Introduction
Acute kidney injury (AKI) is an acute disorder
marked by rapid failure in renal function, associated with an
increased incidence and fatality rate (1). AKI is caused by decreased renal
perfusion pressure, trauma, mechanical and nephrotoxic agents.
Ischemia-reperfusion (IR) damage is a primary reason for acute
tubular necrosis in the kidney, resulting IR injury (IRI) of the
kidney (2,3). The involvement of inflammatory
processes, micro-vascular dysfunctions and adaptive responses of
kidney tubules in the pathogenesis of AKI may provide novel
treatment and diagnostic approaches.
During IRI, the reactive oxygen species (ROS)
generation, elevated calcium levels inside the cell and organelle
destruction lead to intrinsic kidney cell damage (4). IRI can trigger an immune cascade of
inflammation in stressed kidneys, which induces numerous mechanisms
of cell death, such as apoptosis, necrosis and ferroptosis
(5). Increased ROS production is a
hallmark of IRI. Hydrogen peroxide (H2O2),
owing to its self-stability and permeability of the membrane
(6), is a principal mediator of
renal tubular destruction in diverse disorders and has been used to
provoke ROS-mediated oxidative destruction in tubular epithelial
cells of the kidney, particularly within the cellular inflammatory
immune cascade triggered by kidney IRI (7).
Previous studies have revealed that targeting the
apoptosis progression in tubular epithelial cells is a valuable
strategy for protecting AKI (8–10).
Activating mitochondrial-dependent pathways is essential for
apoptosis and kidney injury owing to IR (11). One of the key components of this
pathway, Bax, activates and transfers to the mitochondrial
membrane, resulting in activation of effector caspase3, an inducer
of AKI (12). Studies have
demonstrated that inhibition of Bax protects human tubular
epithelial cells of the kidney against damage caused by cisplatin
and lipopolysaccharide (13,14).
Therefore, investigating the role of the aforementioned
mitochondrial-dependent apoptosis pathway on the mechanism or
management of IRI-AKI is important.
β-elemene (ELE) is a sesquiterpene present in
spices, herbs and root vegetables, used for the treatment of
different malignancies, such as lung, liver cancer, esophageal
cancer, nasopharyngeal cancer, brain cancer, and bone metastasis
(15). ELE kills the tumor cells,
whereas, to the best of our knowledge, there is no known impact on
healthy cells, such as the peripheral blood lymphocytes, at
concentrations below 50 µg/ml (16). β-Elemene was found to have
effectively suppressed M2 macrophage recruitment and MCP-1
expression through inhibiting the Prx-1/NF-κB/HIF-1α signaling
pathway in lung cancer (17). In
glioblastoma cells, β-elemene induced ROS production in a dose- and
time-dependent manner, mediated oxidative damage and inhibited
cancer growth (18). Although the
anti-tumor effects of ELE have been extensively reported, the role
of ELE in AKI remains unclear. The present study assessed the
anti-inflammatory role of ELE in rat proximal tubular epithelial
(NRK52E) cells and a mouse model of IRI to support the potential
use of ELE in AKI.
Materials and methods
Reagents and antibodies
ELE (cat. no. 63965) was acquired from Merck KGaA.
Antibodies targeting toll-like receptor (TLR) 4 (cat. no. AF7017),
myeloid differentiation primary response gene 88 (MyD88; cat. no.
AF5195), P65 (cat. no. BF8005), ERK1/2 (cat. no. AF0155), F4/80
(cat. no. DF2789), β-actin (cat. no. AF7018), phosphorylated
(p)-P65 (cat. no. AF2006), Bax (cat. no. AF0120), JNK (cat. no.
AF6318), Bcl-2 (cat. no. AF6139), P38 (cat. no. AF6456), caspase-3
(cat. no. AF6311), cleaved (c-)caspase-3 (cat. no. AF7022), p-JNK
(cat. no. AF3318), p-P38 (cat. no. AF4001) and p-ERK1/2 (cat. no.
AF1015) were acquired from Affinity Biosciences, Ltd.
N-acetylcysteine (NAC; cat. no. S1623) was acquired from Selleck
Chemicals. The blood urea nitrogen (BUN) test (cat. no. C013-2-1)
and creatinine assay kits (cat. no. C011-2-1) were obtained from
the Nanjing Jiancheng Bioengineering Institute.
Animal experiments
The animal experimental ethics committee at Youjiang
Medical University for Nationalities (Baise, China) authorized all
animal experiments. A total 30 of male C57BL/6 mice (age, 8–10
weeks old and body weight of 18–22 g) were obtained from Changzhou
Cavens Experimental Animal Co. Ltd. (cat. no. 202358084) and housed
in the specific-pathogen-free facility of Youjiang Medical
University for Nationalities (certification no. SYXK 2022–0004).
Mice were housed under standard conditions and had unlimited access
to sterilized food and distilled water. Room temperature was
maintained at 25±2°C and relative humidity at 60±10% and a 12-h
light/dark cycle was used. Mice were randomly separated into four
groups (n=6/group): Sham, ELE, IRI and IRI + ELE. The IRI model was
established as previously described (19). Sham group underwent surgical
exposure of the kidney without ischemia induction. ELE group
received intraperitoneal injection of ELE (40 mg/kg/day) without
ischemia induction for 7 days; In the IRI group, ischemia was
induced by clamping both renal arteries for 45 min, followed by
reperfusion. Mice were anesthetized with an intraperitoneal
injection of sodium pentobarbital (50 mg/kg). A midline abdominal
incision was made to expose both renal arteries, which were clamped
with non-invasive vascular clips for 45 min. The clamps were
removed to restore blood flow. Kidney tissue was collected 24 h
post-surgery and 0.5–1.0 ml of blood were collected for analysis
using cardiac puncture. All IRI model mice underwent 24 h
reperfusion after ischemia to simulate the commonly observed IRI
time window in clinical settings (20,21).
The IRI + ELE group was pre-treated with ELE (40 mg/kg/day) for 1
week prior to the IRI procedure. ELE injection continued until
euthanasia.
All mice were anesthetized with 1% (w/v)
pentobarbital sodium solution, administered at a dose of 50 mg/kg
via intraperitoneal injection, before surgery. At the end of the
experiments, all mice were euthanized by carbon dioxide via a gas
anesthesia machine, controlling the CO2 flow rate at 70%
of the chamber volume/min, followed by cervical dislocation. Death
was confirmed by cessation of respiration for >5 min, (2) absence of pedal reflex.
Cell culture
Rat proximal tubular epithelial NRK52E cells were
obtained from American Type Culture Collection. Cells underwent
incubation in a 5% CO2 atmosphere with 10% fetal bovine
serum and Dulbecco's modified eagle medium (both Gibco; Thermo
Fisher Scientific, Inc.) at 37°C. NRK52E cells were treated with
ELE (0, 5, 10, 20, 40 and 80 µM) for 1 day at 37°C. Subsequently,
NRK52E cells were treated with 150, 300, 450 and 600 µM/ml
H2O2 (PeproTech, Inc.) for3, 6 and 12 h at
37°C. In addition, the ROS scavenger NAC was used to intervene in
NRK52E cells at 2, 5 and 10 µM for 12, 24, 48 h at 37°C.
Renal function and histology
Mouse blood samples were allowed to coagulate at
room temperature for 30–60 min, then centrifuged at 14,000 × g for
10 min at room temperature to obtain a serum sample. Levels of BUN
and serum creatinine (Scr) were identified using the aforementioned
commercial kits according to the manufacturer's instructions. For
histological examination, the mouse renal tissues underwent
fixation with 4% formaldehyde at room temperature for 24 h and were
immersed in paraffin for staining with H&E to analyze the renal
morphology, as previously described (22).
Immunohistochemistry
Kidney tissue from mice of different treatment
groups were fixed in 4% paraformaldehyde solution for 24 h and
dehydrated in increasing order of alcohol at room temperature.
After permeabilization with xylene for 30 min, the heart tissue was
embedded in paraffin and cut into 4–5 µm sections. These tissue
sections were deparaffinized with xylene. For antigen repair, they
were placed in staining jars containing citrate buffer and boiled
in a pressure cooker over medium heat for 15 min. To inhibit
endogenous peroxidase activity, a 3% hydrogen peroxide solution was
added for 10 min at room temperature. Sections were incubate with
10% goat serum (No. SAP-9100; Zhongshan Jinqiao Biotechnology Co.,
Ltd., Beijing, China) for 30 min at room temperature, followed by
incubation with anti-F4/80 primary antibody (1:200; No. A2547,
Sigma-Aldrich) for 12 h at 4°C, followed by incubation with
horseradish peroxidase (HRP)-IgG secondary antibody (1:50, No.
A0208; Beyotime Institute of Biotechnology) for 60 min at 24°C. The
expression of F4/80 protein in kidney tissue was observed using DAB
staining. A light microscope (Nikon, Japan) was used. Five visual
field images of tissue were obtained from each histochemical
section. The number of DAB-positive cells was determined by ImageJ
(version 1.8.0; National Institutes of Health).
