e15. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Emanuel G, Moffitt JR and Zhuang X:
High-throughput, image-based screening of pooled genetic-variant
libraries. Nat Methods. 14:1159–1162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Camp JG, Platt R and Treutlein B: Mapping
human cell phenotypes to genotypes with single-cell genomics.
Science. 365:1401–1405. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Xie S, Duan J, Li B, Zhou P and Hon GC:
Multiplexed engineering and analysis of combinatorial enhancer
activity in single cells. Mol Cell. 66:285–299.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lawson MJ, Camsund D, Larsson J, Baltekin
Ö, Fange D and Elf J: In situ genotyping of a pooled strain library
after characterizing complex phenotypes. Mol Syst Biol. 13:9472017.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Marusyk A, Janiszewska M and Polyak K:
Intratumor heterogeneity: The rosetta stone of therapy resistance.
Cancer Cell. 37:471–484. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Shembrey C, Huntington ND and Hollande F:
Impact of tumor and immunological heterogeneity on the anti-cancer
immune response. Cancers (Basel). 11:12172019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lewis SM, Asselin-Labat ML, Nguyen Q,
Berthelet J, Tan X, Wimmer VC, Merino D, Rogers KL and Naik SH:
Spatial omics and multiplexed imaging to explore cancer biology.
Nat Methods. 18:997–1012. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Fennell KA, Vassiliadis D, Lam EYN,
Martelotto LG, Balic JJ, Hollizeck S, Weber TS, Semple T, Wang Q,
Miles DC, et al: Non-genetic determinants of malignant clonal
fitness at single-cell resolution. Nature. 601:125–131. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jin X, Demere Z, Nair K, Ali A, Ferraro
GB, Natoli T, Deik A, Petronio L, Tang AA, Zhu C, et al: A
metastasis map of human cancer cell lines. Nature. 588:331–336.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Holcar M, Ferdin J, Sitar S,
Tušek-Žnidarič M, Dolžan V, Plemenitaš A, Žagar E and Lenassi M:
Enrichment of plasma extracellular vesicles for reliable
quantification of their size and concentration for biomarker
discovery. Sci Rep. 10:213462020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Welsh JA, Goberdhan DCI, O'Driscoll L,
Buzas EI, Blenkiron C, Bussolati B, Cai H, Di Vizio D, Driedonks
TAP, Erdbrügger U, et al: Minimal information for studies of
extracellular vesicles (MISEV2023): From basic to advanced
approaches. J Extracell Vesicles. 13:e124042024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lozano-Andrés E, Enciso-Martinez A,
Gijsbers A, Ridolfi A, Van Niel G, Libregts SFWM, Pinheiro C, van
Herwijnen MJC, Hendrix A, Brucale M, et al: Physical association of
low density lipoprotein particles and extracellular vesicles
unveiled by single particle analysis. J Extracell Vesicles.
12:e123762023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Chang W-H, Cerione RA and Antonyak MA:
Extracellular Vesicles and Their Roles in Cancer Progression.
Cancer Cell Signaling. vol. 2174. Robles-Flores M: Springer US; New
York, NY: pp. 143–170. 2021, View Article : Google Scholar
|
|
154
|
Ferguson S, Yang KS and Weissleder R:
Single extracellular vesicle analysis for early cancer detection.
Trends Mol Med. 28:681–692. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Yu W, Hurley J, Roberts D, Chakrabortty
SK, Enderle D, Noerholm M, Breakefield XO and Skog JK:
Exosome-based liquid biopsies in cancer: opportunities and
challenges. Ann Oncol. 32:466–477. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Verweij FJ, Balaj L, Boulanger CM, Carter
DRF, Compeer EB, D'Angelo G, El Andaloussi S, Goetz JG, Gross JC,
Hyenne V, et al: The power of imaging to understand extracellular
vesicle biology in vivo. Nat Methods. 18:1013–1026. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Crescitelli R, Lässer C and Lötvall J:
Isolation and characterization of extracellular vesicle
subpopulations from tissues. Nat Protoc. 16:1548–1580. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ko J, Wang Y, Sheng K, Weitz DA and
Weissleder R: Sequencing-based protein analysis of single
extracellular vesicles. ACS Nano. 15:5631–5638. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ivanova A, Chalupska R, Louro AF, Firth M,
González-King Garibotti H, Hultin L, Kohl F, Lázaro-Ibáñez E,
Lindgren J, Musa G, et al: Barcoded hybrids of extracellular
vesicles and lipid nanoparticles for multiplexed analysis of tissue
distribution. Adv Sci (Weinh). 12:e24078502025. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Kunitake K, Mizuno T, Hattori K, Oneyama
C, Kamiya M, Ota S, Urano Y and Kojima R: Barcoding of small
extracellular vesicles with CRISPR-gRNA enables comprehensive,
subpopulation-specific analysis of their biogenesis and release
regulators. Nat Commun. 15:97772024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Farzad N, Enninful A, Bao S, Zhang D, Deng
Y and Fan R: Spatially resolved epigenome sequencing via Tn5
transposition and deterministic DNA barcoding in tissue. Nat
Protoc. 19:3389–3425. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Pandit S, Duchow M, Chao W, Capasso A and
Samanta D: DNA-barcoded plasmonic nanostructures for activity-based
protease sensing. Angew Chem Int Ed Engl. 63:e2023109642024.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Lareau CA, Ma S, Duarte FM and Buenrostro
JD: Inference and effects of barcode multiplets in droplet-based
single-cell assays. Nat Commun. 11:8662020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Hotinger JA, Campbell IW, Hullahalli K,
Osaki A and Waldor MK: Quantification of Salmonella enterica
serovar typhimurium population dynamics in murine infection using a
highly diverse barcoded library. Elife. 13:RP1013882024. View Article : Google Scholar
|
|
165
|
Karlsson F, Kallas T, Thiagarajan D,
Karlsson M, Schweitzer M, Navarro JF, Leijonancker L, Geny S,
Pettersson E, Rhomberg-Kauert J, et al: Molecular pixelation:
spatial proteomics of single cells by sequencing. Nat Methods.
21:1044–1052. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Eyler CE, Matsunaga H, Hovestadt V,
Vantine SJ, van Galen P and Bernstein BE: Single-cell lineage
analysis reveals genetic and epigenetic interplay in glioblastoma
drug resistance. Genome Biol. 21:1742020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Maetzig T, Morgan M and Schambach A:
Fluorescent genetic barcoding for cellular multiplex analyses. Exp
Hematol. 67:10–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Maetzig T, Ruschmann J, Sanchez Milde L,
Lai CK, Von Krosigk N and Humphries RK: Lentiviral Fluorescent
Genetic Barcoding for Multiplex Fate Tracking of Leukemic Cells.
Mol Ther Methods Clin Dev. 6:54–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Yoon B, Kim H, Jung SW and Park J:
Single-cell lineage tracing approaches to track kidney cell
development and maintenance. Kidney Int. 105:1186–1199. 2024.
View Article : Google Scholar : PubMed/NCBI
|