Introduction
Congenital heart disease (CHD) is the most prevalent
birth defect, markedly impacting neonatal health and affecting 2–8%
of newborns (1,2). CHD has been attributed to
environmental and genetic factors, with the former being considered
predominant (3). However,
advancements in technology have revealed a higher incidence of CHD
in identical twins and individuals with a family history of the
condition, and there is growing evidence that genetic factors serve
a crucial role in the pathogenesis of CHD (4,5).
It is estimated that 400 genes are associated with
CHD, including those encoding transcription factors, cell signaling
molecules, and structural proteins involved in heart development,
as well as proteins associated with ciliary movement; examples
include members of the GATA-binding protein, T-box transcription
factor, forkhead box and dynein axonemal heavy chain (DNAH)
families (4). The normal function
of cilia is necessary for heart development, with key genes
involved including those encoding components of the outer and inner
dynein arms. Defects in these genes are typically associated with
left-right pattern abnormalities and cardiac asymmetry (6). Notable examples include EF hand
calcium binding domain (7),
kinesin family member 3B (8) and
DNAH9. The protein encoded by DNAH9 is an outer
dynein arm component crucial for ciliary movement in embryonic
nodes. Disruption of DNAH9 can impair human development,
potentially leading to abnormal organ alignment along the vertical
axis of the body. To date, biallelic mutations in DNAH9 have
been identified in 24 patients, who presented with non-syndromic
asthenozoospermia (9,10), CHD (10–13),
non-syndromic respiratory diseases (10,14–16)
and heterotaxy (12,17). Among the CHD patients, only a
14-year-old boy presented with isolated CHD (F6-II in ref.11), and
it was unclear whether that patient also had asthenozoospermia
(11). Therefore, the relationship
between DNAH9 and non-syndromic CHD remains unclear.
The present study identified a compound heterozygous
mutation in the DNAH9 gene [NM_001372.3: c.3743+1G>T;
c.11176C>T (p.Arg3726Trp)] in a fetus with complex CHD. The
c.3743+1G>T mutation is novel and its potential pathogenicity
was assessed in vitro. This research expanded the mutation
spectrum of the DNAH9 gene and provides a valuable
foundation for genetic counseling in families affected by this
condition.
Materials and methods
Patients
A naturally conceiving couple comprising a
23-year-old male and a 23-year-old female was recruited for the
present study in September 2023 at the Department of Maternity of
The First Hospital of Changsha (Changsha, China). Early pregnancy
tests, including Down syndrome screening, nuchal translucency
screening and noninvasive prenatal testing returned normal results,
as did tests for the infectious diseases (HIV, hepatitis B and
Treponema pallidum). At 24 weeks and 1 day of pregnancy, an
ultrasound examination revealed abnormal sonographic images of the
fetal heart, including dextroversion of the heart, complete
atrioventricular septal defect, subcardiac total pulmonary venous
drainage, pulmonary atresia, arterial catheter blood supply and
C-type collateral circulation (Fig.
1), raising the suspicion of complex CHD. The positions of the
stomach, liver and spleen of the fetus, a male, were all normal.
The pregnant woman later underwent an ultrasound examination at the
Obstetrics Department of The Second Xiangya Hospital of Central
South University (Changsha, China), and the results were consistent
with those obtained at The First Hospital of Changsha. Medical
professionals in the Department of Cardiac Surgery of The Second
Xiangya Hospital of Central South University (Changsha, China)
assessed the situation and recommended early surgical intervention
after birth, despite the high surgical risks and uncertain outcomes
associated with the procedure. After careful consideration, the
pregnant woman and her family chose to terminate the pregnancy and
sought to identify a genetic cause. Following an induced abortion,
tissue samples from the fetus were collected at The First Hospital
of Changsha for copy number variation sequencing and whole exome
sequencing (WES) at BGI Genomics Co., Ltd. No chromosomal
aneuploidy variants or microdeletions/microduplications known to be
clearly pathogenic or suspected to be pathogenic were detected. The
present study was approved by the Ethics Committee of The First
Hospital of Changsha and the family members signed an informed
consent form.