TUNEL staining
TUNEL Apoptosis Detection kit (Item No. KGA1401-100,
Jiangsu Kaiji Biotechnology Co., Ltd.) was used to detect apoptosis
in mouse kidney tissues (5 µm) and NRK52E cells (1×105).
NRK52E cells were treated with 600 µM/ml H2O2
and ELE (10 or 20 µM) at 37°C. Briefly, cells and kidney tissue
sections were fixed on coverslips with 4% paraformaldehyde for 30
min at room temperature. Cells were treated with 0.1% Triton X-100
for 10 min at room temperature. The kidney tissue and cells were
washed with PBS. Cells and kidney tissue sections were treated with
proteinase K working solution for 10 min at 37°C and incubated with
50 µl of TUNEL reaction mixture for 1 h at 37°C. Nuclei were
counterstained by mounting with antifade medium containing DAPI
(cat. no. P0131, Beyotime Biotechnology). for 10 min at room
temperature. Images were captured using an inverted fluorescence
microscope (Nikon Corporation) at high magnification (×400).
Quantification of TUNEL-positive cells in at least 3 randomly
selected areas was performed using ImageJ software (version 1.8.0;
National Institutes of Health).
Cell viability
Detection of ELE-induced cytotoxicity in NRK52E
cells was performed using Cell Counting Kit-8 (CCK-8) assay (cat.
no. M4839; Abmole China Branch). NRK52E cells were seeded into
96-well plates at a density of 3,000 cells/well and incubated with
CCK-8 solution for 2 h. Cell absorbance at 450 nm was determined
using a microplate reader (Titertek-Berthold).
RNA analysis
Total RNA was extracted from kidney tissue and
NRK52E cells (1×106) with TRIzol (Thermo Fisher
Scientific, Inc.) and its quality was assessed using a
spectrophotometer (Beckman Coulter, Inc.). RNA was
reverse-transcribed to cDNA using ReverTra Ace qPCR RT premix
(FSQ-201, TOYOBO) according to the manufacturer's instructions.
QuantiNova SYBR Green PCR kit (Qiagen GmbH) and Analytik Jena
qTOWER 3 G Real-Time PCR System (Jena, Germany) was used for RT-PCR
according to the manufacturer's instructions. all PCRs were
performed in triplicate with the following cycling conditions: i)
initial denaturation at 95°C/10 min; ii) 40 cycles each at 95°C 30
sec, 60°C/1 min; and 72°C/30 sec. The 2-ΔΔCq method was used for
all PCRs. Quantification of mRNA levels was performed using the
2-ΔΔCq method and normalised against the internal reference gene
GADPH (23). Primers are listed in
Table SI.
Small interfering RNA (siRNA)
knockdown
To inhibit MyD88 expression, siRNA sequences were
used. MyD88 (5′-GCCAGCGAGCTAATTGAGAAA-3′; cat. no. SC-106986; Santa
Cruz Biotechnology, Inc.) and negative control siRNA
(5′-GCCAGCGAGCTAATTGAGAAA-3′; cat. no. SC-106986; Santa Cruz
Biotechnology, Inc.) were used to transfect cells. NRK52E cells
(1×105) were inoculated in each well of a 6-well plate.
12 h later, cells were transfected with 80 nM control siRNA and
MyD88 siRNA. Lipofectamine® 2000 Reagent (Art. No.
11668019; Invitrogen; Thermo Fisher Scientific, Inc.) was used for
transfection (5 µl/well). After 18 h of incubation at 37°C, the
transfection medium was replaced with fresh Dulbecco's modified
eagle medium and the cells were further incubated at 37°C for 24
h.
Western blot analysis
Total proteins were extracted from kidney tissues
and NRK52E cells (1×107/per) using RIPA buffer (Beyotime
Institute of Biotechnology) and protein concentration was measured
using BCA assay kit (ZJ101, Epizyme). Proteins were separated using
10% SDS-PAGE (50 µg/lane) and transferred to PVDF membrane. After
being closed with 5% skimmed milk for 1 h at room temperature, the
membranes were incubated with F4/80 (1:1,000), TLR4 (1:1,000),
MyD88 (1:2,000), P65 (1:1,000), ERK1/2 (1:1,000), β-actin
(1:10,000), p-P65 (1:1,000), Bax (1:1,000), JNK (1:1,000), Bcl-2
(1:1,000), P38 (1:1,000), caspase-3 (1:1,000), c-caspase-3
(1:1,000), p-JNK (1:1,000), p-P38 (1:1,000) and p-ERK1/2 (all
1:1,000) primary antibodies overnight at 4°C. The membrane is left
at room temperature for 1 h with an HRP coupled secondary antibody
(1:2,000). Finally, the bands were visualized using ECL Reagent
(SQ201L, Epizyme, Shanghai, China). ImageJ software (version 1.8.0,
National Institutes of Health) was used.
Statistical analysis
All data are presented as the mean ± standard
deviation (n=3–6 repeats/group). Multiple comparisons were
conducted using one-way ANOVA with Tukey's post hoc test using
GraphPad Prism (version 8.0, Dotmatics) P<0.05 was considered to
indicate a statistically significant difference.
Results
ELE ameliorates IR-induced kidney
injury
Levels of the inflammatory cytokine TNF-α were
measured at 6, 24 and 48 h post-reperfusion. A pronounced increase
in inflammatory cytokines occurred at 24 h, while there was no
significant difference at 48 h (Fig.
1A). Therefore, 24 h was selected as the optimal timepoint. The
blood concentrations of Scr and BUN in mice subjected to a renal IR
intervention were elevated compared with sham animals. ELE + IRI
significantly prevented increased Scr and BUN levels (Fig. 1B and C). ELE alone did not cause
kidney injury in mice (Fig. 1D and
E); renal tissue demonstrated infiltration of inflammatory
cells and destruction of the tubules, including a loss of a tubular
brush edging and luminal dilatation in the IRI compared with the
sham group, which was ameliorated by ELE pretreatment. The levels
of the macrophage biomarker F4/80 protein were measured to assess
the extent of inflammatory cell interstitial infiltration in the
kidney of mice. Levels of F4/80 protein were significantly elevated
in IRI mice compared with the sham group, while ELE markedly
decreased the F4/80 protein expression levels in IRI mice (Fig. 1F and G).
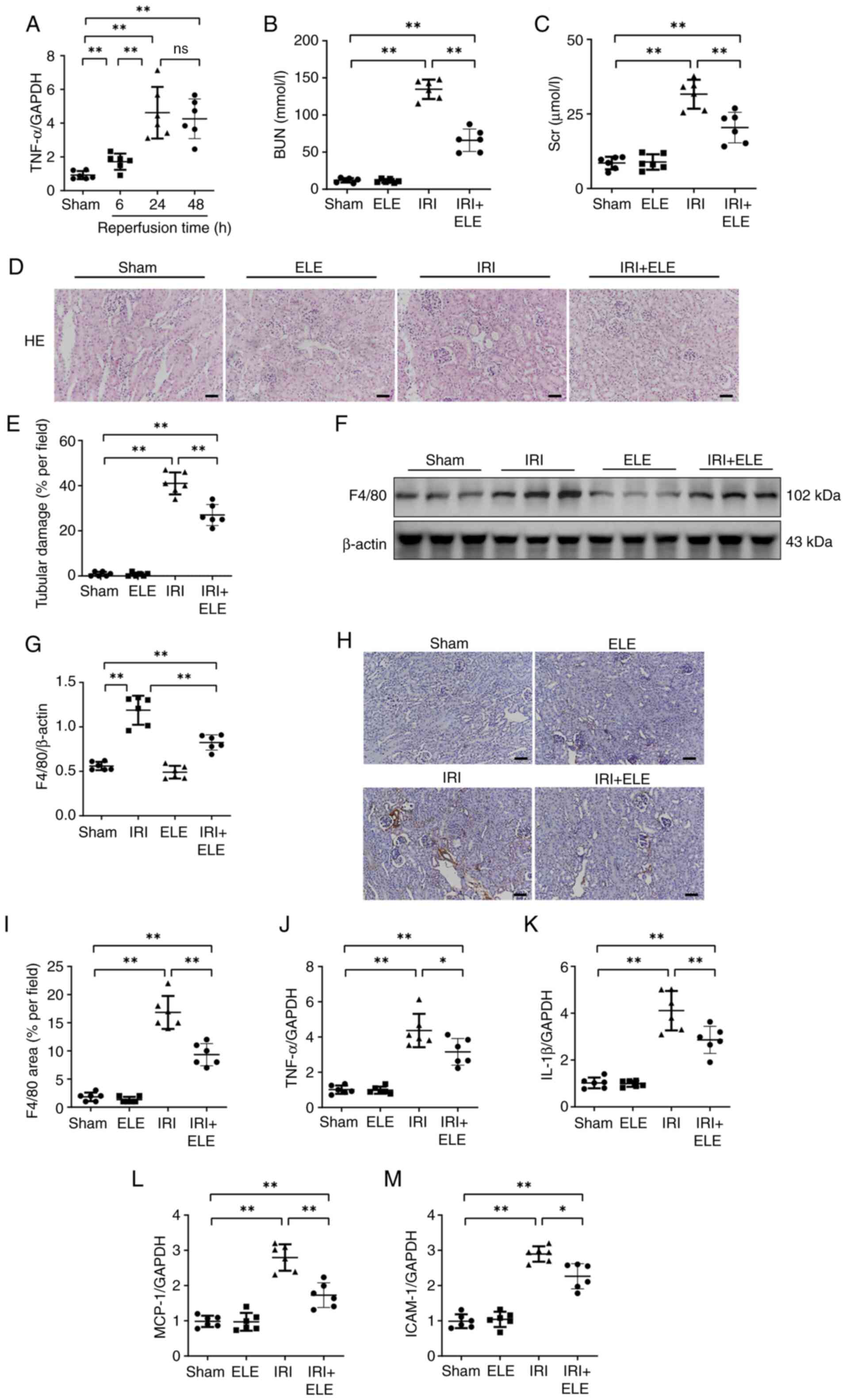 | Figure 1.β-elemene ameliorates kidney damage
due to I/R. (A) RT-qPCR was utilized to identify TNF-α mRNA
expression. The serum (B) BUN and (C) Scr levels. (D)
Representative H&E staining. (E) Tubular damage. (F)
Representative western blotting and (G) F4/80 protein expression in
renal tissue. (H) Representative immunohistochemistry. Scale bar,
50 µm. (I) F4/80 expression in renal tissue. RT-qPCR was used to
identify mRNA expression levels of (J) TNF-α, (K) IL-1β, (L) MCP-1
and (M) ICAM-1. *P<0.05, **P<0.01. RT-q, reverse
transcription-quantitative; ns, not significant; ELE, β-elemene;
IRI, ischemia-reperfusion injury; MCP-1, monocyte chemoattractant
protein-1; ICAM-1, intercellular adhesion molecule 1. |
Immunohistochemical experiments also confirmed that
ELE + IRI mice exhibited significant inhibition in F4/80 deposition
compared with IRI mice (Fig. 1H and
I). IRI mice exhibited increased levels of monocyte
chemoattractant protein-1 (MCP-1), IL-1β, intercellular adhesion
molecule 1 (ICAM-1) and TNF-α expression compared with sham mice
(Fig. 1J-M). By contrast, levels
of MCP-1, TNF-α, IL-1β and ICAM-1 expression were decreased in ELE
+ IRI mice compared with IRI mice. These results suggested that ELE
partially reduced kidney injury by reducing the inflammatory
infiltration in IR-induced AKI.
