Introduction
Ferroptosis is a regulated form of cell death
distinct from necrosis, apoptosis, pyroptosis, and autophagy. It is
characterized by iron-dependent oxidative damage and lipid
peroxidation (1,2). The term ‘ferroptosis’ was first
introduced by Dixon et al (3) in 2012. This form of cell death can be
inhibited by iron chelators such as deferoxamine (DFO) and
radical-trapping antioxidants like ferrostatin-1 (Fer-1) and
liproxstatin-1 (Lip-1) (4,5). A key step in the prevention of
ferroptosis is the synthesis of glutathione (GSH), which requires
intracellular cystine uptake via the system χc- on the cell
membrane (6). Consequently,
inhibitors of system χc- such as erastin are well-established
inducers of ferroptosis (3).
Recently, we reported that 2-deoxy-d-ribose (dRib)
also induces ferroptosis in renal tubular epithelial cells (RTECs)
by increasing the ubiquitination and proteasomal degradation of
solute carrier family 7 member 11 (SLC7A11), a functional subunit
of system χc- (7). Ferroptosis has
been implicated in the pathogenesis of various diseases, including
cancer, ischemia/reperfusion injury, chronic neurodegenerative
diseases, and acute kidney injury (8,9).
Diabetic kidney disease (DKD), which affects 20–40%
of patients with diabetes, is the most common cause of end-stage
kidney disease (10). Although
angiotensin-converting enzyme inhibitors, angiotensin receptor
blockers, sodium-glucose co-transporter 2 inhibitors, and
nonsteroidal mineralocorticoid receptor antagonists have been shown
to slow the progression of DKD, patients often still develop
chronic kidney disease (CKD) and require dialysis (11,12).
This highlights the urgent need for more effective therapies to
treat DKD. Ferroptosis, a regulated form of cell death, has
recently been identified as a key factor in the progression of DKD,
particularly in renal tubular epithelial cells (RTECs) (13,14).
In patients with DKD, serum ferritin levels are elevated, whereas
the mRNA expression of SLC7A11 and GPX4 in kidney tissue is
decreased-a molecular pattern strongly associated with ferroptosis
(15,16). Similarly, in animal models of DKD,
kidney tissues showed increased levels of malondialdehyde (MDA), a
marker of lipid peroxidation, and elevated expression of acyl-CoA
synthetase long-chain family member 4 (ACSL4), a known ferroptosis
marker (17). Moreover, studies by
Wang et al (18) and Li
et al (19) have
demonstrated clear signs of ferroptosis, such as iron overload and
increased reactive oxygen species (ROS) from lipid peroxidation, in
kidney tissues from streptozotocin-induced diabetic mice and in
RTECs exposed to high glucose levels.
Bardoxolone methyl (BM) is a synthetic triterpenoid
derived from the natural compound oleanolic acid and possesses
antioxidant and anti-inflammatory properties. The primary mechanism
of BM involves disrupting the interaction between nuclear factor
erythroid 2-related factor 2 (Nrf2) and Kelch-like ECH-associated
protein 1 (Keap1), thereby activating the intracellular Nrf2
signaling pathway. This prevents Nrf2 degradation, promotes its
nuclear translocation, and enables it to bind to antioxidant
response elements (ARE) in the promoter regions of target genes,
thereby regulating their expression (20,21).
Genes regulated by the Nrf2-ARE pathway include heme oxygenase-1
(HO-1), NADPH quinone oxidoreductase 1 (NQO1), and γ-glutamate
cysteine ligase (GCL) (22). In
addition, SLC7A11, a crucial component in ferroptosis inhibition,
is also regulated by Nrf2 (23).
Clinical studies involving patients with type 2
diabetes and CKD have shown the renoprotective effects of BM. In an
open-label, single-arm study, BM significantly increased estimated
glomerular filtration rate (eGFR) from baseline (24), and randomized controlled trials
also showed improvements in eGFR compared to baseline and placebo
groups (25–27). These findings highlight the
potential of BM as a novel therapeutic option for DKD (28).
The aim of our study was to investigate the
protective mechanisms of BM on RTECs in the context of DKD.
Specifically, we attempted to determine how BM inhibits ferroptosis
in RTECs, which contributes to DKD progression. We explored the
ability of BM to preserve RTEC viability and its mechanisms of
action when ferroptosis is induced by dRib-mediated system χc-
disruption. By elucidating the molecular mechanisms through which
BM suppresses ferroptosis in RTECs, our findings provide valuable
insights into the development of novel therapeutic strategies for
DKD.