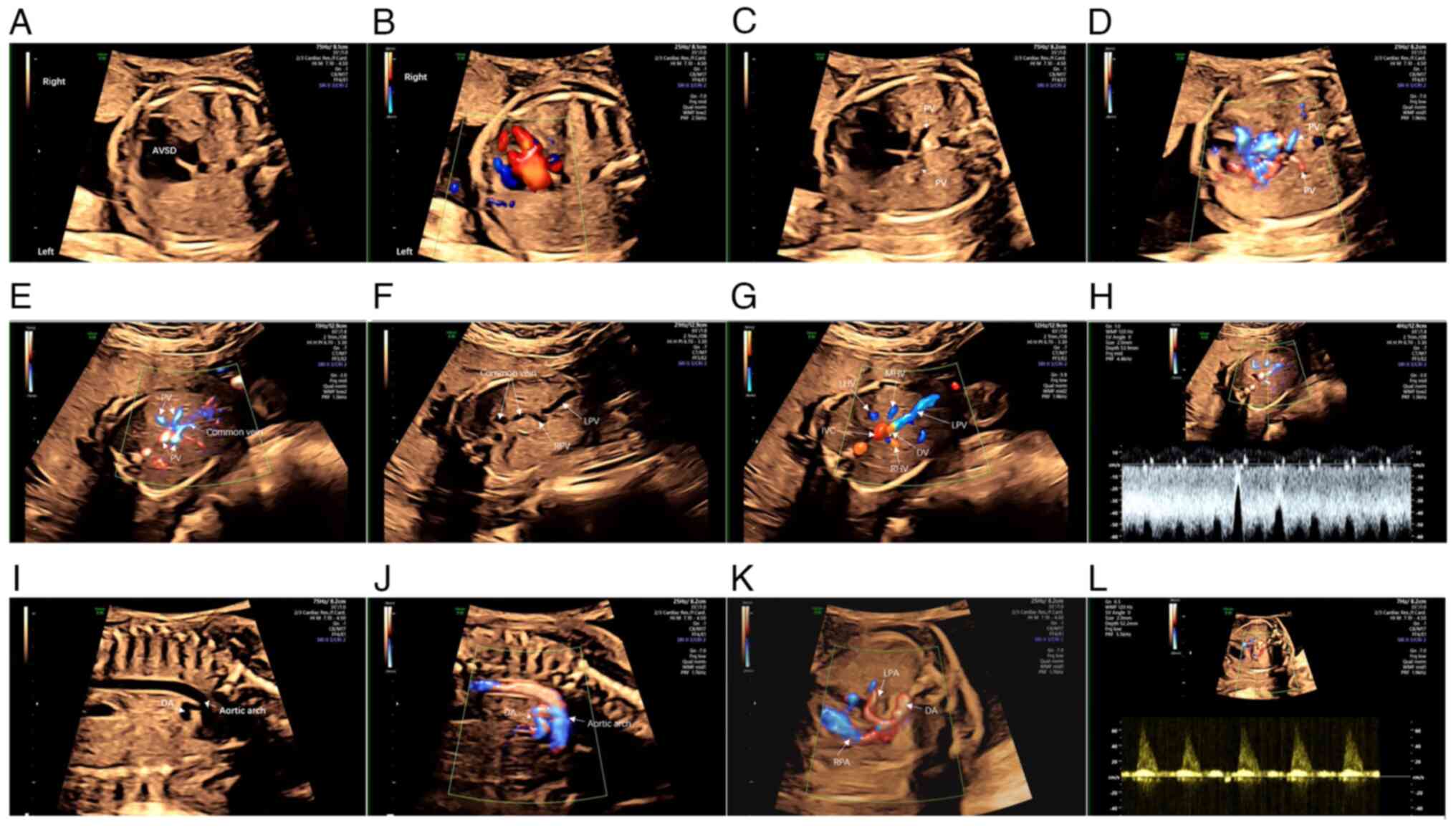 | Figure 1.Cardiac ultrasonography of the
proband reveals a complex congenital heart disorder. (A) In the
four-chamber view of the heart, the apices are oriented toward the
right side of the fetus, and the lower portion of the atrial septum
and the upper section of the ventricular septum are absent, the
width of the defect is ~7 mm. (B) Color Doppler flow imaging
confirms the defect. (C and D) Two PVs fail to drain into the
atrium. (E) Four PVs form a common vein. (F) The common vein
descends and merges with the RPV. (G) The LPV gives rise to a DV
and merges with the LHV, MHV and RHV, ultimately draining into the
IVC. (H) Blood flow spectrum analysis reveals a flow typically
associated with a venous catheter. (I-J) A thick artery is visible
on the left side of the heart. It ascends for a considerable
distance before (K) branching into a vessel in the neck, which then
curves downward and further divides into the RPA and LPA that
supply the right and left lungs, respectively. (L) Blood flow
spectrum analysis confirms that the vessel is a pulmonary artery.
PV, pulmonary vein; AVSD, atrioventricular septal defect; RPV,
right branch of the portal vein; LPV, left branch of the portal
vein; DV, duct vein; LHV, left hepatic vein; MHV, middle hepatic
vein; RHV, right hepatic vein; IVC, inferior vena cava; DA, ductus
arteriosus; RPA, right pulmonary artery; LPA, left pulmonary
artery. |
WES
The genomic DNA of the male proband was extracted
from fetal tissue obtained following pregnancy termination using a
MagPure Buffy Coat DNA Midi KF Kit according to the instructions
provided by the manufacturer (Magen Biotechnology Co., Ltd.).
Shearing enzyme (Enzymatics) and magnetic beads (VAHTS DNA Clean
Beads; cat. no. N411; Vazyme) were used to fragment and purify
genomic DNA to obtain fragments of 200–300 bp. Pre-PCR
amplification was then performed to complete library construction.
The loading concentration of the final library was 29.2 ng/ml. The
DNA of target gene exons and adjacent splicing regions was captured
and enriched using the Roche KAPA HyperExome probe set (cat. no.