ELE protects NRK52E cells from
H2O2-induced inflammation
Effects of ELE on H2O2-induced
inflammation were assessed in NRK52E cells. CCK-8 assay revealed
that 600 µM H2O2 for 12 h significantly
decreased NRK52E cell viability (Fig.
2A). ELE induced cytotoxicity in NRK52E cells. NRK52E cell
viability was not altered by ELE at concentrations of 5–20 µM
(Fig. 2B). Decreased NRK52E cell
viability caused by H2O2 was recovered by ELE
at 5, 10 and 20 µM (Fig. 2C).
Therefore, these concentrations were selected for subsequent
experiments. MCP-1, IL-1β, TNF-α and ICAM-1 mRNA levels in
H2O2-stimulated NRK52E cells increased when
compared with control cells. However, MCP-1, TNF-α, IL-1β and IL-6
levels were suppressed by ELE pretreatment in a dose dependent
manner (Fig. 2D-G).
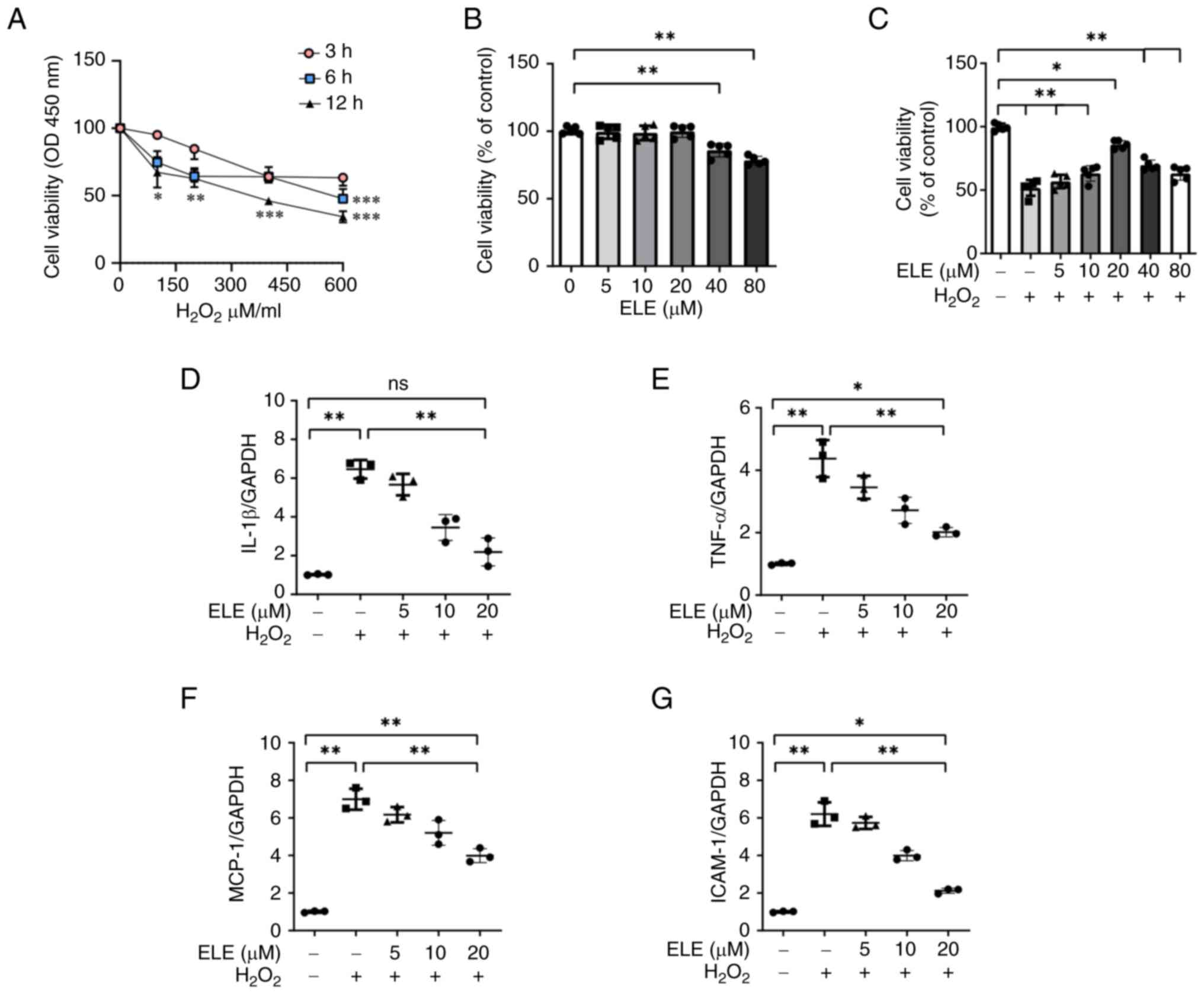 | Figure 2.β-elemene protects NRK52E cells from
H2O2-induced inflammation. (A) Optimal
duration of H2O2 treatment in NRK52E cells
was identified by employing the CCK-8 assay. (B) CCK-8 analysis was
employed to identify the ELE impact on viability of NRK52E cells.
(C) NRK52E cells underwent pretreatment with various doses of ELE
and were treated for 6 h with or without 600 µM
H2O2. CCK-8 analysis was used to determine
cell viability. Reverse transcription-quantitative PCR was employed
to evaluate the mRNA expression of (D) IL-1β, (E) TNF-α, (F) MCP-1
and (G) ICAM-1. *P<0.05, **P<0.01, ***P<0.001. CCK-8, Cell
Counting Kit-8; OD, optical density; ELE, β-elemene; ns, not
significant; MCP-1, monocyte chemoattractant protein-1; ICAM-1,
intercellular adhesion molecule 1. |
ELE inhibits the inflammatory response
by suppressing TLR4/MyD88/NF-κB pathway activation in vivo and in
vitro
As MCP-1, ICAM-1, and TNF-α are cytokine markers for
the NF-κB signal (24), the levels
of p65 and p-p65 protein were detected in mice and NRK52E cells.
Expression of p-p65 protein increased in mice renal tissues after
IRI compared with sham mice (Fig.
3A). ELE prevented the increase in p-p65 expression.
Furthermore, TLR4 and MyD88 protein expression upstream of the
NF-κB signaling pathway was significantly elevated in IRI compared
with the sham mice. ELE decreased the TLR4 and MyD88 expression in
IRI mice. In NRK52E cells, ELE inhibited
H2O2-induced increases in MyD88, TLR4 and
p-P65 protein levels in a dose-dependent manner (Fig. 3B). These findings demonstrated that
ELE exerts anti-inflammatory effects, at least partially, via the
TLR4/MyD88/NF-κB pathway.
