Stroke is the second leading cause of death and the
third leading cause of disability worldwide and a great burden to
patients and society (1). Ischemic
stroke, the most common type of stroke, occurs when cerebral artery
occlusion leads to critical reduction in blood flow. Early
reperfusion remains paramount in acute ischemic stroke (AIS)
management. Intravenous thrombolysis with tissue plasminogen
activator (tPA) is one of the most effective reperfusion
therapeutic strategies and is able to rapidly restore cerebral
perfusion and alleviate the neurological deficits of patients
(2). Despite its clinical
efficacy, tPA thrombolysis remains underused, with only 5–10% of
ischemic stroke patients receiving treatment (3). This limited application primarily
stems from two key constraints: the narrow 4.5-h therapeutic window
and the risk of tPA-induced hemorrhagic transformation (HT)
(4). Therefore, early
identification of the risk of HT and timely prevention strategies
might be helpful for physicians to make more useful decisions on
the treatment for AIS.
The pathophysiological mechanisms of HT are
complicated, involving blood-brain barrier (BBB) disruption,
ischemia-reperfusion injury, oxidative stress, neuroinflammation,
tPA administration and brain cell death (3). These processes interact with each
other to promote the disruption of vascular integrity and BBB
dysfunction (5). BBB is a highly
selective functional structure that acts as a protective interface
between the central nervous system (CNS) and the circulatory
system, playing a critical role in maintaining the
microenvironmental homeostasis of CNS (6). BBB disruption is a prominent
pathophysiological feature of ischemic stroke, accompanied by worse
functional prognosis and higher mortality rate (7). The components of erythrocytes,
neurotoxic plasma components and pathogens enter the brain tissue
through the compromised BBB, markedly contributing to the
pathogenesis of HT (8).
Matrix metalloproteinases (MMPs) play a key role in
the pathological process of BBB damage by mediating the degradation
of extracellular matrix (ECM) and tight junction proteins (TJPs)
(6,9). MMPs comprise a family of
calcium-dependent zinc-containing endopeptidases and play pivotal
roles in tissue remodeling and ECM degradation, including gelatin,
elastins, collagens, matrix glycoproteins and proteoglycans
(10). These enzymes are
synthesized by neurons, astrocytes, endothelial cells, microglia
and peripheral immune cells (11).
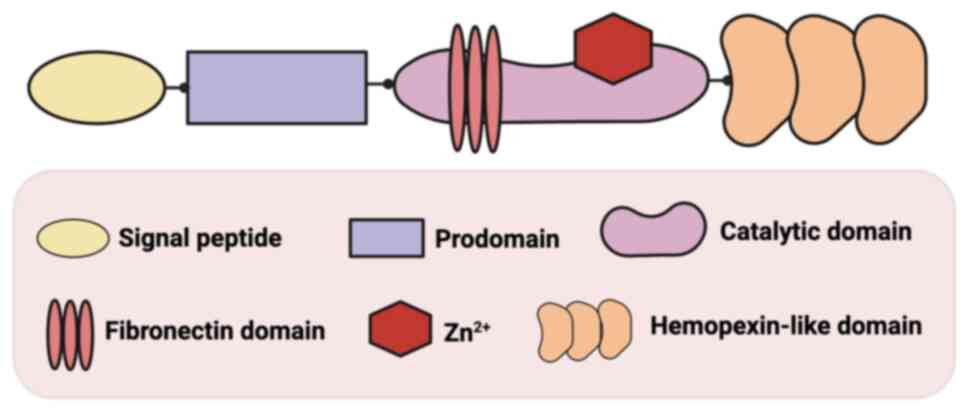
Structurally, MMPs exhibit a highly conserved domain architecture
consisting of four principal components: The amino-terminal signal
peptide domain, the propeptide region (predomain), the catalytic
domain containing zinc and variable C-terminal domains that mediate
substrate recognition and interaction, including the transmembrane
domain, the hemopexin-like domain and the fibronectin domain
(12,13). At present, at least 26 human MMPs
have been discovered, which are divided into six subgroups
according to their different structures: collagenases, gelatinases,
stromelysins, matrilysins, membrane-type MMPs and others (10,14).
Previous studies have demonstrated that MMPs high expression is
associated with BBB disruption and HT following AIS (15–17).
Among all MMP family members, MMP-9 has emerges as the most
extensively investigated protease, with numerous animal and
clinical studies establishing its crucial role in HT pathogenesis
(11,18–22).
The present review aimed to elaborate the
mechanistic involvement of MMP-9 in HT development following AIS
and evaluate its potential as biomarker and therapeutic target.
Belonging to gelatinase B family, MMP-9 comprises a
signal peptide domain, a prodomain, a catalytic domain with a
Zn2+ binding site, a fibronectin domain and a
hemopexin-like domain (Fig. 1)
(23). The signal peptide domain
mediates MMP-9 excretion from the cell (24). The predomain maintains MMP-9 in an
inactive state through binding its conserved cysteine residue to
Zn2+ in the enzyme's active center (25). The catalytic domain is responsible
for the proteolytic activity of MMP-9 (23). The fibronectin domain acts as a
modulator of substrates recognition, which is able to bind to
gelatins, laminins and collagens (26). The hemopexin-like domain plays a
crucial role in substrates specificity and the specific substrates
mainly comprise ECM components (such as collagens, gelatin and
fibronectin) and non-ECM molecules including tissue inhibitor
metalloproteinases and chemokines (23,26).
In the brain, MMP-9 is mainly produced by astrocytes, microglia and
infiltrating leukocytes (27,28).
The expression of MMP-9 is regulated by transcription factors,
inflammatory factors, chemokines and growth factors (29). MMP-9 is usually secreted in an
inactive form and activated by thrombin, MMP-3, tPA and reactive
oxygen/nitrogen species (ROS/RNS) (29–32).
Notably, zinc plays a dual role in MMP-9 regulation: As a
structural cofactor required for its proteolytic function at
physiological levels and as a pro-oxidant stimulus that indirectly
activates MMP-9 via ROS generation when accumulated in excess
(33).
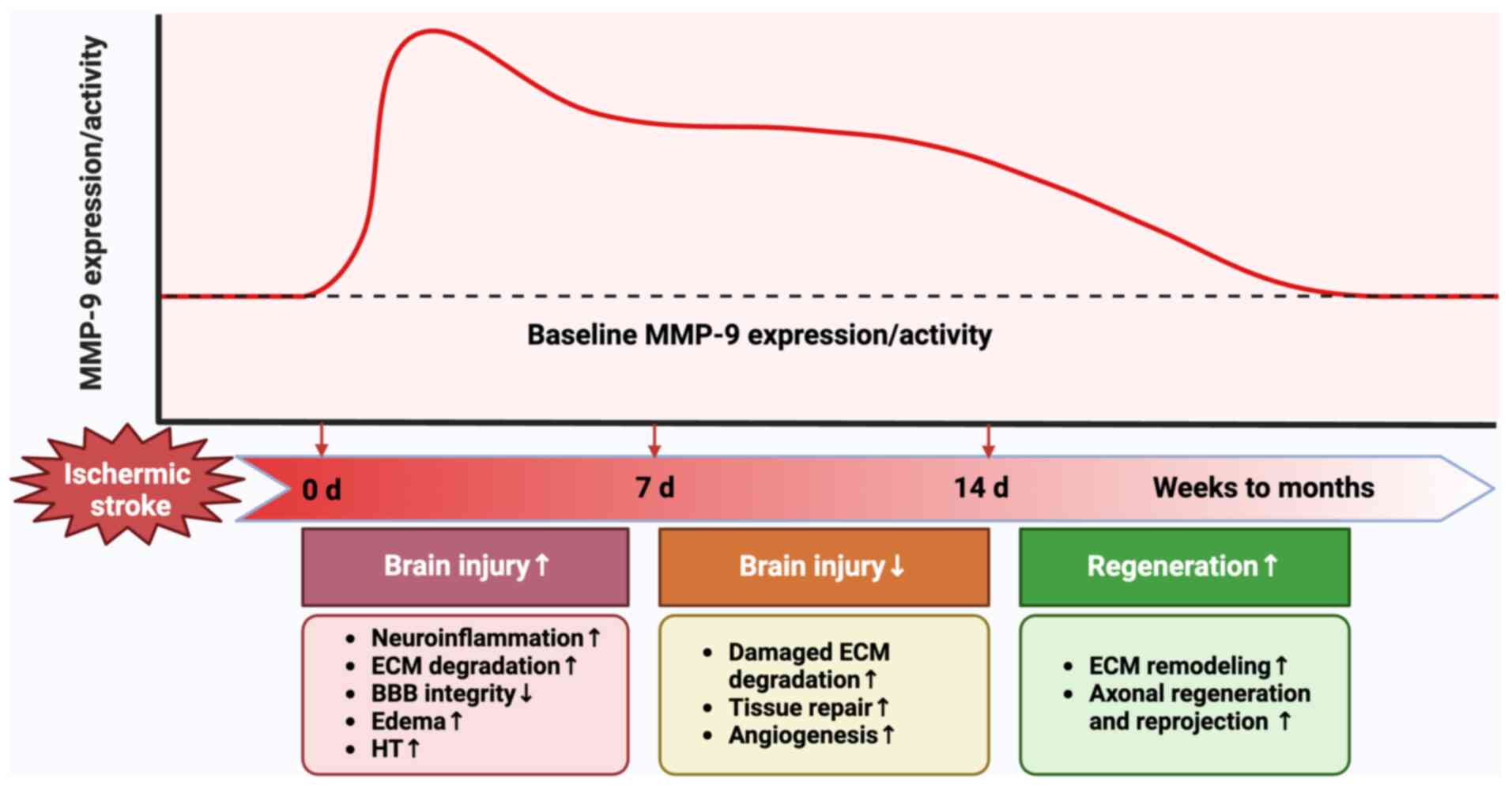
Following ischemic stroke, MMP-9 undergoes a dynamic
change, with its expression and activity varying markedly over time
(Fig. 2). The significant
upregulation of MMP-9 can be found in the acute phase of ischemic
stroke (within 7 days from onset). It has been demonstrated that
the expression and activity of MMP-9 increases rapidly and peaks
within 24 h after ischemic stroke (34,35).
During this time, MMP-9 exacerbates brain injury by damaging BBB
integrity, aggravating edema, enhancing neuroinflammation and brain
cell death and promoting HT (36).
At 7–14 days after ischemic stroke, the activity and expression of
MMP-9 still remains elevated. In this delayed phase following
ischemic stroke, MMP-9 facilitates the tissue repair and
angiogenesis by degrading damaged ECM, thereby mitigating ischemic
brain injury and prompting functional recovery (37). From weeks to months after ischemic
stroke, the expression and activity of MMP-9 gradually decreases.
This phase is characterized by ECM remodeling, in which
impenetrable scar tissues formed by gliosis hinders the
regeneration and reprojection of axons. At this time, the BBB
opening mediated by MMP-9 allows cells to enter the CNS,
facilitating functional recovery (38).