9718630001; Roche Diagnostics, Ltd.). Gel electrophoresis was used
to assess the integrity of DNA, with high-quality DNA displaying a
single, clear band. Sequencing Reaction General Kit (T7 SM FCL
PE100) v2.0 (cat. no. 1000028455; MGI Tech Co., Ltd.) was used on
the MGISEQ-2000 platform (MGI Tech Co., Ltd.) for sequencing. The
quality of the raw sequencing data was assessed using SOAPnuke
software v2.1.2 (18), and the
clean reads were aligned to the hg19 reference genome using
Burrows-Wheeler Aligner software BWA-0.7.17 (r1188) (19). Single nucleotide variants and
insertions/deletions were identified using the Genome Analysis
Toolkit (20), generating results
for base polymorphisms in the target region. Subsequently, 1,000
GEA, 1,000 Genomes (East Asian; www.internationalgenome.org/home), gnomAD_exome (East
Asian; http://gnomad.broadinstitute.org/) and the Exome
Aggregation Consortium (ExAC; East Asian; http://exac.broadinstitute.org), were used to identify
the frequency and context of the identified mutations. The
BGI-varanno algorithm (21,22)
(BGICG_ANNO 0.39; BGI Genomics Co., Ltd) was employed for variant
screening and annotation, with the integration of disease data from
Clin-Var (https://www.ncbi.nlm.nih.gov/clinvar/), Online
Mendelian Inheritance in Man (https://www.omim.org/) and the Human Gene Mutation
Database (http://www.hgmd.cf.ac.uk/). Variant
filtering was performed as described in our previous study
(20), and the American College of
Medical Genetics and Genomics (ACMG)/Association for Molecular
Pathology guidelines for mutation pathogenicity were referred to
for interpretation (23,24). PCR was performed to amplify the
target regions containing the putative variants in the fetus and
the parents: DNA was extracted using the QIAamp DNA Blood Mini Kit
(Qiagen GmbH), PCR amplification using GoTaq Green Master Mix
(Promega Corporation) under the following conditions: 95°C for 30
sec (denaturation), 59°C for 30 sec (annealing) and 72°C for 30 sec
(extension), for a total of 35 cycles, the PCR products were
analyzed via gel (2% agarose) electrophoresis to confirm the
successful amplification of the target regions. Finally, the PCR
products were directly sequenced using Sanger sequencing. The
primers used are listed in Table
SI.
Bioinformatics analysis
Evolutionary conservation analysis was conducted by
aligning the amino acid sequences of DNAH9 proteins across various
species using the BLAST tool available on the NCBI website
(https://blast.ncbi.nlm.nih.gov/Blast.cgi). The
potential pathogenicity of the DNAH9 mutations was assessed
through in silico analysis utilizing four online tools:
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster
(v2021 for GRCh37; http://www.mutationtaster.org/), Sorting Intolerant
from Tolerant (SIFT; http://sift.bii.a-star.edu.sg/) and Combined
Annotation Dependent Depletion (CADD; v1.7; http://cadd.gs.washington.edu/). The RNA splicing
prediction model from the Rare Disease Data Center (RDDC) tool of
the Guangzhou Rare Disease Gene Therapy Alliance (https://rddc.tsinghua-gd.org/tool) was used to
forecast splicing variations. The mutated sequence was derived from
the prediction results. Protein structural analysis of the
DNAH9 (NM_001372.3) mutant was performed using the online
SWISS-MODEL tool (https://swissmodel.expasy.org), using sequences
identified through bioinformatics and minigene experimental
products of the splice mutation. Protein structure visualization
was performed using PyMol (v3.1.0a0 Open-Source; http://github.com/cgohlke/pymol-open-source-wheels/releases).
Minigene analysis
Due to the absence or low levels of DNAH9 in
peripheral blood, obtaining DNAH9 cDNA from this source for
splicing pattern analysis was not feasible. Therefore, minigene
technology was used to analyze the splicing pattern of the
c.3743+1G>T mutation, which is located in intron 19.
Bioinformatics analysis using the RDDC tool predicted that this
mutation could potentially affect the splicing of exon 19. To
investigate the splicing pattern, the target fragment containing
exons 18, 19, and 20, along with a partial sequence (150–200 bp) of
intron 20, was integrated into a pCMV-MYC vector (Takara
Biotechnology Co., Ltd.) using homologous recombination technology.
Briefly, primers with homologous arms were designed to amplify the
target fragment, which then carried overlapping sequences
homologous to the ends of the linearized vector. The integration
was achieved through a homologous recombination reaction
(ClonExpress MultiS One Step Cloning Kit; Vazyme). Genomic DNA from
a control individual and the proband were used as templates to
amplify the target sequence, with the necessary primers listed in
Table SI. PrimerSTAR MAX DNA
Polymerase (Takara Biotechnology Co., Ltd.) was used for
amplification under the following thermal cycling conditions: 98°C
for 10 sec (denaturation), 58°C for 5 sec (annealing) and 72°C for
30 sec (extension), for a total of 30 cycles. After the plasmid
construction was completed, Sanger sequencing was performed on the
plasmid. The cells were cultured in a 6-well cell culture plate
(CellPro) containing medium composed of 90% DMEM, 10% fetal bovine
serum (FBS), and 1% penicillin-streptomycin mixture. They were then
incubated in a cell culture incubator at 37°C with 5%
CO2. When the cell density reached 60–70%, 2 µg of the
plasmid DNA was transfected into 293T cells using Neofect DNA
transfection reagent (Neofect). At two days following transfection,
cellular RNA was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed into cDNA under the following thermal cycling
conditions: 37°C for 15 min, 85°C for 5 sec (PrimeScript RT reagent
Kit with gDNA Eraser, Takara Biotechnology Co., Ltd.). The
resulting cDNA was then subjected to PCR amplification using GoTaq
Green Master Mix (Promega Corporation) under the following
conditions: 95°C for 30 sec (denaturation), 59°C for 30 sec
(annealing) and 72°C for 30 sec (extension), for a total of 35
cycles. The PCR products were subsequently analyzed by Sanger
sequencing. The primers used for amplification are listed in
Table SI.