Materials and methods
Cell culture
The NRK-52E cell line, a proximal tubular epithelial
cell line derived from rats, was provided by Prof. Sang-Ho Lee from
Kyung Hee University College of Medicine. The cells were cultured
in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 100 mg/ml penicillin, and 100 mg/ml
streptomycin under a 5% CO2 and 95% O2
environment. The culture medium was replaced with fresh medium
every 2 days. When the cells reached 70% confluence, they were
subcultured after treatment with trypsin.
To confirm the reproducibility of the experimental
results obtained from NRK-52E cells, primary cultures of rat
proximal tubular epithelial cells were also established and used
for experiments. Sprague Dawley rats (approximately 10 weeks old)
were euthanized using carbon dioxide (CO2) inhalation in
accordance with the protocol approved by the Institutional Animal
Care and Use Committee of Jeju National University (protocol no.
2024–0088; approval no. 2022–0014). The animals were placed in a
dedicated sealed chamber, and CO2 was introduced at a
displacement rate of 30% of the chamber volume per minute until the
concentration reached 60–70%. After the concentration was raised,
the chamber was gently shaken up and down to maintain an
appropriate CO2 level. Following euthanasia, death was
confirmed by checking for the cessation of heartbeat, after which
the kidneys were removed. The kidneys were washed with Hanks'
Balanced Salt Solution (HBSS), the renal capsule was removed, and
the kidneys were dissected into four pieces, isolating only the
cortical tissue. The HBSS was then removed, and the tissue was
washed with cold phosphate-buffered saline (PBS). The washed tissue
was transferred to a trypsin-treated flask, and 20 ml of a solution
containing DNase I (1 mg/ml) and Collagenase Type I (2 mg/ml) in
HBSS was added. The tissue was then sequentially passed through
sieves of different pore sizes to remove distal tubule and
glomerular cells. The proximal tubule cell suspension in HBSS was
centrifuged at 228 × g (1,000 rpm) for 10 min, and the collected
cells were cultured in rat tail collagen-coated plastic ware. After
1 week, the cells were harvested for intracellular
l-[14C]cystine uptake assays, intracellular GSH content
measurement, and lactate dehydrogenase assays.
Measurement of intracellular cystine
transport
NRK-52E cells (3×105 cells/well) or an
equivalent number of isolated RTECs were seeded into 24-well cell
culture plates and cultured in DMEM medium containing 10% FBS for 2
days. After removing the culture medium, the cells were washed with
500 µl of extracellular fluid (ECF) buffer, pre-warmed to 37°C, and
composed of 122 mM NaCl, 25 mM NaHCO3, 3 mM KCl, 1.4 mM
CaCl2, 1.2 mM MgSO4, 0.4 mM
K2HPO4, 10 mM d-glucose, and 10 mM HEPES (pH
7.4). The cells were then stimulated for 4 h with dRib, BM, ML385,
or brusatol. During the final 1 h of stimulation, 500 µl of 37°C
ECF buffer containing 0.1 µCi l-[14C]cystine (1.7 µM)
was added to each well to induce intracellular uptake of
l-[14C]cystine. After incubation, the buffer was
completely removed, and the cells were washed with cold,
radioisotope-free ECF buffer to terminate the uptake process. The
cells were lysed with 750 µl of 1% Triton X-100 in Dulbecco's
phosphate-buffered saline, after which 500 µl of the lysate was
mixed with 5 ml of scintillation cocktail for radioactivity
measurement using the Wallac MicroBeta TriLux 1450 Liquid
Scintillation and Luminescence Counter (PerkinElmer, San Juan, PR,
USA). The remaining cell lysate was used to determine protein
concentration using bicinchoninic acid protein assay kit (Pierce,
Rockford, IL, USA). Intracellular l-[14C]cystine uptake
was expressed as counts per minute (cpm) per microgram of protein,
normalized to total protein concentration.
Measurement of intracellular GSH
levels
Intracellular GSH levels were measured using a GSH
Assay Kit (Cayman, Ann Arbor, MI, USA) based on the enzymatic
recycling method using glutathione reductase. Briefly, NRK-52E
cells (3×105 cells/well) or an equivalent number of
isolated RTECs were seeded into 24-well cell culture plates and
cultured in DMEM medium containing 10% FBS for 2 days. The cells
were then stimulated for 6 h with dRib, BM, ML385, or brusatol.