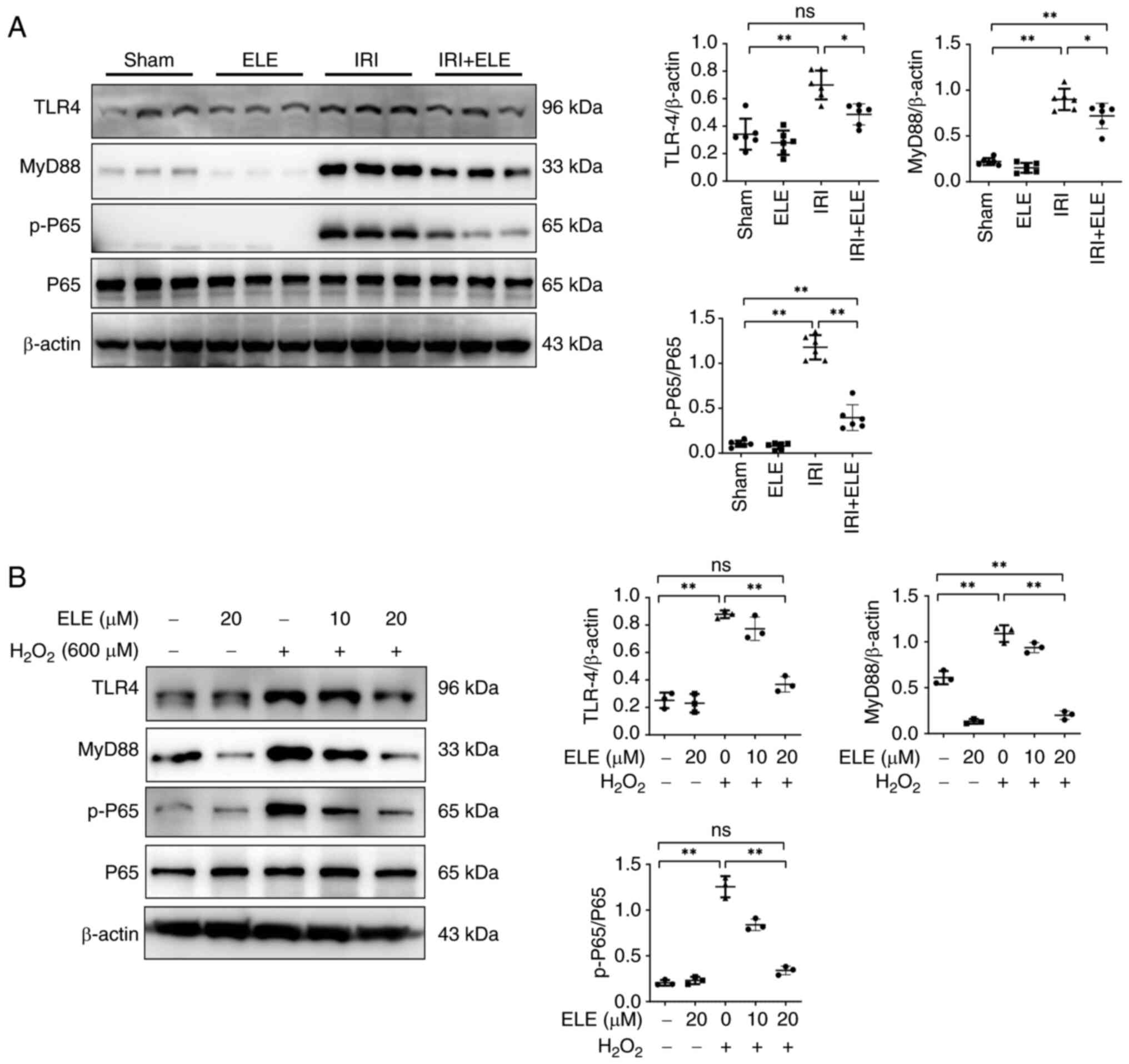 | Figure 3.ELE inhibits the inflammatory
response by suppressing TLR4/MyD88/NF-κB pathway activation in
vivo and in vitro. (A) Renal p-P65, MyD88, TLR4 and P65
expression. (B) Western blotting of the p-P65, MyD88, TLR4 and P65
expression in NRK52E cells treated with H2O2
and various concentrations of ELE. *P<0.05, **P<0.01. ELE,
β-elemene; IRI, ischemia-reperfusion injury; TLR4, toll-like
receptor 4; MyD88, myeloid differentiation primary response gene
88; p-, phosphorylated-; ns, not significant. |
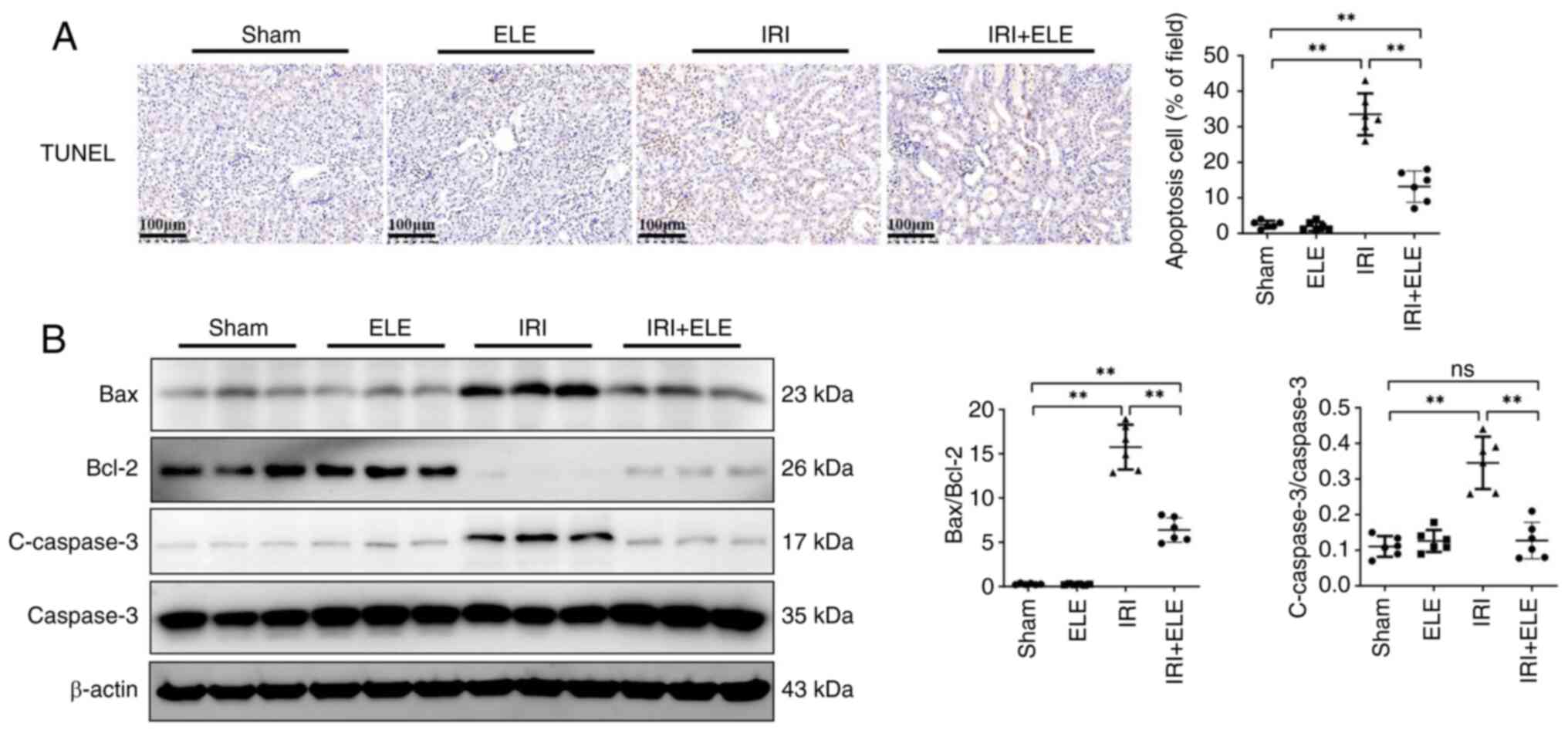
ELE suppresses apoptosis in IRI
mice
Apoptosis serves a key function in renal IR as it
can cause cell death, and the extent of the apoptosis may be
directly associated with the intensity of the damage (25). TUNEL staining in the renal tubule
increased in IRI compared with the sham mice (Fig. 4A). ELE pretreatment decreased
IRI-induced kidney cell apoptosis. Bax/Bcl-2 ratio and c-caspase3
protein expression were downregulated by ELE pretreatment compared
with IRI-alone (Fig. 4B). TUNEL
staining revealed that ELE exhibited an anti-apoptotic effect in
the IRI model.