Results
WES and bioinformatics analysis
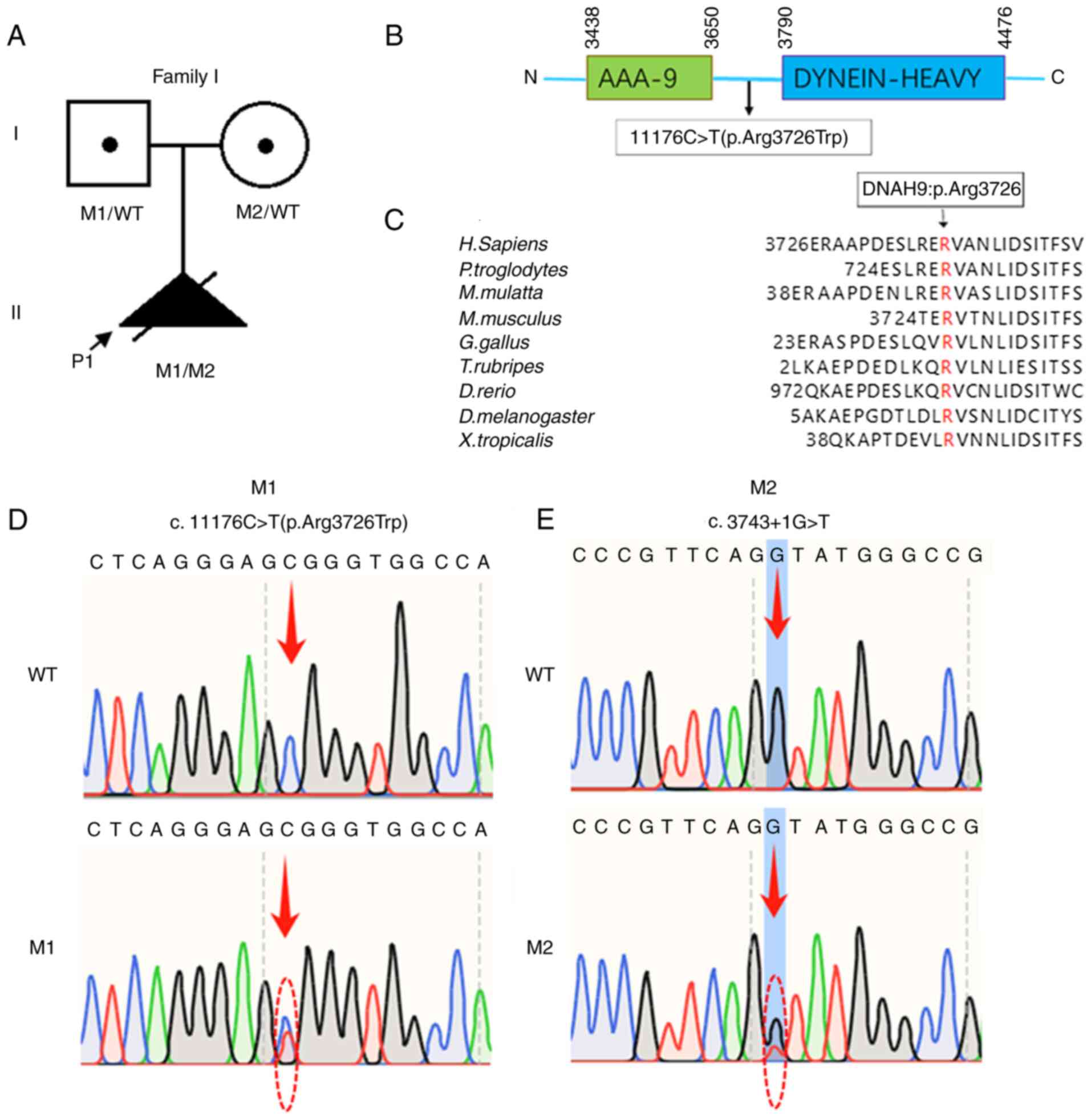
A compound heterozygous mutation in DNAH9
[NM_001372.3: c.11176C>T (p.Arg3726Trp); c.3743+1G>T] was
identified in the fetus by WES and Sanger sequencing. No pathogenic
mutations were identified in other genes known to be associated
with primary ciliary dyskinesia (PCD). The mother of the proband
was a carrier of the c.3743+ 1G>T mutation and the father was a
carrier of the c.11176C>T mutation (Fig. 2). The 1000 Genomes Project (East
Asian), ESP6500, gnomAD exome and ExAC databases indicate that the
c.11176C>T (p.Arg3726Trp) and c.3743+1G>T mutations are
extremely rare or absent, respectively, in the general population.