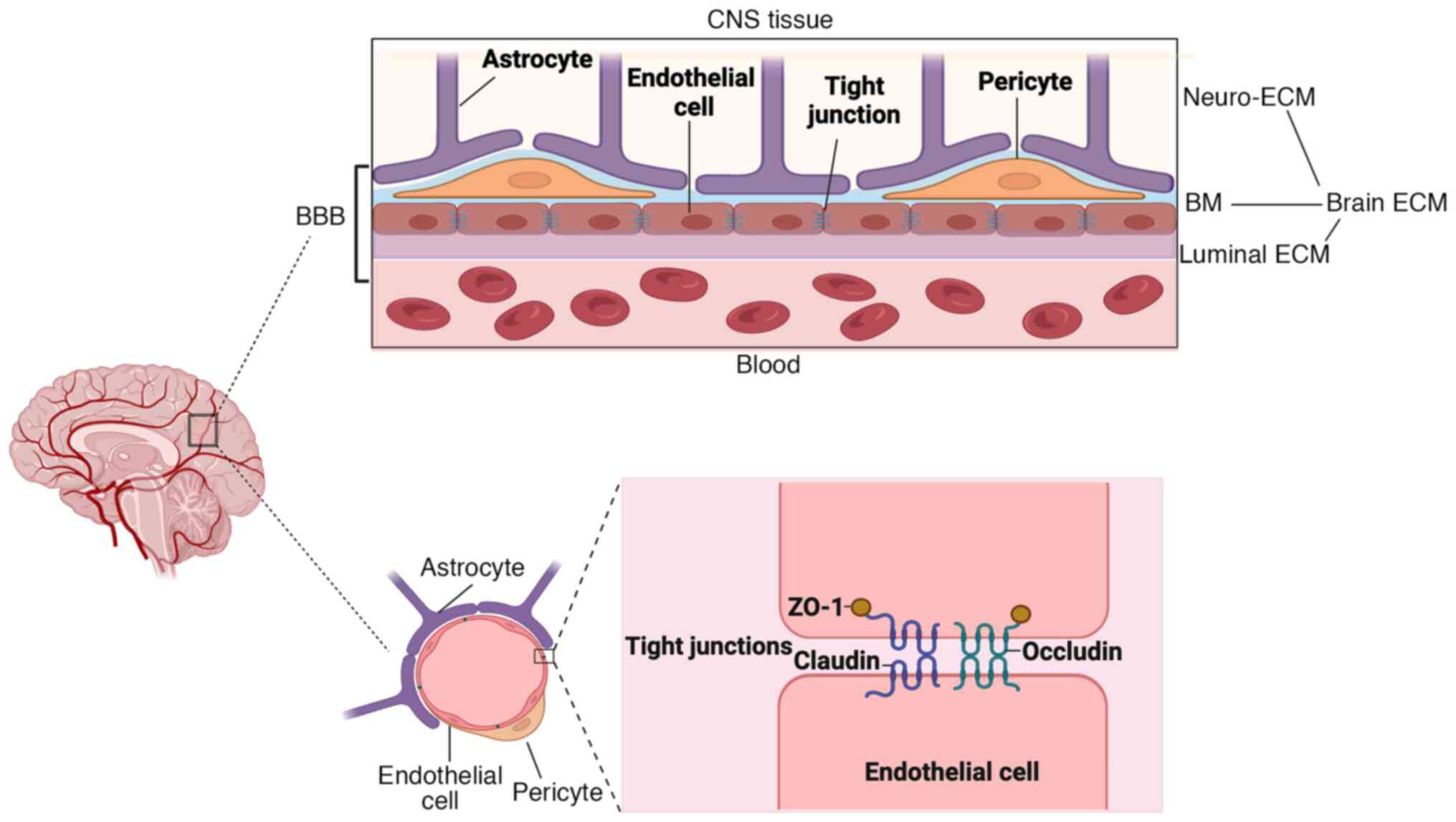
BBB is a unique structure formed by blood vessels in
the CNS, maintaining neuronal activity and blocking pathogenic
damage (39). The maintenance of
BBB function primarily relies on the interaction of endothelial
cells, astrocytes, pericytes and ECM around the vessels (Fig. 3). Endothelial cells express high
levels of TJPs, which help to prevent blood entering brain by
limiting para-cellular permeability (40). TJPs mainly include the
transmembrane proteins (claudin-5, claudin-3, claudin-12 and
occludin) and zonula occludens proteins (ZO-1, ZO-2 and ZO-3), in
which transmembrane proteins, especially claudin-5, interact with
the similar proteins in the adjacent cell to form tight junctions
and zonula occludens proteins connect the aforementioned proteins
to the cytoskeleton (41,42). The basement membrane (BM),
surrounding the lumen surface of endothelial cells, is composed of
collagen IV, fibronectin, heparin sulfate and laminins secreted by
endothelial cells, astrocytes and pericytes (43,44).
Pericytes and astrocytic endfeet are embedded in the BM, playing a
significant role in maintaining BBB integrity (45). The brain ECM consists of neuro-ECM,
BM and luminal ECM. The neuro-ECM serves as the connective
framework of the brain, forming a dynamic scaffold that supports
neurons and glia, while both the BM and the luminal ECM are
involved in forming BBB (Fig. 3)
(46). These integrated cells and
structures collectively promote the integrity of BBB, thereby
playing a critical role in regulating the brain homeostasis and
protecting the CNS.
BBB dysfunction initiates from the onset of ischemic
stroke and aggravates with the sustained hyperfusion. The
disruption of TJPs and ECM is the main reason underlying the
increased permeability of BBB after ischemic stroke, in which MMP-9
plays a critical role (47). MMP-9
primarily disrupts BBB through proteolytic degradation of key ECM
components, including collagen IV, laminin and fibronectin
(48,49). Beyond its role in ECM degradation,
MMP-9 also targets TJPs, with evidence demonstrating its specific
proteolytic effects on occludin, claudin-5 and ZO-1 (50). In addition, MMP-9 mediates BBB
disruption by degrading dystroglycan, a critical ECM receptor on
astrocyte endfeet that anchors them to the BM through the
high-affinity interactions with laminin (51,52).
HT is a common complication of ischemic stroke, with
an incidence of 10–40%, leading to increased stroke mortality
(53). The classification of HT
following ischemic stroke, as defined by the European Cooperative
Acute Stroke Study criteria, encompasses two main categories with
distinct radiographic and prognostic implications: Hemorrhagic
infarction (HI) and parenchymal hematoma (PH) (54). They are further subclassified into
four clinically relevant subtypes based on computed tomography
characteristics: HI-1, HI-2, PH-1 and PH-2 (Table I). The clinical prognosis of HT
exhibits marked heterogeneity across different subtypes. Compared
with ischemic stroke patients without HT, PH-2 markedly increases
the risk of early neurological deterioration, 3-month mortality and
disability, whereas HI-1, HI-2 and PH-1 show no statistically
significant differences in these outcomes (55). HT can be also classified into
symptomatic HT and asymptomatic HT on basis of the presence or
absence of neurological decline (56). While the pathogenesis of HT
following ischemic stroke is complicated, current evidence points
to three interconnected mechanisms, including oxidative stress
induced by ischemia-reperfusion, neuroinflammation and thrombolytic
therapy-associated toxicity (53,57,58).
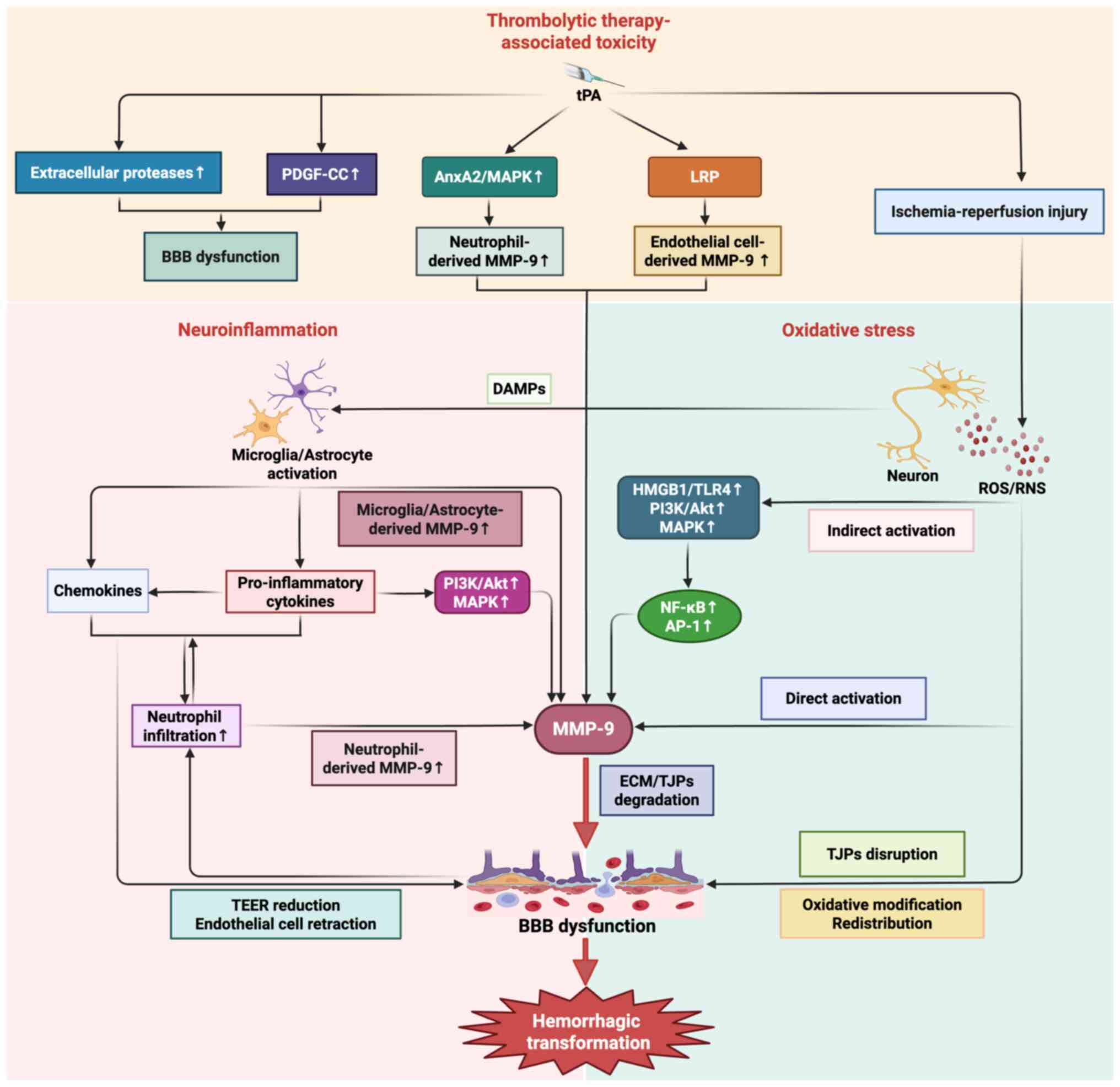
MMP-9 functions as a pivotal effector molecule in these
pathological processes (Fig.
4).