Following treatment, the cells were lysed by sonication, and the
lysates were centrifuged to obtain the supernatant. The GSH
concentration in the supernatant was measured according to the
manufacturer's instructions. Intracellular GSH levels were
normalized to total protein concentration and expressed as nmol/mg
protein.
Measurement of intracellular iron
levels
Intracellular iron levels were measured using the
Iron Assay Kit (Sigma-Aldrich, St. Louis, MO, USA), a colorimetric
assay. NRK-52E cells were seeded at a density of 1×106
cells/well in a six-well culture plate and maintained in DMEM
supplemented with 10% FBS. The cells were treated simultaneously
with dRib, BM, ML385, or brusatol and incubated for 6 h.
Subsequently, the cells were lysed using sonication, and the
supernatant was collected after centrifugation. Total iron levels
were measured according to the manufacturer's protocol.
Intracellular iron levels were normalized to the total protein
concentration of the cells and expressed as nmol/mg·protein.
Assessment of cell viability
Cytotoxicity was first assessed using a lactate
dehydrogenase assay. NRK-52E cells were seeded at a density of
1×105 cells/well in a 96-well cell culture plate,
whereas isolated RTECs were plated at an equal number of cells per
well in the same type of plate. The cells were maintained in DMEM
supplemented with 10% FBS and treated simultaneously with dRib, BM,
ML385, or brusatol for 6 h. Cytotoxicity was then measured using
the Cytotoxicity Detection KitPLUS (Roche, Mannheim, Germany), and
calculated using the following formula: (((experimental value -
background control)-(low control - background control))/((high
control - background control)-(low control - background control)))
× 100. Finally, cell viability (% control) was expressed as: Cell
viability (% control)}=100 - cytotoxicity (%).
Measurement of intracellular MDA
levels
We selected MDA as a marker to evaluate
intracellular lipid peroxidation. NRK-52E cells were seeded in a
six-well culture plate at a density of 1×106 cells/well
and cultured in DMEM medium containing 10% FBS for 48 h. The cells
were then stimulated with dRib, BM, ML385, or brusatol
simultaneously for 6 h. Subsequently, MDA levels were measured from
the cell lysate using the EZ-Lipid Peroxidation Assay Kit
(DoGenBio, Seoul, South Korea). The intracellular MDA levels were
normalized to the total protein concentration of the cells and
expressed as nmol/mg·protein.
Assessment of lipid ROS levels
Intracellular lipid peroxide levels were measured
using flow cytometry with C11-BODIPY dye (Molecular Probes, Eugene,
OR, USA). NRK-52E cells were seeded in a six-well culture plate at
a density of 1×106 cells/well and cultured in DMEM
medium containing 10% FBS. The cells were then stimulated
simultaneously with dRib, BM, ML385, or brusatol and incubated for
6 h. During the last 30 min, the cells were treated with 4 µM
C11-BODIPY, followed by harvesting using 0.05% trypsin. The
collected cells were centrifuged, resuspended in PBS, and analyzed
for intracellular lipid ROS levels using a FACScan instrument (BD
Bioscience, San Jose, CA, USA). A total of 10,000 cells per sample
were analyzed, and the results were expressed as the ratio (fold)
of the mean fluorescence intensity relative to the unstimulated
control group.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
All RT-qPCR experiments were performed in accordance
with the Minimum Information for Publication of Quantitative
Real-Time PCR Experiments (MIQE) guidelines (29). Total RNA was extracted from cells
using TRIzol® (Gibco Invitrogen, Grand Island, NY, USA)
according to the manufacturer's instructions. Reverse transcription
was performed using 2 µg of RNA with MMLV reverse transcriptase
(MGmed Corporation, Seoul, Korea), oligo (dT)15 primer, dNTP (10
mM), and 40 units/µl RNase inhibitor (MGmed Corporation). RT-qPCR
was conducted using the KAPA SYBR® FAST qPCR Master Mix
(KAPA Biosystems, MA, USA) and an iQTM 5 Multicolor Real-Time PCR