ELE ameliorates
H2O2-induced NRK52E cell apoptosis
To detect the ELE-induced anti-apoptotic effect on
H2O2-treated NRK52E cells, a TUNEL assay was
performed. H2O2 markedly upregulated the
proportion of TUNEL-positive cells compared with the control group
(Fig. 5A). The proportion of
TUNEL-positive cells was downregulated in ELE 10 and 20 µm +
H2O2 groups compared with the
H2O2 group, with the most pronounced decrease
in ELE 20 µm + H2O2 group. ELE pretreatment
dose-dependently inhibited the ratio of Bax/Bcl-2 and protein
expression of c-caspase3 in H2O2-treated
NRK52E cells (Fig. 5B).
Additionally, to investigate whether ELE exerts its effects through
the oxidative stress pathway, the ROS scavenger NAC was used. NAC
at 5 mM inhibited proliferation after 24 h in NRK52E cells compared
to the control group (Fig. 5C). In
H2O2-treated NRK52E cells, the combination of
ELE + NAC significantly suppressed the Bax/Bcl-2 ratio and the
protein expression of c-caspase3 compared with ELE alone (Fig. 5D). The data suggested that ELE may
exert its anti-apoptotic effects through the oxidative stress
pathway.
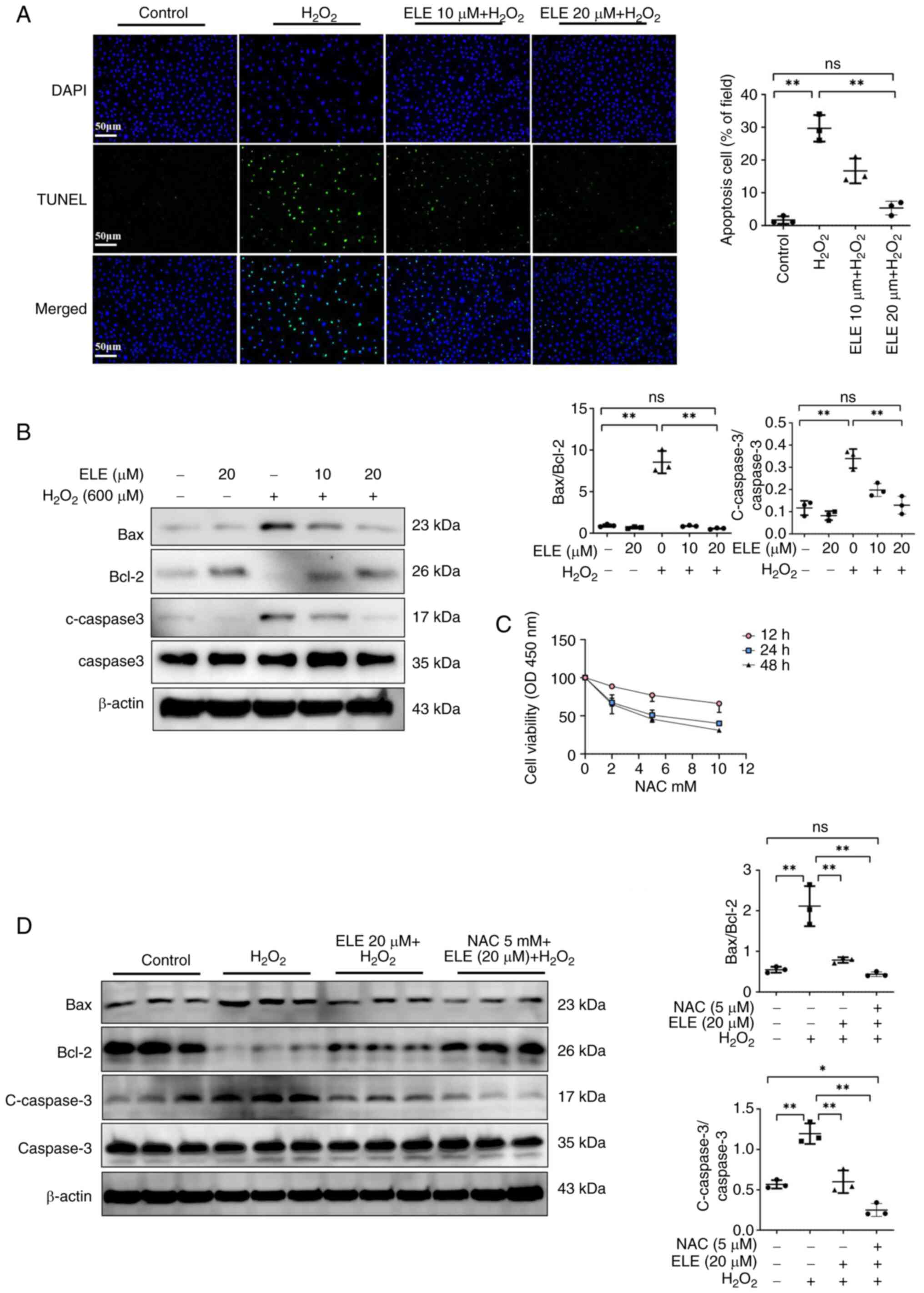 | Figure 5.ELE ameliorates
H2O2-induced NRK52E cell apoptosis. (A) TUNEL
assay; scale bar, 50 µm. (B) Bax, c-caspase3, Bcl-2 and caspase-3
protein expression were measured using western blotting. (C) CCK-8
analysis was performed to identify the NAC impact on viability of
NRK52E cells. (D) Identification and semi-quantification of Bax,
c-caspase3, Bcl-2 and caspase-3 protein expression in NRK52E cells.
*P<0.05, **P<0.01. ELE, β-elemene; c-, cleaved; ns, not
significant; OD, optical density; NAC, N-Acetylcysteine. |
ELE decreases inflammatory and
apoptosis signaling by inhibiting MAPK signal activation
MAPK pathway signaling is affected by the
pro-apoptosis and pro-inflammatory cytokine IL-1β, TNFα (26,27).
Expression of MAPK signaling pathway members was assessed in the
kidney of IRI mice and H2O2-treated NRK52E
cells. The IRI group revealed increased p-ERK, p-JNK and p-p38
protein expression compared with the sham group, while ELE
pretreatment decreased ERK, JNK and p38 protein phosphorylation
levels in the kidney (Fig. 6A).
Similarly, p-JNK, p-ERK and p-p38 protein levels were increased in
NRK52E cells stimulated by H2O2; levels of
p-JNK, p-ERK and p-p38 diminished with elevated ELE levels
(Fig. 6B). These data suggest that
ELE may function by inhibiting MAPK signaling pathway
activation.
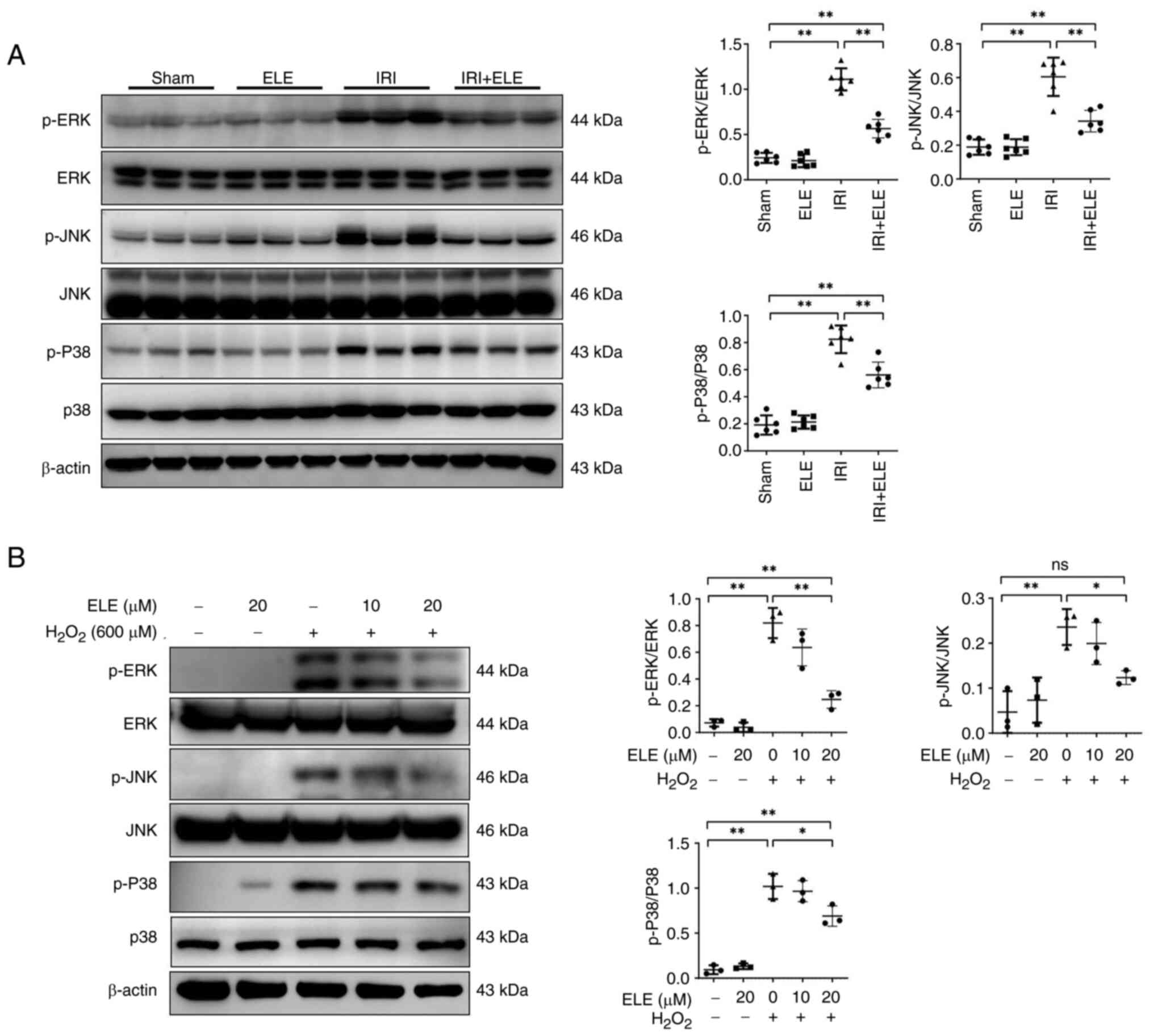 | Figure 6.ELE decreases inflammatory and
apoptosis signaling by inhibiting MAPK signaling pathway
activation. (A) ERK, p-JNK, p-P38, JNK, p-ERK and P38 in the
kidney. (B) Identification and semi-quantification of p-JNK,
p-ERK1/2, p-P38, JNK, ERK1/2 and P38 expression in NRK52E cells.