Oxidative stress is a crucial pathological process
mediating ischemia-reperfusion injury and HT, in which excess
production and activation of ROS/RNS play an important role
(32,59). After stroke, ischemia and hypoxia
of certain brain tissue induces the release of excitotoxic
substance-glutamate, which subsequently activates the calcium
channel-related receptors, especially N-methyl-D-aspartate
receptor, leading to the influx of calcium (22,60).
Calcium influx initiates a series of deleterious cascades, such as
the mitochondrial depolarization and nicotinamide adenine
dinucleotide phosphate oxidase activation, subsequently triggering
an excessive generation and activation of ROS/RNS (61,62).
Mounting evidence indicates that oxidative stress mediates BBB
dysfunction through both direct and indirect mechanisms, with MMP-9
serving as a key downstream mediator in this pathological cascade.
Direct effects involve disrupting TJPs (occludin, ZO-1 and
claudin-5) through oxidative modification and redistribution and
activating MMP-9 to catalyze the ECM degradation (63,64).
Indirectly, ROS/RNS modulate BBB permeability by oxidatively
activating multiple signaling pathways and transcriptional
regulators. Primally, ROS/RNS-mediated activation of the
high-mobility group box1 (HMGB1)/toll-like receptor 4 (TLR4)
signaling axis induces nuclear factor-κB (NF-κB)-dependent
transcriptional upregulation of MMP-9, which subsequently
exacerbates BBB disruption (32).
Furthermore, ROS activate phosphatidylinositol-3 kinase
(PI3K)/protein kinase B (Akt) and mitogen-activated protein kinase
(MAPK) signaling cascades, which promote nuclear translocation of
transcription factors, such as NF-κB and activator protein-1
(AP-1), thereby enhancing MMP-9 expression through specific
promoter binding (65,66).
Neuroinflammation following ischemic stroke is
another widely recognized contributor to BBB breakdown and HT
development. After ischemic stroke, dying brain cells release
damage-associated molecular pattern (DAMP) molecules, including
HMGB1, heat shock protein and DNA, thereby triggering microglia and
astrocytes activation and pro-inflammatory cytokines, chemokines
and MMPs production (67–69). The pro-inflammatory cytokines such
as IL-1β, IL-6 and TNF-α play a profound role in mediating BBB
breakdown (70). IL-1β enhances
chemokines production, promoting neutrophil infiltration and
subsequent MMP-9 release (71,72).
IL-6 directly compromises BBB integrity by reducing the
trans-endothelial electrical resistance value in cerebral
endothelial cells (73). TNF-α
activates MAPK and PI3K/Akt signaling pathways, stimulating
MMP-9-mediated degradation of ECM and TJPs (74). Meanwhile, upregulated chemokines,
such as monocyte chemokine protein-1 (MCP-1), macrophage
inflammatory protein-1α and stromal cell-derived factor 1, mediate
BBB disruption by inducing the endothelial cells retraction and
intercellular gap formation (75).
These pro-inflammatory factors and proteases (especially MMP-9)
collectively contribute to BBB dysfunction, promoting the
infiltration of peripheral immune cells, mainly neutrophils,
through the compromised BBB (70).
The infiltrated and activated neutrophils release more
pro-inflammatory cytokines and chemokines to enhance the activation
of microglia and astrocytes. Additionally, the upregulation of
neutrophil-derived MMP-9 further aggravates BBB dysfunction. These
interconnected pathological cascades synergistically exacerbate BBB
disruption and promote the progression of HT.
Thrombolytic therapy with tPA is a well-established
risk factor for HT. As the only US Food and Drug
Administration-approved agent for AIS, tPA can markedly improve
functional outcomes by restoring cerebral blood flow when
administered within 4.5 h of symptom onset (76). However, the fibrinolytic effect of
tPA simultaneously increases the risk of hemorrhagic complications.
Compelling clinical evidence indicates that intravenous tPA
administration in AIS patients markedly elevates HT risk,
particularly when treatment is delayed beyond the 4.5-h therapeutic
window, compared with non-thrombolytic treatment approaches
(77). Multiple mechanisms are
involved in the development of tPA-mediated HT. First, tPA promotes
the formation of HT through its pharmacological effect. Beyond its
fibrinolytic activity in clot dissolution, tPA also activates some
extracellular proteases and mediates the dysregulation of ECM
proteolysis, leading to BBB leakage (78). Second, tPA can act on the
platelet-derived growth factor receptor alpha (PDGFRα) on
perivascular astrocyte end feet and promote the expression of
PDGF-CC, leading to the upregulation of vascular permeability
(79). Last but not least, tPA can
also increase the risk of HT via multiple MMP-9-dependent pathways
(80–83): i) tPA mobilizes and activates
neutrophils, via annexin A2-dependent MAPK signaling pathway,
promoting neutrophil-derived MMP-9 secretion; ii) tPA binds the
low-density lipoprotein receptor-related protein receptor on
endothelial cells to enhance endothelial cell-derived MMP-9
expression. It is worth mentioning that although the role of tPA in
promoting HT has been well established, it is not the sole
contributing factor to HT during ischemia-reperfusion. The
excessive ROS/RNS production during ischemia-reperfusion also plays
a critical role in HT through activating MMP-9 and mediating BBB
dysfunction (53). Of particular
clinical relevance, the delayed administration of tPA (>4.5 h
post-ischemia) potentiates this pathological process, thereby
amplifying BBB disruption and HT risk during cerebral
ischemia-reperfusion (84). This
pathophysiological cascade elucidates the clinical observation of
elevated HT risks when thrombolysis is administered beyond the
4.5-h therapeutic window.
The role of MMP-9 in mediating BBB disruption
highlights its potential as a predictive biomarker for HT. Previous
reviews revealed that MMP-9 concentration in peripheral blood was
positively associated with larger infarct size, worse prognosis and
higher HT risk after AIS (85,86).
There was a clinical study supporting the aforementioned viewpoints
as well. With 168 patients with ischemic stroke and 40 healthy
controls included in this study, Yuan et al (87) determined the plasma MMP-9
concentrations of these patients through enzyme-linked
immunosorbent assay, finding that higher plasma MMP-9 concentration
was markedly associated with increased spontaneous HT risk and
MMP-9 value >181.7 ng/ml within 24 h after stroke onset was an
independent marker to predict HT risk in ischemic stroke patients.
However, Montaner et al (16) noted that the predictive value of
plasma MMP-9 level differed in different HT subtypes. The authors
found that baseline plasma MMP-9 levels positively corelated to HT
severity following tPA administration: Lowest MMP-9 levels in HI-1
group (lower than non-HT group), higher but almost normal levels in
HI-2 group, markedly higher in PH-1 group and highest in PH-2
group, which highlights the predictive value of baseline plasma
MMP-9 in severe HT. This phenomenon may be attributable to the role
of MMP-9 in mediating BBB dysfunction and promoting HT following
ischemic stroke. Although substantial clinical evidence has
established the specific predictive role of MMP-9 for HT following
ischemic stroke, most clinical trials to date rely exclusively on
peripheral blood MMP-9 measurements. This methodological limitation
primarily reflects the significant ethical constraints and
technical difficulties involved in accurately quantifying MMP-9
concentrations in the human brain. The results from an animal study
clarified that MMP-9 activity within brain was positively
associated with edema and infarct volume after ischemic stroke
(88). Another study performed in
mice and endothelial cells cultured in vitro reported that
tPA-induced HT could be markedly attenuated when suppressing the
expression of brain MMP-9 (89).
These experimental findings from animal models and cell cultures
provide indirect evidence supporting the predictive role of brain
MMP-9 in HT. Additionally, some researchers suggested that an
imbalance of MMP-9 and TIMP-1 contributed to the occurrence of HT
(90). TIMP-1 is secreted together
with MMP-9 and inhibits its proteolytic activity. The higher
MMP-9/TIMP-1 ratio usually indicates the higher MMP-9 activity and
HT risk. Notably, preclinical evidence from rat AIS models revealed
the parallel MMP-9/TIMP-1 ratio changes in serum and brain tissue
(91), indicating their comparable
predictive capacity for HT.
As an important factor regulating MMP-9 expression,
MMP-9 gene polymorphism represents a promising biomarker for
predicting HT risk after ischemic stroke. In a case-control study
involving 1,274 ischemic stroke patients and 1,258 healthy
controls, researchers investigated four critical polymorphic sites
of MMP-9 (rs17156, rs3787268, rs3918241 and rs3918242), finding
that rs3918242 (−1562C/T) polymorphism was markedly associated with
serum MMP-9 levels (92). In
another clinical study including 222 ischemic stroke patients
stratified by magnetic resonance imaging findings into HT and
non-HT groups, researchers genotyped the −1562C/T polymorphism
using PCR-restriction fragment length polymorphism analysis,
revealing that 1562C/T polymorphism was markedly associated with HT
risk, with the T allele potentially serving as a predictive
biomarker for HT susceptibility (93). Contrary to previous findings,
another study performed in the cerebrovascular disease patients
found no correlation between −1562C/T polymorphism and spontaneous
HT (94). Additionally,
Fernández-Cadenas et al (95) genotyped 14 single nucleotide
polymorphisms (SNPs) in MMP-9 gene, demonstrating no correlation
between MMP-9 genetic variations and HT. These inconsistent results
can be explained by several contributing factors. First,
population-specific genetic differences play a crucial role in
these observed variations. A comprehensive meta-analysis revealed
the correlation between MMP-9 gene −1562C/T polymorphism and stroke
risk among Asians, but not among Caucasians (96). Second, the pathophysiological
mechanisms underlying HT encompass a complex cascade of molecular
and cellular events, including BBB disruption, inflammatory
responses, oxidative stress and MMPs activation, which collectively
contribute to the development and progression of this condition.
Therefore, genetic polymorphisms in MMP-9 may not markedly
influence the occurrence of HT, as the development of HT is
mediated through complex interactions among multiple genetic,
molecular and environmental factors. Third, the interaction of
different genetic variants in MMP-9 gene should not be ignored. Yi
et al (97) demonstrated
that the increased HT risk was attributable to the synergistic
interaction between rs3918242 and rs3787268 genetic variants,
rather than being caused by individual variant alone. Finally,
variations in sample size and statistical methodologies may also
contribute to the inconsistencies in research findings.
Reducing HT following ischemic stroke may be
beneficial to extend the therapeutic window of tPA, increase
eligibility for thrombolytic therapy and improve the prognosis of
patients. As a key mediator in HT pathogenesis, MMP-9 may be a
prospective pharmacological target for HT. Although the predictive
role of MMP-9 in severe HT following ischemic stroke has been well
established, whether therapies targeting MMP-9 have varying
efficacy based on HT subtypes remains unknown. To date, no
selective MMP-9 inhibitors have passed clinical trials. The present
review evaluated clinically available neuroprotective agents that
demonstrate indirect regulatory effects on MMP-9 activity or
expression, aiming to elucidate their molecular pathways of MMP-9
regulation and provide evidence-based recommendations for
optimizing therapeutic strategies to mitigate post-ischemic HT risk
(Table II).