Detection System (Bio-Rad, Hercules, CA, USA). Synthesized cDNA was
amplified for 40 cycles at 95°C for 3 sec, 60°C for 30 sec, and
72°C for 30 sec, with an initial cycle at 95°C for 1 min, using
specific primers. All primers were custom-synthesized by Bioneer
Corporation (Daejeon, Korea), and their sequences are listed in
Table I. Amplification specificity
was confirmed by melt curve analysis, which showed a single
distinct peak for each primer set. Standard curves generated from
5-fold serial dilutions of cDNA templates were used to determine
amplification efficiency. All primer pairs showed amplification
efficiencies ranging 92–105%, with R2 values of
>0.99. β-actin was used as a reference gene, and its stability
was validated across experimental conditions using NormFinder
analysis. All qPCR reactions were performed in triplicate, and
results represent the mean of three independent biological
replicates.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| SLC7A11 |
GACAGTGTGTGCATCCCCTT |
GCATGCATTTCTTGCACAGTTC |
| ACSL4 |
ATATTCGTCACCACTCACA |
AACCTTGCTCATAACATTCTT |
| CHAC1 |
GCCCTGTGGATTTTCGGGTA |
ATCTTGTCGCTGCCCCTATG |
| PTGS2 |
GGGAGTCTGGAACATTGTGAA |
GTGCACATTGTAAGTAGGTGGACT |
| HO-1 |
ACCCCACCAAGTTCAAACAG |
GAGCAGGAAGGCGGTCTTAG |
| NQO1 |
AGAAGCGTCTGGAGACTGTCTGG |
GATCTGGTTGTCGGCTGGAATGG |
| GCLC |
GCACATCTACCACGCAGTCAAGG |
TCAAGAACATCGCCGCCATTCAG |
| GCLM |
CTGGACTCTGTCATCATGGCTTCC |
TCCGAGGTGCCTATAGCAACAATC |
| β-actin |
TCCTGGCCTCACTGTCCAC |
GGGCCGGACTCATCGTACT |
Western blotting (immunoblotting)
Cells were lysed using a lysis buffer, and equal
amounts of protein from each group were separated using 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
onto a nitrocellulose membrane. The membrane was incubated in
Tris-buffered saline containing 0.1% Tween and 3% bovine serum
albumin to block non-specific antibody binding. It was then
incubated with primary antibodies specific to Nrf2 (#ab92946,
Abcam), SLC7A11 (#175186, Abcam), ACSL4 (#MA5-42523, Invitrogen),
CHAC1 (#217808, Abcam), and PTGS2 (#12282, Cell Signaling) at 4°C
for 2 h. β-Actin (#A2228, Sigma) was used as a loading control.
Afterward, the membrane was incubated with horseradish
peroxidase-conjugated secondary anti-rabbit antibody (#PI-1000-1,
Vector Laboratories) or anti-mouse antibody (#PI-2000-1, Vector
Laboratories) for 2 h. Bands were visualized using Western
Lightning™ Plus-Enhanced Chemiluminescence (PerkinElmer).
Co-immunoprecipitation (co-IP)
assay
IP was performed using Dynabeads Protein G (#10003D,
Thermo Fisher Scientific Inc., Waltham, MA, USA). NRK-52E cells
were stimulated and cultured under the specified conditions, then
lysed using ultrasonication. After centrifugation, the supernatant
containing the cell lysate was collected. Dynabeads were conjugated
with anti-Nrf2 antibody (#ab92946, Abcam) and anti-rabbit IgG
antibody (#2729s, Cell Signaling) to prepare a bead-antibody
mixture. This mixture was incubated with the cell lysate overnight
at 4°C under rotation. The bead-antibody-antigen complex was washed
four times with Tris-buffered saline with Tween 20. Lithium dodecyl
sulfate sample buffer was then added to the beads, followed by
incubation at 92°C for 5 min to elute the immunoprecipitant. The
eluted immunoprecipitant was analyzed and quantified using western
blot using anti-Keap1 and anti-Nrf2 antibodies.
Immunofluorescence (IF) staining
NRK-52E cells were cultured on coverslips and
divided into three groups: an untreated control group, a group
treated with 50 mM dRib for 6 h, and a group treated with 0.2 µM BM
and 50 mM dRib for 6 h. The cells were fixed with 4%
paraformaldehyde at room temperature for 20 min and then
permeabilized using a 0.1% Triton X-100 solution. Subsequently,
blocking was performed with 5% bovine serum albumin at room
temperature for 45 min. The cells were then incubated overnight at
4°C with primary antibodies: anti-Nrf2 antibody (1:100, #ab92946,
Abcam) and anti-Keap1 antibody (1:100, #D6B12, Cell Signaling).