*P<0.05, **P<0.01. ELE, β-elemene; IRI, ischemia-reperfusion
injury; ns, not significant; p-, phosphorylated. |
MyD88 knockdown inhibits apoptosis and
MAPK signaling pathway activation in
H2O2-treated NRK52E cells
Effects of H2O2 in MyD88
knockdown NRK52E cells was examined. MyD88 protein expression was
not affected by the negative control siRNA (Fig. 7). Following siRNA-MyD88
transfection, a reduction in MyD88 protein expression was observed.
MyD88 knockdown downregulated the ratio of Bax/Bcl-2 and protein
expression of c-caspase3 in NRK52E cells following
H2O2 treatment. The impact of MyD88 knockdown
on MAPK signaling pathway member expression in NRK52E cells was
assessed. p-JNK, p-ERK and p-p38 protein expression levels were
significantly decreased following H2O2
stimulation, while MyD88 knockdown reversed the effects of
H2O2.
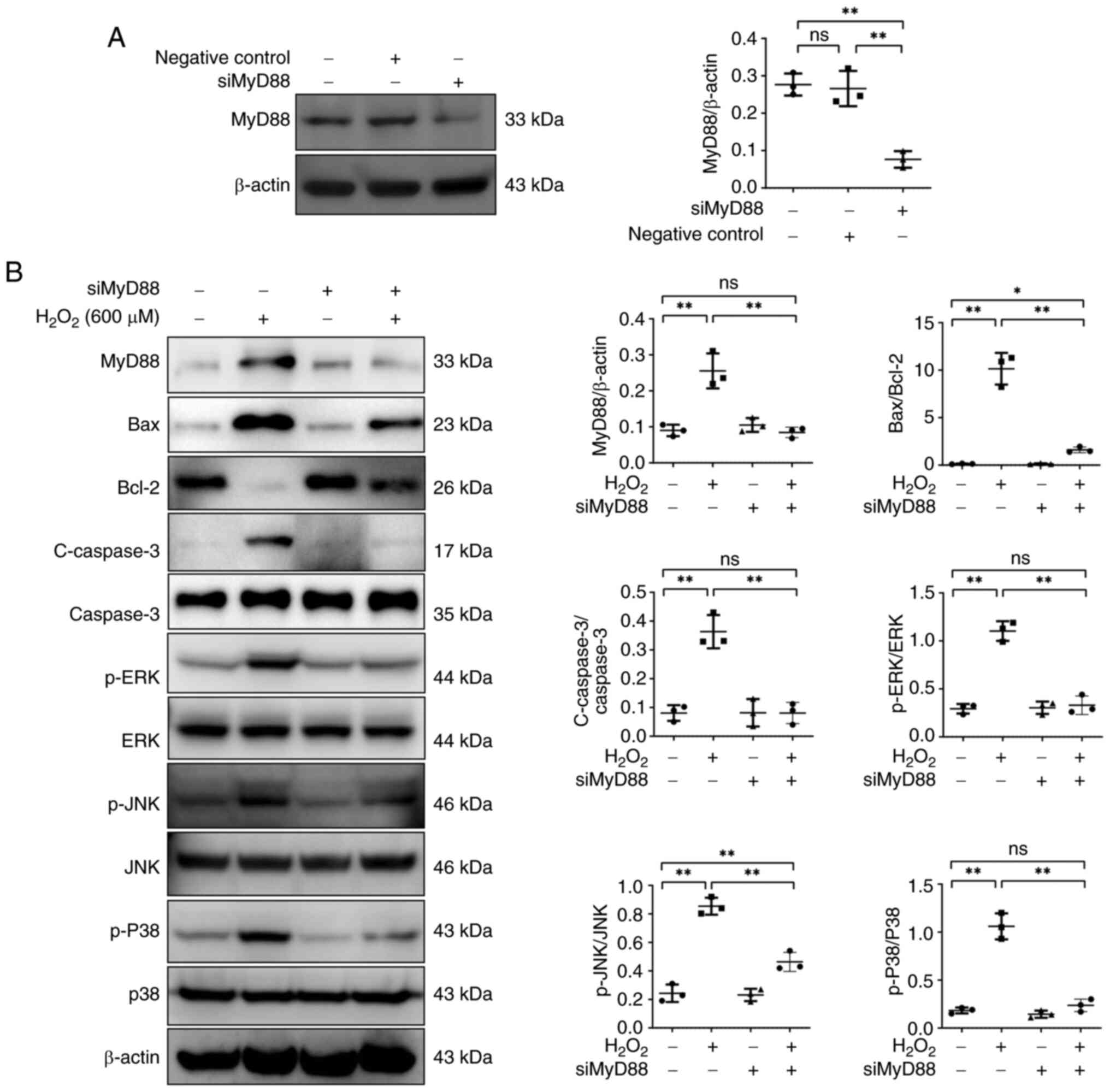 | Figure 7.MyD88 knockdown suppresses apoptosis
and MAPK signaling pathway activation in
H2O2-treated NRK52E cells. (A) MyD88
expression in NRK52E cells. (B) MyD88, Bax, c-caspase3, Bcl-2, JNK,
caspase-3 p-ERK, p-JNK, ERK, p-P38 and P38 expression in NRK52E
cells treated with H2O2. *P<0.05,
**P<0.01. c-, cleaved; p-, phosphorylated; ns, not significant;
si, small interfering. |
Discussion
Previous studies and pathogenic mechanisms support a
key role for renal tubules in postischemic AKI (28–30).
Apoptosis of renal tubule cells causes lethal damage to renal
epithelium cells, aggravating kidney damage (31). Accordingly, the discovery of a
therapeutic drug that exhibits anti-inflammatory and anti-apoptosis
activity may contribute to IRI-AKI treatment. Studies have
demonstrated that ELE has anti-inflammatory and anti-apoptosis
activity (32–34). Nevertheless, the protective
function of ELE in renal IRI has not been explored. In the present
experiments, in vivo and in vitro models of renal IRI
revealed that ELE could alleviate renal damage by
TLR4/MyD88/NF-κB/MAPK pathway downregulation and suppression of
apoptosis.
ELE was initially reported as an adjunctive medicine
for anti-tumor agents due to its ability to cause apoptosis in
malignant cells (35). Gan et
al (36) revealed that ELE
improves bladder cancer cell apoptosis by targeting Bcl-2 family
protein. Lee et al (37)
demonstrated that ELE directly suppresses ovarian malignancy cell
proliferation by inducing cell cycle arrest. ELE has been explored
in inflammatory disorders: Zhou et al (38) revealed that ELE treats chronic
inflammation caused by obesity by enabling the migration of
Foxp3CD4T+ cells to the adipose tissue. Our previous
study demonstrated that ELE decreases renal fibrosis by inhibiting
MyD88/JAK/STAT signaling pathway activation (22). Nevertheless, the mechanism
underlying the effects of ELE on renal IRI are unknown. The
inflammatory reaction is initiated after IRI and worsening the
condition during disease progression. Chemotaxis in inflammatory
cells is an important feature of IRI (39). In IRI, damaged renal tubule
epithelial cells produce chemokines, which release inflammatory
factors that contribute to inflammation, causing renal tissue
damage (40). In the present
study, ELE decreased the Scr and BUN serum levels in IRI mice, as
well as morphological changes such as infiltration of inflammatory
cells in the kidney and renal tubule damage. In addition, ELE
significantly inhibited the F4/80 infiltration in IRI renal
interstitial tissue. Additionally, ELE resulted in the inhibition
of inflammatory cytokine (TNF-α and IL-1β), MCP-1 and ICAM-1
expression in vivo and in vitro. ELE may suppress
inflammatory responses and restore renal function caused by
IRI.