Statins, 3-hydroxy-3-methylglutaryl coenzyme A
reductase inhibitors, are used for the primary and secondary
prevention of AIS for its role of plaque stabilization,
anti-inflammatory and neuroprotective effects (98). In a previous study using an
embolized rabbit model, tPA was administered 1 h following
embolization. The results suggested that stains treatment markedly
reduced MMP-9 levels, preserved cerebrovascular integrity and
lowered the hemorrhagic incidence associated with intravenous
thrombolysis (99). Another study
revealed that lipophilic statins (such as simvastatin and
atorvastatin) markedly reduced MMP-9/TIMP-1 ratio and suppressed
MMP-9 activity in human endothelial cells (90). Furthermore, both in vivo and
in vitro studies demonstrated that early administration of
atorvastatin or simvastatin attenuated the risk of HT by inhibiting
MMP-9 expression and preserving BBB integrity (100,101).
Simvastatin is an inhibitor of the Ras homolog
family member A (RhoA)/Rho-associated coiled-coil containing
kinases (ROCK) signaling pathway. In a rat model of AIS where t-PA
was administered 3 h after occlusion, simvastatin pretreatment
markedly alleviated tPA-induced HT (91). A randomized controlled trial has
demonstrated that the protective effects of simvastatin are
mediated through the suppression of MMP-9 activation and the
reduction of the MMP-9/TIMP-1 ratio (102). The potentially mechanisms may be
associated with the inhibition of RhoA/ROCK signaling pathway.
Simvastatin has been found to inhibit geranylgeranylation of RhoA,
prevent its translocation to plasma membrane and suppress the
subsequent activation of ROCK (103). RhoA/ROCK pathway mediates myosin
light chain phosphorylation and activation, which is necessary for
vesicular transport and Cyclophilin A secretion. Cyclophilin A
might promote the secretion and activation of MMP-9 via enhancing
free radical generation and upregulating extracellular matrix
metalloproteinase inducer (EMMPRIN) (104). Concerning atorvastatin, in the
middle cerebral artery occlusion (MCAO) rat model, Liu et al
(105) found that atorvastatin
markedly suppressed tPA-induced MMP-9 mRNA upregulation. This
finding is further supported by clinical evidence from a randomized
trial in non-ST-elevation acute coronary syndrome patients, where
atorvastatin markedly suppressed serum MMP-9 activity (106). A preclinical study conducted in
embolic stroke rat model indicated that combined therapy of
atorvastatin administered at 4 h and delayed tPA at 6 h after
stroke decreased tPA-induced MMP-9 upregulation and HT, indicating
that atorvastatin could help extend the therapeutic window of tPA
for ischemic stroke (107). In
addition, Bellosta et al (108) investigated the effects of
fluvastatin on MMP-9 activity in mouse and human macrophages and
found that fluvastatin markedly suppressed MMP-9 activity in a
dose-dependent manner.
Despite these promising preclinical findings,
clinical evidence supporting the efficacy of statins in reducing HT
risk following ischemic stroke remains limited. Although stains
have demonstrated an established safety in clinical practice, its
potential adverse effects should not be ignored, particularly
myopathy, new-onset diabetes mellitus and cognitive dysfunction
(109). These adverse events may
occur under specific clinical circumstances, but their absolute
risks remain substantially lower than the demonstrated therapeutic
benefits.
Edaravone, a potent free radical scavenger, has been
widely used in AIS patients due to its anti-inflammatory and
neuroprotective properties. Clinical data have highlighted that
edaravone markedly reduces neurological deficits and improve the
life quality in ischemic stroke patients, with an excellent safety
profile (110). A minimal
proportion of edaravone-treated patients experienced mild adverse
events, such as headache and dizziness, both of which were
self-limiting and did not require treatment withdrawal (111). Growing evidence suggests that
edaravone may play a crucial role in maintaining BBB integrity
(112,113). A number of clinical studies have
demonstrated its potential in reducing HT risk. A randomized
controlled trial involving 65 AIS patients with diabetes showed
markedly lower HT incidence in the edaravone-treated group Compared
with the controls (114). Further
evidence revealed that edaravone administration not only decreased
the incidence of HT, but also markedly attenuated its severity in
ischemic stroke patients (115).
These clinical findings are supported by robust preclinical
evidence. Okamura et al (116) reported that edaravone markedly
reduced the hematoma volumes in a rat ischemic stroke model.
Further animal study revealed that edaravone downregulated the
expression and activity of MMP-9, thereby decreasing tPA-induced HT
risk (117). Similar protective
effects were observed in a cerebral hypoperfusion mouse model,
where edaravone preserved BBB integrity through MMM-9 inhibition
(118). The underlying mechanism
appears to involve the suppression of the NF-κB pathway, leading to
reduced MMP-9 expression and enhanced BBB stabilization (119,120). NF-κB is a crucial transcriptional
regulator that becomes rapidly activated during the acute phase of
ischemic stroke. This activation occurs primarily through the
canonical pathway, where inflammatory stimuli, such as TNF-α, IL-1β
and ROS, trigger the phosphorylation and subsequent degradation of
inhibitor κB (IκB) by the IκB kinase (IKK) complex (121). The liberated NF-κB dimers
(predominantly p50/p65) then translocate to the nucleus, where they
bind to specific κB sites in promoter regions to upregulate the
expression of numerous pro-inflammatory mediators, including
cytokines (TNF-α and IL-6), chemokines (MCP-1) and MMP-9 (122). Edaravone exerts its inhibitory
effect on NF-κB activation through multiple mechanisms (119,123): i) Edaravone scavenges ROS that
serve as upstream activators of the NF-κB pathway; ii) Edaravone
suppresses the phosphorylation and subsequent proteasomal
degradation of IκBα, thereby preventing the nuclear translocation
of liberated NF-κB dimers (particularly p50/p65), ultimately
inhibiting their DNA-binding activity and transcriptional
regulation of downstream target genes. Contrary to previous
findings, a large case-control study (n=613) found increased HT
incidence in ischemic stroke patients with edaravone treatment
(124). These discrepancies
highlight the need for further large-scale studies to clarify the
precise pharmacological effects of edaravone on HT following
ischemic stroke.
Minocycline, a semi-synthetic second-generation
tetracycline antibiotic, demonstrates significant neuroprotective
properties and exhibits therapeutic potential for multiple
neurological disorders, such as ischemic stroke, intracerebral
hemorrhage (ICH), epilepsy, multiple sclerosis, Parkinson's
disease, Alzheimer's disease and spinal cord injury (125,126). Multiple randomized controlled
trials have consistently established the favorable safety profile
of minocycline in acute stroke management, while its therapeutic
efficacy requires further validation (127–129). In a clinical trial evaluating the
efficacy and safety of minocycline as an adjunct to tPA therapy in
ischemic stroke patients within 6 h of onset, no severe hemorrhages
were observed in minocycline-treated cohort (130). Further clinical investigation
demonstrated that combining minocycline with tPA appeared to
mitigate thrombolytic therapy-associated complications, primarily
through the inhibition of MMP-9 activity. Meanwhile, comprehensive
preclinical studies have established that the neuroprotective
properties of minocycline are largely attributable to its
inhibitory effects on MMP-9 activity or expression. A previous
study has revealed that minocycline, at concentrations ranging from
20 nM-20 µM, effectively alleviated oxygen-glucose
deprivation-induced cell cytotoxicity by suppressing both MMP-9
expression and enzymatic activity (131). Minocycline also exerted a
protective role in reducing ischemic lesion volume and decreasing
HT risk by inhibiting MMP-9 (132). Combined administration of
minocycline with tPA could lower the tPA-induced HT risk and extend
the narrow treatment time windows of tPA in experimental stroke
models, which was mediated by plasma MMP-9 downregulation (133). Our previous research demonstrated
that the administration of minocycline could inhibit the expression
of EMMPRIN, a key inflammatory mediator promoting MMPs production,
thereby downregulating MMP-9 expression and alleviating BBB injury
(134,135). Minocycline likely regulates
EMMPRIN expression through attenuating IKK/IκBα phosphorylation,
impeding NF-κB nuclear translocation and subsequently suppressing
EMMPRIN transcriptional activation (136,137). While this proposed mechanism is
plausible, rigorous experimental validation through approaches such
as chromatin immunoprecipitation assays to demonstrate the reduced
binding of NF-κB to the EMMPRIN promoter following minocycline
treatment is currently lacking in the literature. In addition,
minocycline also protected the integrity of BBB and reduced the
risk of HT by targeting neuroinflammation via suppressing the
migration and infiltration of neutrophils into the brain and
inhibiting the activation of microglia (57,138).
Diabetes mellitus is characterized by hyperglycemia
and microvascular damage of multiple organs. Clinical evidence
indicates that hyperglycemia is closely associated with an
increased risk of symptomatic ICH, particularly in patients with
blood glucose levels >200 mg/dl at stroke onset, who might face
a substantially greater risk (139). The pathophysiological basis for
this clinical observation may involve hyperglycemia-induced BBB
disruption. It has been demonstrated that hyperglycemia disrupts
BBB homeostasis by impairing the cerebral endothelial cell function
through dysregulating redox signaling, inflammatory mediators and
vasoactive factors (140).
Furthermore, both in vitro and in vivo studies have
revealed that hyperglycemia impaired BBB integrity via suppressing
TJPs expression in the neurovascular unit (141,142). Additionally, clinical data form a
study enrolling 287 patients demonstrated higher HT incidence among
those with stress hyperglycemia, suggesting its potential role in
post-stroke HT development (143).
Considering the contribution of hyperglycemia to BBB
damage and HT, antidiabetic drugs might represent promising
therapeutic candidates to prevent the HT following ischemic stroke.
Thiazolidinediones, as peroxisome proliferator-activated receptor
(PPAR) agonists, have been extensively studied, among which
rosiglitazone shows promise in lowering HT risk after ischemic
stroke (144). In a rat MCAO
model, rosiglitazone treatment effectively protected against BBB
disruption and mitigated tPA-induced HT after stroke (145). While the precise mechanisms of
the BBB protective effects of rosiglitazone remain incompletely
understood, accumulating evidence suggests that MMP-9
downregulation may play a pivotal role. Clinical data from type 2
diabetic patients demonstrated a markedly reduction of MMP-9 serum
levels in those treated with rosiglitazone (146). In rabbit ICH models treated with
minimally invasive evacuation, rosiglitazone was shown to decrease
MMP-9 expression and protect BBB integrity (147). Another animal study using rat
embolic stroke models indicated that rosiglitazone treatment
prevented the reduction of collagen type IV and stabilized BBB
function by inhibiting MMP-9 activation (148). Furthermore, experimental studies
have elucidated the molecular mechanisms by which rosiglitazone
modulates MMP-9 expression. Rosiglitazone activates PPARγ-mediated
transcription, which enhances adiponectin production and initiates
downstream signaling pathways, leading to glycogen synthase
kinase-3 β (GSK-3β) activation (149). The activated GSK-3β exerts its
regulatory effects by stabilizing IκBα, thereby maintaining NF-κB
in an inactive cytoplasmic state (150). This GSK-3β-mediated suppression
of NF-κB nuclear translocation and DNA binding capacity results in
significant downregulation of MMP-9 expression through the
inhibition of promoter activity and subsequent attenuation of gene
transcription (151). Notably,
clinical evidence indicates that rosiglitazone therapy is
associated with an increased risk of heart failure in patients with
type 2 diabetes mellitus (152).