After washing, the cells were incubated at room temperature for 1 h
with the secondary antibody, donkey anti-rabbit AlexaFluor 488
(1:300, #ab150073, Abcam). Nuclear counterstaining was performed
using 4′,6-diamidino-2-phenylindole at a 1:5,000 dilution. The
stained cells were visualized using a STELLARIS 5 confocal
microscope (Leica, Deerfield, IL, USA). To determine the ratio of
Nrf2 fluorescence expressed in the nucleus and cytoplasm, images
were analyzed using ImageJ software and displayed the values as a
bar graph.
Statistical analysis
All data are presented as mean ± standard deviation.
Statistical analyses were performed using SPSS software (version
14.0; SPSS Inc.), and P<0.05 was considered to indicate a
statistically significant difference. Homogeneity of variance was
assessed using Levene's test. When the assumption of equal
variances was met (P>0.05), one-way ANOVA followed by Tukey's
post hoc test was used. When the assumption was violated
(P<0.05), Welch's ANOVA followed by Dunnett's T3 post hoc test
was applied. For comparisons between two groups, an unpaired
Student's t-test was used.
Results
BM enhances cystine uptake, GSH
content, and cell viability, while reducing intracellular iron and
lipid peroxidation in dRib-treated RTECs
When NRK-52E cells were stimulated with dRib, there
was a significant, dose-dependent decrease in intracellular cystine
uptake, GSH content, and cell viability. However, co-treatment with
BM significantly prevented these reductions (Fig. 1). Similarly, in primary cultured
rat RTECs, dRib led to decreased cystine uptake, GSH levels and
cell viability. These impairments were effectively restored by BM
treatment (Fig. S1), indicating
its protective effect against dRib-induced cellular damage. This
shows that BM prevents cell death by increasing the intracellular
transport of cystine in RTECs, thereby restoring GSH levels.
Furthermore, intracellular iron levels, a key mediator of
ferroptosis, were increased by dRib but were reduced to control
levels by BM (Fig. 2C). Similarly,
intracellular MDA levels and C11-BODIPY fluorescence, indicators of
lipid peroxidation, were elevated by dRib but were nearly restored
to control levels by BM (Figs. 2D,
2E and S2). Therefore, BM prevents dRib-induced
ferroptosis in RTECs by counteracting the reduction in cystine
uptake.
![Effects of BM treatment on the
dRib-induced decreases in (A) l-[14C]cystine uptake, (B)
intracellular GSH content and (C) cell viability. (A) NRK-52E cells
were co-stimulated with 0, 10, 30 and 50 mM dRib with or without
0.2 µM BM for 4 h in the extracellular fluid buffer containing 1.7
µM l-[14C]cystine (0.1 µCi/ml) at 37°C. The
radioactivity incorporated into the cells was determined by a
liquid scintillation counter. (B and C) The cells were
co-stimulated with 0, 10, 30 and 50 mM dRib with or without 0.2 µM
BM for 6 h in DMEM containing 10% FBS. (B) Intracellular GSH
concentration was measured using a GSH assay kit. (C) Cell
viability was measured by LDH release assay. The data are presented
as the mean ± SD. These experiments were performed thrice, in
triplicate. *P<0.05 and **P<0.01 vs. control and
††P<0.01 vs. dRib alone, as determined by one-way
analysis of variance and Tukey's post-hoc test or Welch's ANOVA
followed by Dunnett's T3 post hoc test, depending on the result of
Levene's test. BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose; GSH,
glutathione; LDH, lactate dehydrogenase; DMEM, Dulbecco's Modified
Eagle Medium; FBS, fetal bovine serum; SD, standard deviation.](/article_images/mmr/32/4/mmr-32-04-13632-g00.jpg) | Figure 1.Effects of BM treatment on the
dRib-induced decreases in (A) l-[14C]cystine uptake, (B)
intracellular GSH content and (C) cell viability. (A) NRK-52E cells
were co-stimulated with 0, 10, 30 and 50 mM dRib with or without
0.2 µM BM for 4 h in the extracellular fluid buffer containing 1.7
µM l-[14C]cystine (0.1 µCi/ml) at 37°C. The
radioactivity incorporated into the cells was determined by a
liquid scintillation counter. (B and C) The cells were
co-stimulated with 0, 10, 30 and 50 mM dRib with or without 0.2 µM