The innate immune system can recognize cells
undergoing ischemic injury and stimulate inflammatory responses
using pattern recognition receptors. TLR proteins serve a role in
inflammatory responses of the kidney (41) and renal IRI. Chen et al
(42) revealed that TLR4 regulates
leukocytosis infiltration in the kidney during ischemia. Wu et
al (43) revealed that
knockout of TLR4 in IRI-induced mice decreases infiltration of
macrophages and neutrophils. Notably, MyD88 may be a key downstream
protein for TLR4 to regulate renal IRI (43). Zhang et al (44) revealed that MyD88 inhibitor
TJ-M2010-2 markedly alleviates TGF-β-induced renal fibrosis in mice
by inhibiting the TLR-4/MyD88 pathway. In the present study, IRI
mice and H2O2-induced NRK52E cell treated
with ELE exhibited decreased inflammation and TLR4 and MyD88
protein levels, indicating a feedback loop between ELE and
TLR/MyD88 signaling.
The impairment of renal tubule epithelium function
caused by apoptosis is a key feature of renal IRI progression.
Pro-inflammatory cytokines induce apoptosis, primarily through
inducing the caspase family of proteases (45). Caspase-3 activation aggravates
renal injury by initiating the final enzymatic apoptosis cascade
following ischemia of the kidney (12,46).
Here, ELE mechanisms in apoptosis were assessed by measuring the
activity of caspase-3 protein and using TUNEL staining in IRI
models. The IRI group demonstrated elevated c-caspase3 protein
activity compared with the sham group, but ELE significantly
decreased c-caspase3 protein expression levels in IRI mice. The
increased activity of the c-caspase3 protein was associated with
TUNEL-positive staining of renal tubule epithelium cells. The
proteins of the Bcl-2 family serve a key role in maintaining renal
tubule epithelium cell apoptosis and ameliorating renal dysfunction
(25). ELE pretreatment inhibited
the ratio of Bax/Bcl-2 protein in IRI mice and
H2O2-treated NRK52E cells. Therefore, the
cytoprotective function of ELE may be induced by suppressing
caspase-3 and Bcl-2 family protein activity.
Dysregulated NF-κB activation during renal
ischemia-reperfusion injury promotes tubular cell damage via
inflammatory responses (47).
Ischemia of the kidney induces translocation of nuclear NF-κB in
the renal tubules, promoting ischemia-induced cell death (48). Zou et al (49) revealed that inhibiting the NF-κB
signaling pathway decreases kidney inflammatory responses in IRI
mice. Our previous study revealed that maslinic acid decreases
renal interstitial fibrosis by suppressing NF-κB signal activation
(50). The aforementioned study
revealed that ELE reverses NF-κB signaling activation in the
kidneys of IRI-induced mice and H2O2-treated
NRK52E cells. Furthermore, MyD88 inhibition decreases TLR4
signaling and NF-κB protein levels (22). It was hypothesized that the
TLR4/MyD88/NF-κB pathway creates a directional signal axis and is
involved in the inflammatory reaction of IRI-induced mice. ELE
pretreatment decreased the expression of TLR4 protein and MyD88
protein, to inhibit the downstream NF-κB signaling activation.
Similar to the present study, a previous study demonstrated that
fucoxanthin alleviates lipopolysaccharide-induced acute lung injury
by inhibiting the TLR4/MyD88 signaling axis (51). Although the models differ (lung vs.
kidney injury), both demonstrated targeting of the TLR4/MyD88
pathway, suggesting that this pathway has a general role in organ
ischemia/inflammation (52). ELE
may exert multi-organ protective effects via a similar mechanism,
which requires further validation.
MAPK signaling members (such as ERK, JNK and p38
protein) mediate proximal renal tubular cell injury mediator
(52,53). Activation of MAPK family proteins
is associated with NF-κB signal activity and generally considered
to mediate apoptosis and inflammatory responses (54,55).
In the present study, ELE pretreatment reversed IRI and
H2O2-induced NRE52K cell injury by
suppressing p-JNK, p-ERK and p-p38 protein expression.
Additionally, inhibition of MyD88 markedly reduced the p-ERK, p-p38
and p-JNK protein expression in H2O2-treated
NRE52K cells. It was hypothesized that ELE may decrease the
inflammatory response and apoptosis caused by IR renal damage by
targeting the TLR4/MyD88/NF-κB/MAPK signaling pathway.
The present study had limitations. The present study
did not identify a phenotype of ELE in IRI-AKI. Further research is
required to explore potential roles or mechanisms of ELE. Secondly,
the present study revealed the protective effects of ELE on kidney
injury solely in IRI. It is essential to investigate the protective
role of ELE on AKI by establishing different models of AKI,
including sepsis- and nephrotoxin-induced AKI. IRI-AKI is a dynamic
process; different time points following reperfusion may reveal
distinct aspects of injury and repair mechanisms. The present study
focused on the 24 h time point because it is associated with the
acute phase of IRI-AKI and is suitable for evaluating therapeutic
interventions (20,21). Other time points should also be
studied (for example, 6, 48 and 72 h) to gain a more comprehensive
understanding of the injury progression and recovery processes.
In conclusion, ELE suppressed the inflammatory
response and apoptosis by downregulating the stimulation of
TLR4/MyD88/NF-κB/MAPK signaling, which further prevented kidney
dysfunction following IR. ELE may be an anti-inflammatory agent to
treat AKI.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Shanghai Jiao Tong University
Affiliated Sixth People's Hospital Basic Research Youth Cultivation
Project (grant no. ynqn201313), Scientific Research Project of
Shanghai Qingpu District Health and Wellness Committee (grant no.
QWJ2022-19), Shanghai Municipal Health Commission Key Clinical
Medicine Discipline (grant no. 2024ZDXK0008) and Shanghai Qingpu
District High-Level Discipline (grant no. GF2023-7).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WS, SB and QG conceived and designed the study. QG,
YW, FL, YH, LL and DC interpreted data. QG and YW wrote the
manuscript. WS revised the manuscript. WS and QG confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the ethics
committee of Youjiang Medical University (approval no. 2023090601).
All methodologies are documented in compliance with ARRIVE
standards (arriveguidelines.org) for submitting reports of animal
research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Waikar SS, Liu KD and Chertow GM:
Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J
Am Soc Nephrol. 3:844–861. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boratyńska M, Kamińska D and Mazanowska O:
Pathophysiology of ischemia-reperfusion injury in renal
transplantation. Postepy Hig Med Dosw (Online). 58:1–8. 2004.(In
Polish). PubMed/NCBI
|
|
3
|
Dong Y, Zhang Q, Wen J, Chen T, He L, Wang
Y, Yin J, Wu R, Xue R, Li S, et al: Ischemic duration and frequency
determines AKI-to-CKD progression monitored by dynamic changes of
tubular biomarkers in IRI mice. Front Physiol. 10:1532019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Forbes JM, Hewitson TD, Becker GJ and
Jones CL: Ischemic acute renal failure: Long-term histology of cell
and matrix changes in the rat. Kidney Int. 57:2375–2385. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanz AB, Sanchez-Niño MD, Ramos AM and
Ortiz A: Regulated cell death pathways in kidney disease. Nat Rev
Nephrol. 19:281–299. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Phaniendra A, Jestadi DB and Periyasamy L:
Free radicals: Properties, sources, targets, and their implication
in various diseases. Indian J Clin Biochem. 30:11–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim J, Seok YM, Jung KJ and Park KM:
Reactive oxygen species/oxidative stress contributes to progression
of kidney fibrosis following transient ischemic injury in mice. Am
J Physiol Renal Physiol. 297:F461–F470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao L, Hao Y, Tang S, Han X, Li R and
Zhou X: Energy metabolic reprogramming regulates programmed cell
death of renal tubular epithelial cells and might serve as a new
therapeutic target for acute kidney injury. Front Cell Dev Biol.
11:12762172023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gong S, Xiong H, Lei Y, Huang S, Ouyang Y,
Cao C and Wang Y: Usp9× contributes to the development of
sepsis-induced acute kidney injury by promoting inflammation and
apoptosis in renal tubular epithelial cells via activation of the
TLR4/nf-κb pathway. Ren Fail. 46:23610892024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Devarajan P: Update on mechanisms of
ischemic acute kidney injury. J Am Soc Nephrol. 17:1503–1520. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang B, Lan S, Dieudé M, Sabo-Vatasescu
JP, Karakeussian-Rimbaud A, Turgeon J, Qi S, Gunaratnam L, Patey N
and Hébert MJ: Caspase-3 is a pivotal regulator of microvascular
rarefaction and renal fibrosis after ischemia-reperfusion injury. J
Am Soc Nephrol. 29:1900–1916. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang Q, Qian L and Zhang S: Ginsenoside
Rh1 alleviates HK-2 apoptosis by inhibiting ROS and the JNK/p53
pathways. Evid Based Complement Alternat Med. 2020:34010672020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu L, Zhang R, Lin S, Lin M and Wang J:
Silencing CDK6-AS1 inhibits LPS-induced inflammatory damage in HK-2
cells. Open Med (Wars). 16:1256–1264. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bai Z, Yao C, Zhu J, Xie Y, Ye XY, Bai R
and Xie T: Anti-tumor drug discovery based on natural product
β-Elemene: Anti-tumor mechanisms and structural modification.