Therefore, comprehensive cardiovascular evaluation, including
assessment of left ventricular function and fluid status, should be
routinely performed prior to treatment initiation and during
follow-up monitoring.
Cilostazol, a selective phosphodiesterase III
inhibitor, is primarily used as an antiplatelet agent for patients
with intermittent claudication or ischemic stroke. A multicenter
randomized controlled trial involving patients with lacunar stroke
demonstrated that cilostazol markedly reduced the incidence of
recurrent ischemic stroke while maintaining a favorable safety
profile, with no increase in severe or life-threatening bleeding
events (153). Additionally,
clinical data demonstrates that cilostazol is associated with a
significant lower incidence of HT, compared with other antiplatelet
drugs such as aspirin (154).
Meanwhile, extensive experimental evidence from animal studies has
consistently corroborated this finding. Intraperitoneally
administration of cilostazol (10 mg/kg) could effectively protect
against HT in a transient MCAO mouse model (155). Cilostazol might protect against
tPA-induced brain edema and HT by downregulating microglia-derived
MMP-9 in mice with focal cerebral ischemia (156). Consistent with the previous
reports, administration of cilostazol for 7 days before ischemia
markedly decreased the risk of HT following injection of tPA, which
was associated with the inhibition of MMP-9 activity (157). Mechanistically, it has been
revealed that cilostazol inhibited the translocation of NF-κB to
nuclear and decreased the activity of MMP-9 promoter, indicating
that cilostazol suppressed MMP-9 expression at transcriptional
level (158). Although the
detailed mechanisms underlying the inhibitory effects of cilostazol
on NF-κB pathway have not been fully elucidated, AMP-activated
protein kinase (AMPK) appears to play a critical role. As reported
in a previous study, cilostazol promoted AMPK phosphorylation,
which subsequently blocked NF-κB activation (159). Conversely, another study
demonstrated that cilostazol attenuated warfarin-induced HT through
upregulation of TJPs (claudin-5 and ZO-1) and VE-cadherin
expression, while showing no significant effect on MMP-9 levels
(160). These divergent results
indicate that the protective role of cilostazol in HT involves
multiple pathways, warranting systematic exploration of its
underlying mechanisms.
MMP-9 is involved in a number of pathophysiological
processes associated with BBB breakdown and HT after ischemic
stroke, making it a promising predictive biomarker for HT risk.
Accumulating evidence from preclinical and clinical studies
indicates that pharmacological agents, such as statins, edaravone,
minocycline, rosiglitazone and cilostazol, can attenuate MMP-9
expression or activity, thereby preserving BBB integrity and
reducing HT incidence following ischemic stroke. The combined
administration of these therapeutic agents may effectively reduce
tPA-induced HT while potentially extending the therapeutic window
for thrombolytic therapy in ischemic stroke.
Not applicable.
The present study was supported by grant support from National
Key Research and Development Program of China (grant no.
2018YFC1312200) and the National Natural Science Foundation of
China (grant nos. 82071331 and 81870942).
Not applicable.
PG conceived and drafted the manuscript. HL and XZ
performed the literature search. YL and SX revised the manuscript
and created the figures. MX and VY reviewed and revised the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Feigin VL and Owolabi MO; World Stroke
Organization-Lancet Neurology Commission Stroke Collaboration
Group, : Pragmatic solutions to reduce the global burden of stroke:
A orld stroke organization-lancet neurology commission. Lancet
Neurol. 22:1160–1206. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hasan TF, Hasan H and Kelley RE: Overview
of acute ischemic stroke evaluation and management. Biomedicines.
9:14862021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Otsu Y, Namekawa M, Toriyabe M, Ninomiya
I, Hatakeyama M, Uemura M, Onodera O, Shimohata T and Kanazawa M:
Strategies to prevent hemorrhagic transformation after reperfusion
therapies for acute ischemic stroke: A literature review. J Neurol
Sci. 419:1172172020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goncalves A, Su EJ, Muthusamy A,
Zeitelhofer M, Torrente D, Nilsson I, Protzmann J, Fredriksson L,
Eriksson U, Antonetti DA and Lawrence DA: Thrombolytic tPA-induced
hemorrhagic transformation of ischemic stroke is mediated by PKCβ
phosphorylation of occludin. Blood. 140:388–400. 2022.PubMed/NCBI
|
|
5
|
Kovács KB, Bencs V, Hudák L, Oláh L and
Csiba L: Hemorrhagic transformation of ischemic strokes. Int J Mol
Sci. 24:140672023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu W and Wen J: The relationship among
H2S, neuroinflammation and MMP-9 in BBB injury following ischemic
stroke. Int Immunopharmacol. 146:1139022025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Desilles JP, Rouchaud A, Labreuche J,
Meseguer E, Laissy JP, Serfaty JM, Lapergue B, Klein IF, Guidoux C,
Cabrejo L, et al: Blood-brain barrier disruption is associated with
increased mortality after endovascular therapy. Neurology.
80:844–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Z, Nelson AR, Betsholtz C and
Zlokovic BV: Establishment and dysfunction of the blood-brain
barrier. Cell. 163:1064–1078. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosell A, Cuadrado E, Ortega-Aznar A,
Hernandez-Guillamon M, Lo EH and Montaner J: MMP-9-positive
neutrophil infiltration is associated to blood-brain barrier
breakdown and basal lamina type IV collagen degradation during
hemorrhagic transformation after human ischemic stroke. Stroke.
39:1121–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kapoor C, Vaidya S, Wadhwan V, Hitesh,
Kaur G and Pathak A: Seesaw of matrix metalloproteinases (MMPs). J
Cancer Res Ther. 12:28–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Misra S, Talwar P, Kumar A, Kumar P, Sagar
R, Vibha D, Pandit AK, Gulati A, Kushwaha S and Prasad K:
Association between matrix metalloproteinase family gene
polymorphisms and risk of ischemic stroke: A systematic review and
meta-analysis of 29 studies. Gene. 672:180–194. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Y and Rosenberg GA: Matrix
metalloproteinases as therapeutic targets for stroke. Brain Res.
1623:30–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kimura-Ohba S and Yang Y: Oxidative DNA
damage mediated by intranuclear MMP activity is associated with
neuronal apoptosis in ischemic stroke. Oxid Med Cell Longev.
2016:69273282016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui N, Hu M and Khalil RA: Biochemical and
biological attributes of matrix metalloproteinases. Prog Mol Biol
Transl Sci. 147:1–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montaner J, Alvarez-Sabín J, Molina CA,
Anglés A, Abilleira S, Arenillas J and Monasterio J: Matrix
metalloproteinase expression is related to hemorrhagic
transformation after cardioembolic stroke. Stroke. 32:2762–2767.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Montaner J, Molina CA, Monasterio J,
Abilleira S, Arenillas JF, Ribó M, Quintana M and Alvarez-Sabín J:
Matrix metalloproteinase-9 pretreatment level predicts intracranial
hemorrhagic complications after thrombolysis in human stroke.
Circulation. 107:598–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Ghorbani S, Ling CC, Yong VW and Xue
M: The extracellular matrix as modifier of neuroinflammation and
recovery in ischemic stroke and intracerebral hemorrhage. Neurobiol
Dis. 186:1062822023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang W, Li M, Chen Q and Wang J:
Hemorrhagic transformation after tissue plasminogen activator
reperfusion therapy for ischemic stroke: Mechanisms, models, and
biomarkers. Mol Neurobiol. 52:1572–1579. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Wei C, Deng L, Wang Z, Song M,
Xiong Y and Liu M: The accuracy of serum matrix metalloproteinase-9
for predicting hemorrhagic transformation after acute ischemic
stroke: A systematic review and meta-analysis. J Stroke Cerebrovasc
Dis. 27:1653–1665. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barr TL, Latour LL, Lee KY, Schaewe TJ,
Luby M, Chang GS, El-Zammar Z, Alam S, Hallenbeck JM, Kidwell CS
and Warach S: Blood-brain barrier disruption in humans is
independently associated with increased matrix metalloproteinase-9.
Stroke. 41:e123–e128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jha R, Battey TW, Pham L, Lorenzano S,
Furie KL, Sheth KN and Kimberly WT: Fluid-attenuated inversion
recovery hyperintensity correlates with matrix metalloproteinase-9
level and hemorrhagic transformation in acute ischemic stroke.
Stroke. 45:1040–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Zhang Y, Su Q, Liu Y, Li Z, Yong
VW and Xue M: Ion channel dysregulation following intracerebral
hemorrhage. Neurosci Bull. 40:401–414. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mondal S, Adhikari N, Banerjee S, Amin SA
and Jha T: Matrix metalloproteinase-9 (MMP-9) and its inhibitors in
cancer: A minireview. Eur J Med Chem. 194:1122602020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das S, Amin SA and Jha T: Inhibitors of
gelatinases (MMP-2 and MMP-9) for the management of hematological
malignancies. Eur J Med Chem. 223:1136232021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beroun A, Mitra S, Michaluk P, Pijet B,
Stefaniuk M and Kaczmarek L: MMPs in learning and memory and
neuropsychiatric disorders. Cell Mol Life Sci. 76:3207–3228. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cathcart J, Pulkoski-Gross A and Cao J:
Targeting matrix metalloproteinases in cancer: Bringing new life to
old ideas. Genes Dis. 2:26–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizoguchi H, Nakade J, Tachibana M, Ibi D,
Someya E, Koike H, Kamei H, Nabeshima T, Itohara S, Takuma K, et
al: Matrix metalloproteinase-9 contributes to kindled seizure
development in pentylenetetrazole-treated mice by converting
pro-BDNF to mature BDNF in the hippocampus. J Neurosci.
31:12963–12971. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li YJ, Wang ZH, Zhang B, Zhe X, Wang MJ,
Shi ST, Bai J, Lin T, Guo CJ, Zhang SJ, et al: Disruption of the
blood-brain barrier after generalized tonic-clonic seizures
correlates with cerebrospinal fluid MMP-9 levels. J
Neuroinflammation. 10:802013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bronisz E and Kurkowska-Jastrzebska I:
Matrix metalloproteinase 9 in epilepsy: The role of
neuroinflammation in seizure development. Mediators Inflamm.
2016:73690202016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stawarski M, Stefaniuk M and Wlodarczyk J:
Matrix metalloproteinase-9 involvement in the structural plasticity
of dendritic spines. Front Neuroanat. 8:682014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xue M, Hollenberg MD and Yong VW:
Combination of thrombin and matrix metalloproteinase-9 exacerbates
neurotoxicity in cell culture and intracerebral hemorrhage in mice.
J Neurosci. 26:10281–10291. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen H, He Y, Chen S, Qi S and Shen J:
Therapeutic targets of oxidative/nitrosative stress and
neuroinflammation in ischemic stroke: Applications for natural
product efficacy with omics and systemic biology. Pharmacol Res.