Bioinformatics analysis performed using PolyPhen-2, MutationTaster,
SIFT and CADD tools predicted both mutations to be pathogenic
(Table SII). The c.11176C>T
mutation occurs in a residue that is highly conserved across
species (Fig. 2C). In addition,
this mutation has been previously reported as pathogenic, causing
the downregulation of DNAH9 mRNA expression, ultrastructural
defects in ciliary outer dynein arms and reduced ciliary beating
frequency (11). The
c.3743+1G>T mutation is predicted to result in two aberrant
splicing events: Skipping of exon 19 (167 bp) and the insertion of
a 691-bp segment of intron 19. Compared with the structure of the
wild-type DNAH9 protein (Fig. 3A),
each aberrant splicing event is predicted to lead to a frameshift
and the production of truncated DNAH9 proteins (Fig. 3B). In addition, 3-dimensional (3D)
structure prediction indicates that the c.11176C>T
(p.Arg3726Trp) mutation will disrupt the hydrogen bond between the
arginine residue at position 3,726 and the glutamic acid residue at
position 3,594 (Fig. 3C, red
arrow). This disruption is expected to lead to the formation of an
α-helical structural segment (Fig.
3C, dashed red oval) not present in the wild-type protein
(Fig. 3C, dashed blue oval).
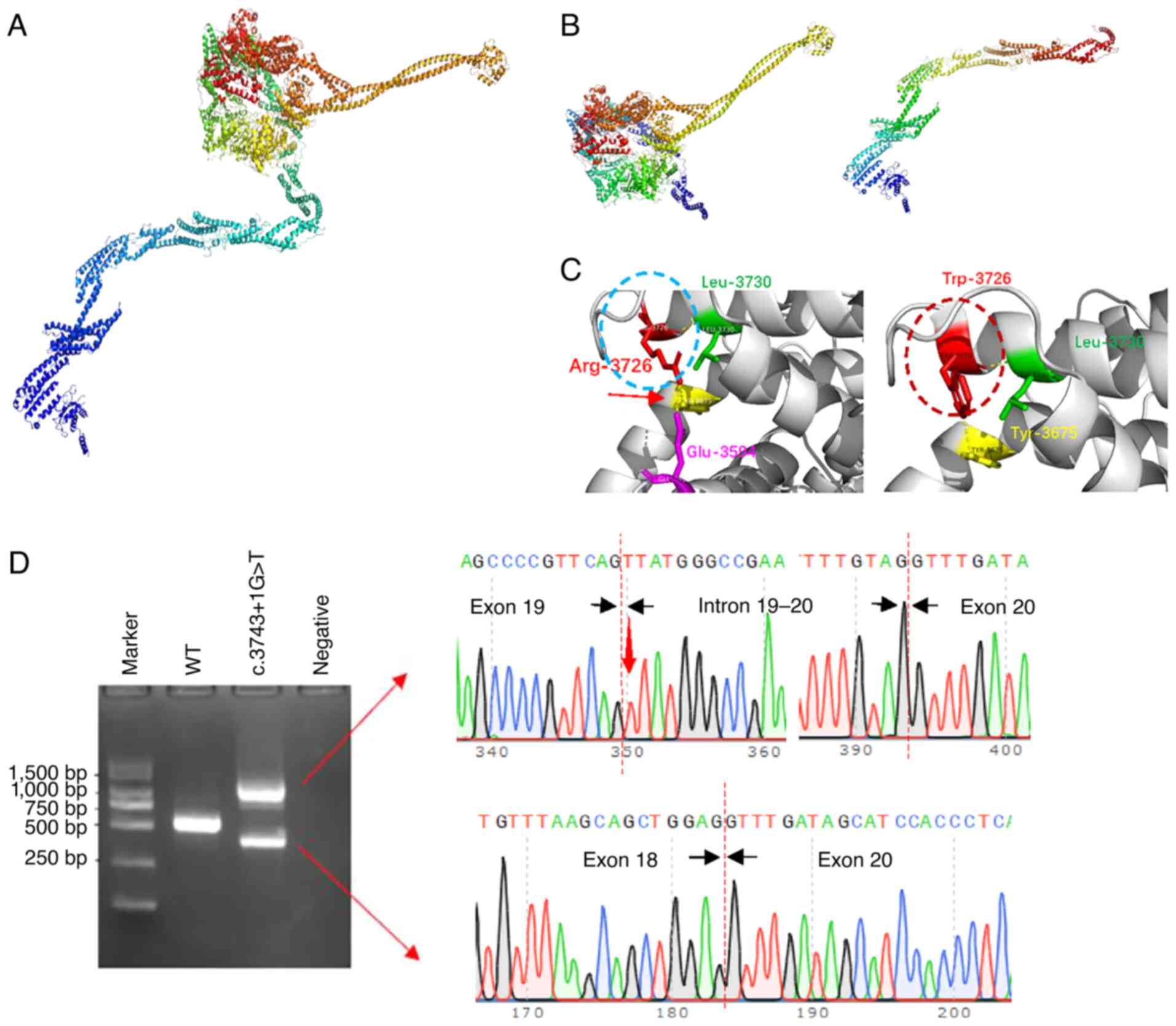 | Figure 3.Structural models of wild-type and
mutant DNAH9, and in vitro minigene analysis of the
c.3743+1G>T mutant. (A) Model of wild-type DNAH9 consists
of a tail comprising the N-terminus (marked in blue) and a
spherical head comprising the C-terminus. The head contains a motor
domain (marked in red, green and yellow) and microtubule binding
sites (marked in orange). (B) Two predicted protein structures
resulting from the splicing of c.3743+1G>T, one of which lacks
the N-terminal tail while the other does not have a spherical head.