Molecules. 26:14492021. View Article : Google Scholar
|
|
16
|
Zhu T, Xu Y, Dong B, Zhang J, Wei Z, Xu Y
and Yao Y: β-elemene inhibits proliferation of human glioblastoma
cells through the activation of glia maturation factor β and
induces sensitization to cisplatin. Oncol Rep. 26:405–413.
2011.PubMed/NCBI
|
|
17
|
Yu X, Li Z, Zhang Y, Xu M, Che Y, Tian X,
Wang R, Zou K and Zou L: β-elemene inhibits radiation and
hypoxia-induced macrophages infiltration via Prx-1/NF-κB/HIF-1α
signaling pathway. Onco Targets Ther. 12:4203–4211. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai SZ, Xiong QW, Zhao L, Ji YT, Luo ZX
and Ma ZR: β-elemene triggers ROS-dependent apoptosis in
glioblastoma cells through suppressing STAT3 signaling pathway.
Pathol Oncol Res. 27:5942992021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun W, Choi HS, Kim CS, Bae EH, Ma SK and
Kim SW: Maslinic acid attenuates ischemia/reperfusion-induced acute
kidney injury by suppressing inflammation and apoptosis through
inhibiting NF-κB and MAPK signaling pathway. Front Pharmacol.
13:8074522022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei Q and Dong Z: Mouse model of ischemic
acute kidney injury: Technical notes and tricks. Am J Physiol Renal
Physiol. 303:F1487–F1494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basile DP, Bonventre JV, Mehta R, Nangaku
M, Unwin R, Rosner MH, Kellum JA and Ronco C; ADQI XIII Work Group,
: ADQI XIII work group. Progression after AKI: Understanding
maladaptive repair processes to predict and identify therapeutic
treatments. J Am Soc Nephrol. 27:687–697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun W, Kim DH, Byon CH, Choi HI, Park JS,
Bae EH, Ma SK and Kim SW: β-Elemene attenuates renal fibrosis in
the unilateral ureteral obstruction model by inhibition of STAT3
and Smad3 signaling via suppressing MyD88 expression. Int J Mol
Sci. 23:55532022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Havasi A and Borkan SC: Apoptosis and
acute kidney injury. Kidney Int. 80:29–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng F, Chen Q, Gu S, Cui R, Ma Q, Cao R
and Zhao M: Inhibition of Circ-Snrk ameliorates apoptosis and
inflammation in acute kidney injury by regulating the MAPK pathway.
Ren Fail. 44:672–681. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kwon O, Hong SM, Sutton TA and Temm CJ:
Preservation of peritubular capillary endothelial integrity and
increasing pericytes may be critical to recovery from postischemic
acute kidney injury. Am J Physiol Renal Physiol. 295:F351–F359.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng Q, Xing J, Li X, Tang X and Zhang D:
PRDM16 suppresses ferroptosis to protect against sepsis-associated
acute kidney injury by targeting the NRF2/GPX4 axis. Redox Biol.
78:1034172024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Yuan F, Xiong Y, Tang Y, Li Z, Ai J,
Miao J, Ye W, Zhou S, Wu Q, et al: FAM3A plays a key role in
protecting against tubular cell pyroptosis and acute kidney injury.
Redox Biol. 74:1032252024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Linkermann A, Chen G, Dong G, Kunzendorf
U, Krautwald S and Dong Z: Regulated cell death in AKI. J Am Soc
Nephrol. 25:2689–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fang Y, Kang Y, Zou H, Cheng X, Xie T, Shi
L and Zhang H: β-elemene attenuates macrophage activation and
proinflammatory factor production via crosstalk with Wnt/β-catenin
signaling pathway. Fitoterapia. 124:92–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Q, Chen L, Zhang X, Yang H, Li Y and
Li P: β-elemene promotes microglial M2-like polarization against
ischemic stroke via AKT/mTOR signaling axis-mediated autophagy.
Chin Med. 19:862024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang G, Xue C and Zeng Y: β-elemene
alleviates airway stenosis via the ILK/Akt pathway modulated by
MIR143HG sponging miR-1275. Cell Mol Biol Lett. 26:282021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhai B, Wu Q, Wang W, Zhang M, Han X, Li
Q, Chen P, Chen X, Huang X, Li G, et al: Preparation,
characterization, pharmacokinetics and anticancer effects of
PEGylated β-elemene liposomes. Cancer Biol Med. 17:60–75. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gan D, He W, Yin H and Gou X: β-elemene
enhances cisplatin-induced apoptosis in bladder cancer cells
through the ROS-AMPK signaling pathway. Oncol Lett. 19:291–300.
2020.PubMed/NCBI
|
|
37
|
Lee RX, Li QQ and Reed E: β-elemene
effectively suppresses the growth and survival of both
platinum-sensitive and -resistant ovarian tumor cells. Anticancer
Res. 32:3103–3113. 2012.PubMed/NCBI
|
|
38
|
Zhou Y, Takano T, Wang Y, Li X, Wang R,
Wakatsuki Y, Nakajima-Adachi H, Tanokura M, Miyakawa T and
Hachimura S: Intestinal regulatory T cell induction by β-elemene
alleviates the formation of fat tissue-related inflammation.
iScience. 24:1018832021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Eltzschig HK and Collard CD: Vascular
ischaemia and reperfusion injury. Br Med Bull. 70:71–86. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ramesh G and Reeves WB: Inflammatory
cytokines in acute renal failure. Kidney Int Suppl. 91:S56–S61.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Delneste Y, Beauvillain C and Jeannin P:
Innate immunity: Structure and function of TLRs. Med Sci (Paris).
23:67–73. 2007.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen J, Hartono JR, John R, Bennett M,
Zhou XJ, Wang Y, Wu Q, Winterberg PD, Nagami GT and Lu CY: Early
interleukin 6 production by leukocytes during ischemic acute kidney
injury is regulated by TLR4. Kidney Int. 80:504–515. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu H, Chen G, Wyburn KR, Yin J, Bertolino
P, Eris JM, Alexander SI, Sharland AF and Chadban SJ: TLR4
activation mediates kidney ischemia/reperfusion injury. J Clin
Invest. 117:2847–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang LM, Liu JH, Xue CB, Li MQ, Xing S,
Zhang X, He WT, Jiang FC, Lu X and Zhou P: Pharmacological
inhibition of MyD88 homodimerization counteracts renal ischemia
reperfusion-induced progressive renal injury in vivo and in vitro.
Sci Rep. 6:269542016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Basnakian AG, Kaushal GP and Shah SV:
Apoptotic pathways of oxidative damage to renal tubular epithelial
cells. Antioxid Redox Signal. 4:915–924. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaushal GP, Basnakian AG and Shah SV:
Apoptotic pathways in ischemic acute renal failure. Kidney Int.
66:500–506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang H and Sun SC: NF-κB in inflammation
and renal diseases. Cell Biosci. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oberbauer R, Schwarz C, Regele HM,
Hansmann C, Meyer TW and Mayer G: Regulation of renal tubular cell
apoptosis and proliferation after ischemic injury to a solitary
kidney. J Lab Clin Med. 138:343–351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zou G, Zhou Z, Xi X, Huang R and Hu H:
Pioglitazone ameliorates renal ischemia-reperfusion injury via
inhibition of NF-κB activation and inflammation in rats. Front
Physiol. 12:7073442021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun W, Byon CH, Kim DH, Choi HI, Park JS,
Joo SY, Kim IJ, Jung I, Bae EH, Ma SK and Kim SW: Renoprotective
effects of maslinic acid on experimental renal fibrosis in
unilateral ureteral obstruction model via targeting MyD88. Front
Pharmacol. 12:7085752021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li X, Huang R, Liu K, Li M, Luo H, Cui L,
Huang L and Luo L: Fucoxanthin attenuates LPS-induced acute lung
injury via inhibition of the TLR4/MyD88 signaling axis. Aging
(Albany NY). 13:2655–2667. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cuarental L, Sucunza-Sáenz D, Valiño-Rivas
L, Fernandez-Fernandez B, Sanz AB, Ortiz A, Vaquero JJ and
Sanchez-Niño MD: MAP3K kinases and kidney injury. Nefrologia (Engl
Ed). 39:568–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chu W, Li M, Li F, Hu R, Chen Z, Lin J and
Feng H: Immediate splenectomy down-regulates the MAPK-NF-κB
signaling pathway in rat brain after severe traumatic brain injury.
J Trauma Acute Care Surg. 74:1446–1453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo X, Jiang H, Chen J, Zhang BF, Hu Q,
Yang S, Yang J and Zhang J: RP105 ameliorates hypoxia/reoxygenation
injury in cardiac microvascular endothelial cells by suppressing
TLR4/MAPKs/NF-κB signaling. Int J Mol Med. 42:505–513.
2018.PubMed/NCBI
|