158:1048772020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi Z, Liang J, Pan R, Dong W, Shen J, Yang
Y, Zhao Y, Shi W, Luo Y, Ji X and Liu KJ: Zinc contributes to acute
cerebral ischemia-induced blood-brain barrier disruption. Neurobiol
Dis. 95:12–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Liu Y, Wei R, Yong VW and Xue M: The
important role of Zinc in neurological diseases. Biomolecules.
13:282022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Foerch C, Montaner J, Furie KL, Ning MM
and Lo EH: Invited article: searching for oracles? Blood biomarkers
in acute stroke. Neurology. 73:393–399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Romanic AM, White RF, Arleth AJ, Ohlstein
EH and Barone FC: Matrix metalloproteinase expression increases
after cerebral focal ischemia in rats: Inhibition of matrix
metalloproteinase-9 reduces infarct size. Stroke. 29:1020–1030.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rosenberg GA and Yang Y: Vasogenic edema
due to tight junction disruption by matrix metalloproteinases in
cerebral ischemia. Neurosurg Focus. 22:E42007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao BQ, Wang S, Kim HY, Storrie H, Rosen
BR, Mooney DJ, Wang X and Lo EH: Role of matrix metalloproteinases
in delayed cortical responses after stroke. Nat Med. 12:441–445.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Candelario-Jalil E, Yang Y and Rosenberg
GA: Diverse roles of matrix metalloproteinases and tissue
inhibitors of metalloproteinases in neuroinflammation and cerebral
ischemia. Neuroscience. 158:983–994. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iadecola C and Nedergaard M: Glial
regulation of the cerebral microvasculature. Nat Neurosci.
10:1369–1376. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cottarelli A, Corada M, Beznoussenko GV,
Mironov AA, Globisch MA, Biswas S, Huang H, Dimberg A, Magnusson
PU, Agalliu D, et al: Fgfbp1 promotes blood-brain barrier
development by regulating collagen IV deposition and maintaining
Wnt/β-catenin signaling. Development. 147:dev1851402020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heinemann U and Schuetz A: structural
features of tight-junction proteins. Int J Mol Sci. 20:60202019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Biswas S, Cottarelli A and Agalliu D:
Neuronal and glial regulation of CNS angiogenesis and
barriergenesis. Development. 147:dev1822792020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Milner R, Hung S, Wang X, Berg GI, Spatz M
and del Zoppo GJ: Responses of endothelial cell and astrocyte
matrix-integrin receptors to ischemia mimic those observed in the
neurovascular unit. Stroke. 39:191–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thomsen MS, Routhe LJ and Moos T: The
vascular basement membrane in the healthy and pathological brain. J
Cereb Blood Flow Metab. 37:3300–3317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kadry H, Noorani B and Cucullo L: A
blood-brain barrier overview on structure, function, impairment,
and biomarkers of integrity. Fluids Barriers CNS. 17:692020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tabet A, Apra C, Stranahan AM and Anikeeva
P: Changes in brain neuroimmunology following injury and disease.
Front Integr Neurosci. 16:8945002022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jiang X, Andjelkovic AV, Zhu L, Yang T,
Bennett MVL, Chen J, Keep RF and Shi Y: Blood-brain barrier
dysfunction and recovery after ischemic stroke. Prog Neurobiol.
163-164:144–171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rashid ZA and Bardaweel SK: Novel matrix
metalloproteinase-9 (MMP-9) inhibitors in cancer treatment. Int J
Mol Sci. 24:121332023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Luchian I, Goriuc A, Sandu D and Covasa M:
The role of matrix metalloproteinases (MMP-8, MMP-9, MMP-13) in
periodontal and peri-implant pathological processes. Int J Mol Sci.
23:18062022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen X and Wang L, Wang N, Li C, Hang H,
Wu G, Ren S, Jun T and Wang L: An apolipoprotein E receptor mimetic
peptide decreases blood-brain barrier permeability following
intracerebral hemorrhage by inhibiting the CypA/MMP-9 signaling
pathway via LRP1 activation. Int Immunopharmacol. 143 (Pt
3):1130072024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hannocks MJ, Zhang X, Gerwien H,
Chashchina A, Burmeister M, Korpos E, Song J and Sorokin L: The
gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory
processes. Matrix Biol. 75-76:102–113. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Könnecke H and Bechmann I: The role of
microglia and matrix metalloproteinases involvement in
neuroinflammation and gliomas. Clin Dev Immunol. 2013:9141042013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fiorelli M, Bastianello S, von Kummer R,
del Zoppo GJ, Larrue V, Lesaffre E, Ringleb AP, Lorenzano S,
Manelfe C and Bozzao L: Hemorrhagic transformation within 36 hours
of a cerebral infarct: relationships with early clinical
deterioration and 3-month outcome in the European Cooperative Acute
Stroke Study I (ECASS I) cohort. Stroke. 30:2280–2284. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hacke W, Kaste M, Fieschi C, Toni D,
Lesaffre E, von Kummer R, Boysen G, Bluhmki E, Höxter G, Mahagne
MH, et al: Intravenous thrombolysis with recombinant tissue
plasminogen activator for acute hemispheric stroke. The European
Cooperative Acute Stroke Study (ECASS). JAMA. 274:1017–1025. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ande SR, Grynspan J, Aviv RI and Shankar
JJS: Imaging for predicting hemorrhagic transformation of acute
ischemic stroke-a narrative review. Can Assoc Radiol J. 73:194–202.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Khatri P, Wechsler LR and Broderick JP:
Intracranial hemorrhage associated with revascularization
therapies. Stroke. 38:431–440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ma G, Pan Z, Kong L and Du G:
Neuroinflammation in hemorrhagic transformation after tissue
plasminogen activator thrombolysis: Potential mechanisms, targets,
therapeutic drugs and biomarkers. Int Immunopharmacol.
90:1072162021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kanazawa M, Takahashi T, Nishizawa M and
Shimohata T: Therapeutic strategies to attenuate hemorrhagic
transformation after tissue plasminogen activator treatment for
acute ischemic stroke. J Atheroscler Thromb. 24:240–253. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang Y, Khan S, Liu Y, Wu G, Yong VW and
Xue M: Oxidative stress following intracerebral hemorrhage: From
molecular mechanisms to therapeutic targets. Front Immunol.
13:8472462022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhao Y, Zhang X, Chen X and Wei Y:
Neuronal injuries in cerebral infarction and ischemic stroke: From
mechanisms to treatment (Review). Int J Mol Med. 49:152022.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Abdullahi W, Tripathi D and Ronaldson PT:
Blood-brain barrier dysfunction in ischemic stroke: targeting tight
junctions and transporters for vascular protection. Am J Physiol
Cell Physiol. 315:C343–C356. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fraser PA: The role of free radical
generation in increasing cerebrovascular permeability. Free Radic
Biol Med. 51:967–977. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun MS, Jin H, Sun X, Huang S, Zhang FL,
Guo ZN and Yang Y: Free radical damage in ischemia-reperfusion
injury: An obstacle in acute ischemic stroke after
revascularization therapy. Oxid Med Cell Longev. 2018:38049792018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shuvalova M, Dmitrieva A, Belousov V and
Nosov G: The role of reactive oxygen species in the regulation of
the blood-brain barrier. Tissue Barriers. May 29–2024.(Epub ahead
of print). View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hong S, Park KK, Magae J, Ando K, Lee TS,
Kwon TK, Kwak JY, Kim CH and Chang YC: Ascochlorin inhibits matrix
metalloproteinase-9 expression by suppressing activator
protein-1-mediated gene expression through the ERK1/2 signaling
pathway: Inhibitory effects of ascochlorin on the invasion of renal
carcinoma cells. J Biol Chem. 280:25202–25209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lee GH, Jin SW, Kim SJ, Pham TH, Choi JH
and Jeong HG: Tetrabromobisphenol A induces MMP-9 expression via
NADPH Oxidase and the activation of ROS, MAPK and Akt pathways in
human breast cancer MCF-7 cells. Toxicol Res. 35:93–101. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Banjara M and Ghosh C: Sterile
Neuroinflammation and strategies for therapeutic intervention. Int
J Inflam. 2017:83859612017.PubMed/NCBI
|
|
69
|
Gülke E, Gelderblom M and Magnus T: Danger
signals in stroke and their role on microglia activation after
ischemia. Ther Adv Neurol Disord. 11:17562864187742542018.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Alsbrook DL, Di Napoli M, Bhatia K, Biller
J, Andalib S, Hinduja A, Rodrigues R, Rodriguez M, Sabbagh SY,
Selim M, et al: Neuroinflammation in acute ischemic and hemorrhagic
stroke. Curr Neurol Neurosci Rep. 23:407–431. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yang C, Hawkins KE, Doré S and
Candelario-Jalil E: Neuroinflammatory mechanisms of blood-brain
barrier damage in ischemic stroke. Am J Physiol Cell Physiol.
316:C135–C153. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
McColl BW, Rothwell NJ and Allan SM:
Systemic inflammation alters the kinetics of cerebrovascular tight
junction disruption after experimental stroke in mice. J Neurosci.
28:9451–9462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
McColl BW, Rothwell NJ and Allan SM:
Systemic inflammatory stimulus potentiates the acute phase and CXC
chemokine responses to experimental stroke and exacerbates brain
damage via interleukin-1- and neutrophil-dependent mechanisms. J
Neurosci. 27:4403–4412. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
de Vries HE, Blom-Roosemalen MC, van
Oosten M, de Boer AG, van Berkel TJ, Breimer DD and Kuiper J: The
influence of cytokines on the integrity of the blood-brain barrier
in vitro. J Neuroimmunol. 64:37–43. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Takata F, Dohgu S, Matsumoto J, Takahashi
H, Machida T, Wakigawa T, Harada E, Miyaji H, Koga M, Nishioku T,
et al: Brain pericytes among cells constituting the blood-brain
barrier are highly sensitive to tumor necrosis factor-α, releasing
matrix metalloproteinase-9 and migrating in vitro. J
Neuroinflammation. 8:1062011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Dimitrijevic OB, Stamatovic SM, Keep RF
and Andjelkovic AV: Effects of the chemokine CCL2 on blood-brain
barrier permeability during ischemia-reperfusion injury. J Cereb
Blood Flow Metab. 26:797–810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kim JS: tPA Helpers in the treatment of
acute ischemic stroke: Are they ready for clinical use? J Stroke.
21:160–174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lees KR, Bluhmki E, von Kummer R, Brott
TG, Toni D, Grotta JC, Albers GW, Kaste M, Marler JR, Hamilton SA,
et al: Time to treatment with intravenous alteplase and outcome in
stroke: An updated pooled analysis of ECASS, ATLANTIS, NINDS, and
EPITHET trials. Lancet. 375:1695–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Fan X, Jiang Y, Yu Z, Yuan J, Sun X, Xiang
S, Lo EH and Wang X: Combination approaches to attenuate
hemorrhagic transformation after tPA thrombolytic therapy in
patients with poststroke hyperglycemia/diabetes. Adv Pharmacol.