(C) Local structure comparison at p.3726 between the wild-type and
c.11176 C>T (p.Arg3726Trp) mutant. In the mutant, the Arg at
position 3726 is replaced with Trp, resulting in the loss of a
hydrogen bond between Arg-3726 and Glu-3594, and the formation of a
new α-helical fragment (shown in the dashed red circle). (D)
Results of the in vitro minigene expression experiment show
that the c.3743+1G>T mutation produces two abnormal splicing
products. One splicing pattern of c.3743+1G>T was the insertion
of a 691-bp fragment of intron 19, leading to frameshift and
premature termination. The other was the deletion of the 167-bp
exon 19, leading to exon jumping, frameshift and premature
termination. DNAH9, dynein axonemal heavy chain 9; Arg,
arginine; Trp, tryptophan; Glu, glutamic acid; Leu, leucine; Tyr,
tyrosine. |
Minigene analysis and structure
prediction
Minigene analysis revealed that the c.3743+1G>T
mutation produced two expression products while the wild-type gene
produced only one (Fig. 3D), which
is consistent with the results of the bioinformatics analysis.
Sanger sequencing confirmed that the product with the higher
molecular weight contained a portion of intron 19 (Fig. 3D), while the product with the lower
molecular weight resulted from the skipping of exon 19 (Fig. 3D). The 3D structure predictions
were consistent with the bioinformatics forecasts, indicating that
the two abnormal splicing events lead to the production of two
truncated DNAH9 proteins, one lacking a tail and the other missing
the spherical head domain (Fig.
3B). According to ACMG guidelines, both the c.3743+1G>T and
c.11176C>T (p.Arg3726Trp) mutations are classified as likely
pathogenic.
Literature analysis
The 9 previous publications on DNAH9
mutations (9–17), describe findings for 24 patients in
total (Table SIII; Fig. S1). These include 10 patients who
presented with congenital heart disease (P3, P7-11, P13, P14, P18
and P20), 7 patients with respiratory diseases (P3-6, P21-P23), 3
patients with asthenozoospermia (P1, P2 and P6), and 5 patients who
exhibited only situs anomalies (P12, P15-17 and P19). In addition,
1 patient exhibited heterotaxy of abdominal organs and intrauterine
fetal death (P24); this patient also carried the RSPH1,
c.121G>A (p.G41R) mutation. P6 presented with both respiratory
diseases and asthenozoospermia, and P3 had both congenital heart
disease and respiratory diseases. Mutation sites associated with
different phenotypes were widely distributed across the
DNAH9 gene locus, with no obvious pattern discernible.
Discussion
The present study identified a compound heterozygous
mutation in the DNAH9 gene associated with structural heart
abnormalities. Multiple public databases, including gnomAD, ExAC
and the 1000 Genomes Project, indicate that the c.11176C>T
(p.Arg3726Trp) and c.3743+1G>T mutations are extremely rare in
the general population. The c.11176C>T mutation has previously
been reported as pathogenic, and minigene analysis suggests that
the c.3743+1G>T mutation is also pathogenic. No other family
members of the proband are known to have similar cardiac or other
ciliary-related diseases. A multidisciplinary consultation
involving cardiac surgeons, geneticists and obstetricians took
place to comprehensively evaluate the echocardiographic results of
the fetus. The experts unanimously concluded that the cardiac
abnormalities of the fetus were highly associated with the
DNAH9 gene mutations and recommended genetic counseling and
reproductive intervention.
In vertebrates, the fluid flow generated by the
ciliary movement of the embryonic node is crucial for the proper
positional distribution of organs. Ciliary defects, whether in
motile or primary cilia, can impair fluid flow or disrupt
left-right positional signaling, leading to abnormal organ
distribution (25–30). DNAH9, which is associated
with ciliary motility, may cause organ positional abnormalities
when defective. The analysis of 24 known cases with DNAH9
mutations in the present study revealed that 3 patients carrying
biallelic loss-of-function (LOF) mutations (P16-18) exhibited mild
symptoms, such as mirror-image distribution of organs, while 8
patients (P3, P7-11, P13 and P14) carried at least one missense
mutation manifesting as CHD. In the present study, a fetus carrying
biallelic DNAH9 mutations, specifically a LOF mutation and a
missense mutation, also presented with CHD. Therefore, we
hypothesize that biallelic LOF mutations, which result in a
complete loss of ciliary function, lead to mirror-image organ
distribution, whereas other mutations that partially impair ciliary
function lead to more severe phenotypes.