71:391–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Su EJ, Fredriksson L, Geyer M, Folestad E,
Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, et al:
Activation of PDGF-CC by tissue plasminogen activator impairs
blood-brain barrier integrity during ischemic stroke. Nat Med.
14:731–737. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cuadrado E, Ortega L, Hernández-Guillamon
M, Penalba A, Fernández-Cadenas I, Rosell A and Montaner J: Tissue
plasminogen activator (t-PA) promotes neutrophil degranulation and
MMP-9 release. J Leukoc Biol. 84:207–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang X, Lee SR, Arai K, Lee SR, Tsuji K,
Rebeck GW and Lo EH: Lipoprotein receptor-mediated induction of
matrix metalloproteinase by tissue plasminogen activator. Nat Med.
9:1313–1317. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cheng T, Petraglia AL, Li Z, Thiyagarajan
M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernández JA, et
al: Activated protein C inhibits tissue plasminogen
activator-induced brain hemorrhage. Nat Med. 12:1278–1285. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Shi K, Zou M, Jia DM, Shi S, Yang X, Liu
Q, Dong JF, Sheth KN, Wang X and Shi FD: tPA mobilizes immune cells
that exacerbate hemorrhagic transformation in stroke. Circ Res.
128:62–75. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Mashaqi S, Mansour HM, Alameddin H, Combs
D, Patel S, Estep L and Parthasarathy S: Matrix metalloproteinase-9
as a messenger in the cross talk between obstructive sleep apnea
and comorbid systemic hypertension, cardiac remodeling, and
ischemic stroke: A literature review. J Clin Sleep Med. 17:567–591.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
di Biase L, Bonura A, Pecoraro PM, Carbone
SP and Di Lazzaro V: Unlocking the potential of stroke blood
biomarkers: Early diagnosis, ischemic vs. haemorrhagic
differentiation and haemorrhagic transformation risk: A
comprehensive review. Int J Mol Sci. 24:115452023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yuan R, Tan S, Wang D, Wu S, Cao X, Zhang
S, Wu B and Liu M: Predictive value of plasma matrix
metalloproteinase-9 concentrations for spontaneous haemorrhagic
transformation in patients with acute ischaemic stroke: A cohort
study in Chinese patients. J Clin Neurosci. 58:108–112. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Arkelius K, Wendt TS, Andersson H, Arnou
A, Gottschalk M, Gonzales RJ and Ansar S: LOX-1 and MMP-9
inhibition attenuates the detrimental effects of delayed rt-PA
therapy and improves outcomes after acute ischemic stroke. Circ
Res. 134:954–969. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sun X, Liu Z, Zhou L, Ma R, Zhang X, Wang
T, Fu F and Wang Y: Escin avoids hemorrhagic transformation in
ischemic stroke by protecting BBB through the AMPK/Cav-1/MMP-9
pathway. Phytomedicine. 120:1550712023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Izidoro-Toledo TC, Guimaraes DA, Belo VA,
Gerlach RF and Tanus-Santos JE: Effects of statins on matrix
metalloproteinases and their endogenous inhibitors in human
endothelial cells. Naunyn Schmiedebergs Arch Pharmacol.
383:547–554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yin B, Li DD, Xu SY, Huang H, Lin J, Sheng
HS, Fang JH, Song JN and Zhang M: Simvastatin pretreatment
ameliorates t-PA-induced hemorrhage transformation and MMP-9/TIMP-1
imbalance in thromboembolic cerebral ischemic rats. Neuropsychiatr
Dis Treat. 15:1993–2002. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li Y, Chen L, Yao S, Chen J, Hu W, Wang M,
Chen S, Chen X, Li S, Gu X, et al: Association of polymorphisms of
the matrix metalloproteinase 9 gene with ischaemic stroke in a
southern Chinese population. Cell Physiol Biochem. 49:2188–2199.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang X, Cao X, Xu X, Li A and Xu Y:
Correlation between the −1562C/T polymorphism in the matrix
metalloproteinase-9 gene and hemorrhagic transformation of ischemic
stroke. Exp Ther Med. 9:1043–1047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Szczudlik P and Borratyńska A: Association
between the −1562 C/T MMP-9 polymorphism and cerebrovascular
disease in a Polish population. Neurol Neurochir Pol. 44:350–357.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fernández-Cadenas I, Del Río-Espínola A,
Carrera C, Domingues-Montanari S, Mendióroz M, Delgado P, Rosell A,
Ribó M, Giralt D, Quintana M, et al: Role of the MMP-9 gene in
hemorrhagic transformations after tissue-type plasminogen activator
treatment in stroke patients. Stroke. 43:1398–1400. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang B, Wang Y and Zhao L: MMP-9 gene
rs3918242 polymorphism increases risk of stroke: A meta-analysis. J
Cell Biochem. 119:9801–9808. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yi X, Sui G, Zhou Q, Wang C, Lin J, Chai Z
and Zhou J: Variants in matrix metalloproteinase-9 gene are
associated with hemorrhagic transformation in acute ischemic stroke
patients with atherothrombosis, small artery disease, and
cardioembolic stroke. Brain Behav. 9:e012942019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kytö V, Åivo J and Ruuskanen JO: Intensity
of statin therapy after ischaemic stroke and long-term outcomes: A
nationwide cohort study. Stroke Vasc Neurol. 10:142–145. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lapchak PA and Han MK: The
3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor
simvastatin reduces thrombolytic-induced intracerebral hemorrhage
in embolized rabbits. Brain Res. 1303:144–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Reuter B, Rodemer C, Grudzenski S, Meairs
S, Bugert P, Hennerici MG and Fatar M: Effect of simvastatin on
MMPs and TIMPs in human brain endothelial cells and experimental
stroke. Transl Stroke Res. 6:156–159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Fang X, Tao D, Shen J, Wang Y, Dong X and
Ji X: Neuroprotective effects and dynamic expressions of MMP9 and
TIMP1 associated with atorvastatin pretreatment in
ischemia-reperfusion rats. Neurosci Lett. 603:60–65. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kurzepa J, Szczepanska-Szerej A,
Stryjecka-Zimmer M, Malecka-Massalska T and Stelmasiak Z:
Simvastatin could prevent increase of the serum MMP-9/TIMP-1 ratio
in acute ischaemic stroke. Folia Biol (Praha). 52:181–183. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Turner NA, Aley PK, Hall KT, Warburton P,
Galloway S, Midgley L, O'Regan DJ, Wood IC, Ball SG and Porter KE:
Simvastatin inhibits TNFalpha-induced invasion of human cardiac
myofibroblasts via both MMP-9-dependent and -independent
mechanisms. J Mol Cell Cardiol. 43:168–176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Skrzypiec-Spring M, Kaczorowski M,
Rak-Pasikowska A, Sapa-Wojciechowska A, Kujawa K, Żuryń A, Bil-Lula
I, Hałoń A and Szeląg A: RhoA/ROCK pathway is upregulated in
experimental autoimmune myocarditis and is inhibited by simvastatin
at the stage of myosin light chain phosphorylation. Biomedicines.
12:5962024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu XS, Zhang ZG, Zhang L, Morris DC,
Kapke A, Lu M and Chopp M: Atorvastatin downregulates tissue
plasminogen activator-aggravated genes mediating coagulation and
vascular permeability in single cerebral endothelial cells captured
by laser microdissection. J Cereb Blood Flow Metab. 26:787–796.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Gómez-Hernández A, Sánchez-Galán E, Ortego
M, Martín-Ventura JL, Blanco-Colio LM, Tarín-Vicente N,
Jiménez-Nacher JJ, López-Bescos L, Egido J and Tuñón J: Effect of
intensive atorvastatin therapy on prostaglandin E2 levels and
metalloproteinase-9 activity in the plasma of patients with
non-ST-elevation acute coronary syndrome. Am J Cardiol. 102:12–18.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhang L, Chopp M, Jia L, Cui Y, Lu M and
Zhang ZG: Atorvastatin extends the therapeutic window for tPA to 6
h after the onset of embolic stroke in rats. J Cereb Blood Flow
Metab. 29:1816–1824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bellosta S, Via D, Canavesi M, Pfister P,
Fumagalli R, Paoletti R and Bernini F: HMG-CoA reductase inhibitors
reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc
Biol. 18:1671–1678. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Collins R, Reith C, Emberson J, Armitage
J, Baigent C, Blackwell L, Blumenthal R, Danesh J, Smith GD, DeMets
D, et al: Interpretation of the evidence for the efficacy and
safety of statin therapy. Lancet. 388:2532–2561. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Sun Z, Xu Q, Gao G, Zhao M and Sun C:
Clinical observation in edaravone treatment for acute cerebral
infarction. Niger J Clin Pract. 22:1324–1327. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Batino LKJ, Escabillas CG and Navarro JC:
Edaravone's safety profile in acute ischemic stroke. Brain Behav.
14:e701582024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu J, Jiang Y, Zhang G, Lin Z and Du S:
Protective effect of edaravone on blood-brain barrier by affecting
NRF-2/HO-1 signaling pathway. Exp Ther Med. 18:2437–2442.
2019.PubMed/NCBI
|
|
113
|
Barna L, Walter FR, Harazin A, Bocsik A,
Kincses A, Tubak V, Jósvay K, Zvara Á, Campos-Bedolla P and Deli
MA: Simvastatin, edaravone and dexamethasone protect against
kainate-induced brain endothelial cell damage. Fluids Barriers CNS.
17:52020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zheng J and Chen X: Edaravone offers
neuroprotection for acute diabetic stroke patients. Ir J Med Sci.
185:819–824. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Toyoda K, Fujii K, Kamouchi M, Nakane H,
Arihiro S, Okada Y, Ibayashi S and Iida M: Free radical scavenger,
edaravone, in stroke with internal carotid artery occlusion. J
Neurol Sci. 221:11–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Okamura K, Tsubokawa T, Johshita H,
Miyazaki H and Shiokawa Y: Edaravone, a free radical scavenger,
attenuates cerebral infarction and hemorrhagic infarction in rats
with hyperglycemia. Neurol Res. 36:65–69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yagi K, Kitazato KT, Uno M, Tada Y,
Kinouchi T, Shimada K and Nagahiro S: Edaravone, a free radical
scavenger, inhibits MMP-9-related brain hemorrhage in rats treated
with tissue plasminogen activator. Stroke. 40:626–631. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Miyamoto N, Pham LD, Maki T, Liang AC and
Arai K: A radical scavenger edaravone inhibits matrix
metalloproteinase-9 upregulation and blood-brain barrier breakdown
in a mouse model of prolonged cerebral hypoperfusion. Neurosci
Lett. 573:40–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Harada K, Suzuki Y, Yamakawa K, Kawakami J
and Umemura K: Combination of reactive oxygen species and
tissue-type plasminogen activator enhances the induction of
gelatinase B in brain endothelial cells. Int J Neurosci. 122:53–59.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang CC, Hsiao LD, Tseng HC, Kuo CM and
Yang CM: Pristimerin inhibits MMP-9 expression and cell migration
through attenuating NOX/ROS-dependent NF-κB activation in rat brain
astrocytes challenged with LPS. J Inflamm Res. 13:325–341. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang H and Sun SC: NF-κB in inflammation
and renal diseases. Cell Biosci. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ridder DA and Schwaninger M: NF-κB
signaling in cerebral ischemia. Neuroscience. 158:995–1006. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Amirshahrokhi K and Imani M: Edaravone
reduces brain injury in hepatic encephalopathy by upregulation of
Nrf2/HO-1 and inhibition of NF-κB, iNOS/NO and inflammatory
cytokines. Mol Biol Rep. 52:2222025. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Mishina M, Komaba Y, Kobayashi S, Kominami
S, Fukuchi T, Mizunari T, Teramoto A and Katayama Y: Administration
of free radical scavenger edaravone associated with higher
frequency of hemorrhagic transformation in patients with
cardiogenic embolism. Neurol Med Chir (Tokyo). 48:292–297. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Liao TV, Forehand CC, Hess DC and Fagan
SC: Minocycline repurposing in critical illness: Focus on stroke.