Previous research has demonstrated that biallelic
mutations in DNAH9 can result in asthenozoospermia,
respiratory diseases and CHD. The present study performed an
analysis of the mutation sites and phenotypes of DNAH9
mutations for 24 patients reported in nine articles; however, no
obvious association between the mutation sites and phenotypes was
identified (Fig. S1). This
suggests that the function of the DNAH9 gene is complex, and
the impact of its mutations on phenotypes may be influenced by
multiple factors. However, the small sample size of 24 patients is
a limitation of this analysis. Future research integrating gene
function studies with analyses of individual genetic backgounds is
necessary to more accurately reveal the relationship between
mutation sites and phenotypes.
When analyzing the WES results, all genes previously
reported to be responsible for PCD were considered as candidate
genes, including DNAH9. However, after mutation filtering,
only two mutations in DNAH9 were retained. Additionally,
ultrasound showed no positional abnormalities of PCD-related organs
such as the liver and spleen in the fetus in the present study.
However, the possibility that the fetus suffered from syndromic CHD
cannot be excluded. Since the fetal respiratory and reproductive
systems were not yet fully developed, it was not possible to assess
the impact of the mutations on these two systems through existing
clinical means. Therefore, it is necessary to develop novel
strategies to verify the effects of mutations on these two systems.
The lack of a differential diagnosis to rule out other potential
ciliary dysfunctions is a limitation of the present study.
Congenital heart defects resulting from genetic
abnormalities can be severe and often require medical management
and surgical intervention; they can even be life-threatening,
imposing a significant financial and psychological burden on
families. Reproductive interventions, such as preimplantation
genetic testing (PGT) and prenatal diagnosis, are essential tools
for preventing and managing birth defects by helping to avoid the
birth of children with CHD caused by genetic factors. The DNAH9
protein is an outer dynein arm protein that serves a crucial role
in the movement of embryonic nodal cilia and is essential for
cardiac development. The present study identified DNAH9
mutations as the underlying cause of CHD in a specific family,
providing the couple with the opportunity to have a healthy child
through PGT or prenatal diagnosis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the open research
fund of National Key Research and Development Program of China
(grant no. 2023YFC2705605), Hunan Provincial Key Laboratory of
Regional Hereditary Birth Defects Prevention and Control (grant no.
HPKL2023032), the National Natural Science Foundation of China
(grant no. 82201773), Hunan Provincial Natural Science Foundation
(2023JJ30716) and the Hunan Provincial Science and Technology
Innovation Plan Project (grant no. 2021SK53204).
Availability of data and materials
The original data generated using whole-exome
sequencing in this study have been deposited into Mendeley Data
(V1) with the accession number doi: 10.17632/tgdng3bt9y.1
(https://data.mendeley.com/datasets/tgdng3bt9y/1). In
addition, the data generated in the present study may be requested
from the corresponding author.
Authors' contributions
JY and CYY were responsible for performing clinical
work, recruiting the patients and obtaining informed consent. HYZ
conducted the ultrasound examinations. JY and WBH conceived and
designed the experiments, while XL and JLZ performed the
experiments. XL authored the primary manuscript, while JY and WBH
reviewed and revised the manuscript. WBH also acquired funding. All
authors read and approved the final version of the manuscript. JY
and WBH confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
This study was approved by the institutional ethics
committee of The First Hospital of Changsha (Changsha, China), and
written informed consent was obtained from all participants.
Patient consent for publication
Patient consent for publication was obtained from
all participants.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hoffman JI, Kaplan S and Liberthson RR:
Prevalence of congenital heart disease. Am Heart J. 147:425–439.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lambrechts D, Devriendt K, Driscoll DA,
Goldmuntz E, Gewillig M, Vlietinck R, Collen D and Carmeliet P: Low
expression VEGF haplotype increases the risk for tetralogy of
Fallot: A family-based association study. J Med Genet. 42:519–522.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalisch-Smith JI, Ved N and Sparrow DB:
Environmental risk factors for congenital heart disease. Cold
Spring Harb Perspect Biol. 12:a0372342020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Williams K, Carson J and Lo C: Genetics of
congenital heart disease. Biomolecules. 9:8792019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van der Bom T, Zomer AC, Zwinderman AH,
Meijboom FJ, Bouma BJ and Mulder BJ: The changing epidemiology of
congenital heart disease. Nat Rev Cardiol. 8:50–60. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shaikh Qureshi WM and Hentges KE:
Functions of cilia in cardiac development and disease. Ann Hum
Genet. 88:4–26. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang X, Wang Q, Li T, Zhou Y, Gao J, Ma W,
Zhao N, Liu X, Ai Z, Cheng SY, et al: A splicing variant in EFCAB7
hinders ciliary transport and disrupts cardiac development. J Biol
Chem. 301:1082492025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saric N and Ishibashi N: The role of
primary cilia in congenital heart defect-associated neurological
impairments. Front Genet. 15:14602282024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang D, Sha Y, Gao Y, Zhang J, Cheng H,
Zhang J, Ni X, Wang C, Xu C, Geng H, et al: Novel variants in Dnah9
lead to nonsyndromic severe asthenozoospermia. Reprod Biol
Endocrinol. 19:272021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fassad MR, Shoemark A, Legendre M, Hirst
RA, Koll F, le Borgne P, Louis B, Daudvohra F, Patel MP, Thomas L,
et al: Mutations in outer dynein arm heavy chain DNAH9 cause motile
cilia defects and situs inversus. Am J Hum Genet. 103:984–994.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen W, Zhang Y, Shen L, Zhu J, Cai K, Lu
Z, Zeng W, Zhao J and Zhou X: Biallelic DNAH9 mutations are
identified in Chinese patients with defective left-right patterning
and cilia-related complex congenital heart disease. Hum Genet.
141:1339–1353. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loges NT, Antony D, Maver A, Deardorff MA,
Güleç EY, Gezdirici A, Nöthe-Menchen T, Höben IM, Jelten L, Frank
D, et al: Recessive DNAH9 loss-of-function mutations cause
laterality defects and subtle respiratory ciliary-beating defects.
Am J Hum Genet. 103:995–1008. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang T, Yuan H, Zhu H, Ying Y, Ding J,
Ding H, Shi X, He Y, Pan H and Zhong Y: Fetal congenital heart
disease caused by compound heterozygous mutations in the DNAH9
gene: A case report. Front Genet. 12:7717562021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng J, Li J, Du Y, Shi T, Sharma L and
Jie Z: Case report: Rare dynein axonemal heavy chain 9 mutations in
a han-Chinese patient with kartagener syndrome. Front Med
(Lausanne). 9:8939682022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takeuchi K, Xu Y, Ogawa S, Ikejiri M,
Nakatani K, Gotoh S, Usui S, Masuda S, Nagao M and Fujisawa T: A
pediatric case of productive cough caused by novel variants in
DNAH9. Hum Genome Var. 8:32021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Isa HM, Alkharsi FA, Busehail MY and
Haider F: A Novel DNAH9 gene mutation causing primary ciliary
dyskinesia with an unusual association of jejunal atresia in a
bahraini child. Cureus. 14:e329642022.PubMed/NCBI
|
|
17
|
Tate G: Whole-exome sequencing reveals a
combination of extremely rare single-nucleotide polymorphism of
DNAH9 and RSPH1 genes in a Japanese fetus with situs viscerum
inversus. Med Mol Morphol. 54:275–280. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Chen Y, Shi C, Huang Z, Zhang Y,
Li S, Li Y, Ye J, Yu C, Li Z, et al: SOAPnuke: A MapReduce
acceleration-supported software for integrated quality control and
preprocessing of high-throughput sequencing data. Gigascience.
7:1–6. 2018. View Article : Google Scholar
|
|
19
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He WB, Tu CF, Liu Q, Meng LL, Yuan SM, Luo
AX, He FS, Shen J, Li W, Du J, et al: DMC1 mutation that causes
human non-obstructive azoospermia and premature ovarian
insufficiency identified by whole-exome sequencing. J Med Genet.
55:198–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qi H, Pan D, Zhang Y, Zhu Y, Zhang X and
Fu T: NEXMIF combined with KIDINS220 gene mutation caused
neurodevelopmental disorder and epilepsy: One case report. Actas
Esp Psiquiatr. 52:588–594. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li G, Chen Y, Han X, Li N and Li S:
Concurrent of compound heterozygous variant of a novel in-frame
deletion and the common hypomorphic haplotype in TBX6 and inherited
17q12 microdeletion in a fetus. BMC Pregnancy Childbirth.
24:4562024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hamada H, Meno C, Watanabe D and Saijoh Y:
Establishment of vertebrate left-right asymmetry. Nat Rev Genet.
3:103–113. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Komatsu Y and Mishina Y: Establishment of
left-right asymmetry in vertebrate development: The node in mouse
embryos. Cell Mol Life Sci. 70:4659–4666. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nonaka S, Shiratori H, Saijoh Y and Hamada
H: Determination of left-right patterning of the mouse embryo by
artificial nodal flow. Nature. 418:96–99. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Essner JJ, Vogan KJ, Wagner MK, Tabin CJ,
Yost HJ and Brueckner M: Conserved function for embryonic nodal
cilia. Nature. 418:37–38. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Djenoune L, Mahamdeh M, Truong TV, Nguyen
CT, Fraser SE, Brueckner M, Howard J and Yuan S: Cilia function as
calcium-mediated mechanosensors that instruct left-right asymmetry.
Science. 379:71–78. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Katoh TA, Omori T, Mizuno K, Sai X,
Minegishi K, Ikawa Y, Nishimura H, Itabashi T, Kajikawa E, Hiver S,
et al: Immotile cilia mechanically sense the direction of fluid
flow for left-right determination. Science. 379:66–71. 2023.
View Article : Google Scholar : PubMed/NCBI
|