Curr Top Med Chem. 13:2283–2290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Singh T, Thapliyal S, Bhatia S, Singh V,
Singh M, Singh H, Kumar A and Mishra A: Reconnoitering the
transformative journey of minocycline from an antibiotic to an
antiepileptic drug. Life Sci. 293:1203462022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chang JJ, Kim-Tenser M, Emanuel BA, Jones
GM, Chapple K, Alikhani A, Sanossian N, Mack WJ, Tsivgoulis G,
Alexandrov AV and Pourmotabbed T: Minocycline and matrix
metalloproteinase inhibition in acute intracerebral hemorrhage: A
pilot study. Eur J Neurol. 24:1384–1391. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fouda AY, Newsome AS, Spellicy S, Waller
JL, Zhi W, Hess DC, Ergul A, Edwards DJ, Fagan SC and Switzer JA:
Minocycline in acute cerebral hemorrhage: an early phase randomized
trial. Stroke. 48:2885–2887. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kohler E, Prentice DA, Bates TR, Hankey
GJ, Claxton A, van Heerden J and Blacker D: Intravenous minocycline
in acute stroke: A randomized, controlled pilot study and
meta-analysis. Stroke. 44:2493–2499. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Fagan SC, Waller JL, Nichols FT, Edwards
DJ, Pettigrew LC, Clark WM, Hall CE, Switzer JA, Ergul A and Hess
DC: Minocycline to improve neurologic outcome in stroke (MINOS): A
dose-finding study. Stroke. 41:2283–2287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Chen X, Chen S, Jiang Y, Zhu C, Wu A, Ma
X, Peng F, Ma L, Zhu D, Wang Q and Pi R: Minocycline reduces
oxygen-glucose deprivation-induced PC12 cell cytotoxicity via
matrix metalloproteinase-9, integrin β1 and phosphorylated Akt
modulation. Neurol Sci. 34:1391–1396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Knecht T, Borlongan C and Dela Peña I:
Combination therapy for ischemic stroke: Novel approaches to
lengthen therapeutic window of tissue plasminogen activator. Brain
Circ. 4:99–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Murata Y, Rosell A, Scannevin RH, Rhodes
KJ, Wang X and Lo EH: Extension of the thrombolytic time window
with minocycline in experimental stroke. Stroke. 39:3372–3377.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liu Y, Li Z, Khan S, Zhang R, Wei R, Zhang
Y, Xue M and Yong VW: Neuroprotection of minocycline by inhibition
of extracellular matrix metalloproteinase inducer expression
following intracerebral hemorrhage in mice. Neurosci Lett.
764:1362972021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Liu Y, Mu Y, Li Z, Yong VW and Xue M:
Extracellular matrix metalloproteinase inducer in brain ischemia
and intracerebral hemorrhage. Front Immunol. 13:9864692022.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Song ZP, Xiong BR, Guan XH, Cao F,
Manyande A, Zhou YQ, Zheng H and Tian YK: Minocycline attenuates
bone cancer pain in rats by inhibiting NF-κB in spinal astrocytes.
Acta Pharmacol Sin. 37:753–762. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Fan Y, Meng S, Wang Y, Cao J and Wang C:
Visfatin/PBEF/Nampt induces EMMPRIN and MMP-9 production in
macrophages via the NAMPT-MAPK (p38, ERK1/2)-NF-κB signaling
pathway. Int J Mol Med. 27:607–615. 2011.PubMed/NCBI
|
|
138
|
Chen Y, Won SJ, Xu Y and Swanson RA:
Targeting microglial activation in stroke therapy: Pharmacological
tools and gender effects. Curr Med Chem. 21:2146–2155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kase CS, Furlan AJ, Wechsler LR, Higashida
RT, Rowley HA, Hart RG, Molinari GF, Frederick LS, Roberts HC,
Gebel JM, et al: Cerebral hemorrhage after intra-arterial
thrombolysis for ischemic stroke: the PROACT II trial. Neurology.
57:1603–1610. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Arcambal A, Taïlé J, Rondeau P,
Viranaïcken W, Meilhac O and Gonthier MP: Hyperglycemia modulates
redox, inflammatory and vasoactive markers through specific
signaling pathways in cerebral endothelial cells: Insights on
insulin protective action. Free Radic Biol Med. 130:59–70. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Liu Y, Zhang H, Wang S, Guo Y, Fang X,
Zheng B, Gao W, Yu H, Chen Z, Roman RJ and Fan F: Reduced pericyte
and tight junction coverage in old diabetic rats are associated
with hyperglycemia-induced cerebrovascular pericyte dysfunction. Am
J Physiolo Heart Circ Physiol. 320:H549–H562. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Rom S, Heldt NA, Gajghate S, Seliga A,
Reichenbach NL and Persidsky Y: Hyperglycemia and advanced
glycation end products disrupt BBB and promote occludin and
claudin-5 protein secretion on extracellular microvesicles. Sci
Rep. 10:72742020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Yuan C, Chen S, Ruan Y, Liu Y, Cheng H,
Zeng Y, Chen Y, Cheng Q, Huang G, He W and He J: The stress
hyperglycemia ratio is associated with hemorrhagic transformation
in patients with acute ischemic stroke. Clin Interv Aging.
16:431–442. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang FH, Lin YH, Huang HG, Sun JZ, Wen SQ
and Lou M: Rosiglitazone attenuates hyperglycemia-enhanced
hemorrhagic transformation after transient focal ischemia in rats.
Neuroscience. 250:651–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li Y, Zhu ZY, Lu BW, Huang TT, Zhang YM,
Zhou NY, Xuan W, Chen ZA, Wen DX, Yu WF and Li PY: Rosiglitazone
ameliorates tissue plasminogen activator-induced brain hemorrhage
after stroke. CNS Neurosci Ther. 25:1343–1352. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Marx N, Froehlich J, Siam L, Ittner J,
Wierse G, Schmidt A, Scharnagl H, Hombach V and Koenig W:
Antidiabetic PPAR gamma-activator rosiglitazone reduces MMP-9 serum
levels in type 2 diabetic patients with coronary artery disease.
Arterioscler Thromb Vasc Biol. 23:283–288. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wu G, Wu J, Jiao Y, Wang L, Wang F and
Zhang Y: Rosiglitazone infusion therapy following minimally
invasive surgery for intracerebral hemorrhage evacuation decreases
matrix metalloproteinase-9 and blood-brain barrier disruption in
rabbits. BMC Neurol. 15:372015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Wang CX, Ding X, Noor R, Pegg C, He C and
Shuaib A: Rosiglitazone alone or in combination with tissue
plasminogen activator improves ischemic brain injury in an embolic
model in rats. J Cereb Blood Flow Metab. 29:1683–1694. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhou M, Xu A, Lam KS, Tam PK, Che CM, Chan
L, Lee IK, Wu D and Wang Y: Rosiglitazone promotes fatty acyl CoA
accumulation and excessive glycogen storage in livers of mice
without adiponectin. J Hepatol. 53:1108–1116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Medunjanin S, Schleithoff L, Fiegehenn C,
Weinert S, Zuschratter W and Braun-Dullaeus RC: GSK-3β controls
NF-kappaB activity via IKKγ/NEMO. Sci Rep. 6:385532016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Lee CS, Kwon YW, Yang HM, Kim SH, Kim TY,
Hur J, Park KW, Cho HJ, Kang HJ, Park YB and Kim HS: New mechanism
of rosiglitazone to reduce neointimal hyperplasia: Activation of
glycogen synthase kinase-3beta followed by inhibition of MMP-9.
Arterioscler Thromb Vasc Biol. 29:472–479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Home PD, Pocock SJ, Beck-Nielsen H, Curtis
PS, Gomis R, Hanefeld M, Jones NP, Komajda M and McMurray JJ;
RECORD Study Team, : Rosiglitazone evaluated for cardiovascular
outcomes in oral agent combination therapy for type 2 diabetes
(RECORD): A multicentre, randomised, open-label trial. Lancet.
373:2125–2135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Nishiyama Y, Kimura K, Otsuka T, Toyoda K,
Uchiyama S, Hoshino H, Sakai N, Okada Y, Origasa H, Naritomi H, et
al: Dual antiplatelet therapy with cilostazol for secondary
prevention in lacunar stroke: Subanalysis of the CSPS.com trial.
Stroke. 54:697–705. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Uchiyama S: Results of the Cilostazol
Stroke Prevention Study II (CSPS II): A randomized controlled trial
for the comparison of cilostazol and aspirin in stroke patients.
Rinsho Shinkeigaku. 50:832–834. 2010.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Nonaka Y, Tsuruma K, Shimazawa M,
Yoshimura S, Iwama T and Hara H: Cilostazol protects against
hemorrhagic transformation in mice transient focal cerebral
ischemia-induced brain damage. Neurosci Lett. 452:156–161. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Hase Y, Okamoto Y, Fujita Y, Kitamura A,
Nakabayashi H, Ito H, Maki T, Washida K, Takahashi R and Ihara M:
Cilostazol, a phosphodiesterase inhibitor, prevents no-reflow and
hemorrhage in mice with focal cerebral ischemia. Exp Neurol.
233:523–533. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Kasahara Y, Nakagomi T, Matsuyama T, Stern
D and Taguchi A: Cilostazol reduces the risk of hemorrhagic
infarction after administration of tissue-type plasminogen
activator in a murine stroke model. Stroke. 43:499–506. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chuang SY, Yang SH, Chen TY and Pang JH:
Cilostazol inhibits matrix invasion and modulates the gene
expressions of MMP-9 and TIMP-1 in PMA-differentiated THP-1 cells.
Eur J Pharmacol. 670:419–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
da Motta NA and de Brito FC: Cilostazol
exerts antiplatelet and anti-inflammatory effects through AMPK
activation and NF-kB inhibition on hypercholesterolemic rats.
Fundam Clin Pharmacol. 30:327–337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Kitashoji A, Egashira Y, Mishiro K, Suzuki
Y, Ito H, Tsuruma K, Shimazawa M and Hara H: Cilostazol ameliorates
warfarin-induced hemorrhagic transformation after cerebral ischemia
in mice. Stroke. 44:2862–2868. 2013. View Article : Google Scholar : PubMed/NCBI
|