BM for 6 h in DMEM containing 10% FBS. (B) Intracellular GSH
concentration was measured using a GSH assay kit. (C) Cell
viability was measured by LDH release assay. The data are presented
as the mean ± SD. These experiments were performed thrice, in
triplicate. *P<0.05 and **P<0.01 vs. control and
††P<0.01 vs. dRib alone, as determined by one-way
analysis of variance and Tukey's post-hoc test or Welch's ANOVA
followed by Dunnett's T3 post hoc test, depending on the result of
Levene's test. BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose; GSH,
glutathione; LDH, lactate dehydrogenase; DMEM, Dulbecco's Modified
Eagle Medium; FBS, fetal bovine serum; SD, standard deviation. |
![Effects of ML385 and brusatol on the
protective effects of BM treatment on dRib-induced changes in (A)
l-[14C]cystine uptake, (B) intracellular GSH and (C)
iron contents, (D) intracellular MDA, (E) lipid ROS levels and (F)
cell viability. (A) NRK-52E cells were co-stimulated with 0.2 µM
BM, 100 µM ML385, or 100 µM brusatol and 50 mM dRib for 4 h in the
extracellular fluid buffer containing 1.7 µM
l-[14C]cystine (0.1 µCi/ml) at 37°C. The radioactivity
incorporated into the cells was determined by a liquid
scintillation counter. (B-D and F) NRK-52E cells were co-stimulated
with 0.2 µM BM, 100 µM ML385, or 100 µM brusatol and 50 mM dRib for
6 h in DMEM media containing 10% FBS. The intracellular GSH and
iron levels, intracellular MDA levels, and cell viability were
measured using a GSH assay kit, an iron assay kit, MDA assay kit,
and LDH release assay kit, respectively. These experiments were
performed thrice, in triplicate. (E) Intracellular lipid ROS level
was quantified by flow cytometry using the lipophilic fluorescent
dye C11-BODIPY. Cells were incubated with 4 µM C11-BODIPY during
the final 30 min. Fold control of the mean fluorescence intensity
of experimental groups. This experiment was performed four times.
Data are presented as the mean ± SD. **P<0.01 vs. control;
††P<0.01 vs. 50 mM dRib-alone group;
‡‡P<0.01 vs. 50 mM dRib plus 0.2 µM BM group, as
determined by one way analysis of variance and Tukey's post hoc
test. BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose; GSH,
glutathione; MDA, malondialdehyde; ROS, reactive oxygen
species.](/article_images/mmr/32/4/mmr-32-04-13632-g01.jpg) | Figure 2.Effects of ML385 and brusatol on the
protective effects of BM treatment on dRib-induced changes in (A)
l-[14C]cystine uptake, (B) intracellular GSH and (C)
iron contents, (D) intracellular MDA, (E) lipid ROS levels and (F)
cell viability. (A) NRK-52E cells were co-stimulated with 0.2 µM
BM, 100 µM ML385, or 100 µM brusatol and 50 mM dRib for 4 h in the
extracellular fluid buffer containing 1.7 µM
l-[14C]cystine (0.1 µCi/ml) at 37°C. The radioactivity
incorporated into the cells was determined by a liquid
scintillation counter. (B-D and F) NRK-52E cells were co-stimulated
with 0.2 µM BM, 100 µM ML385, or 100 µM brusatol and 50 mM dRib for
6 h in DMEM media containing 10% FBS. The intracellular GSH and
iron levels, intracellular MDA levels, and cell viability were
measured using a GSH assay kit, an iron assay kit, MDA assay kit,
and LDH release assay kit, respectively. These experiments were
performed thrice, in triplicate. (E) Intracellular lipid ROS level
was quantified by flow cytometry using the lipophilic fluorescent
dye C11-BODIPY. Cells were incubated with 4 µM C11-BODIPY during
the final 30 min. Fold control of the mean fluorescence intensity
of experimental groups. This experiment was performed four times.
Data are presented as the mean ± SD. **P<0.01 vs. control;
††P<0.01 vs. 50 mM dRib-alone group;
‡‡P<0.01 vs. 50 mM dRib plus 0.2 µM BM group, as
determined by one way analysis of variance and Tukey's post hoc
test. BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose; GSH,
glutathione; MDA, malondialdehyde; ROS, reactive oxygen
species. |
Nrf2 inhibitors abolish the
BM-mediated recovery of cystine uptake, GSH content, and cell
viability, while reversing the BM-induced reduction in iron levels
and lipid peroxidation in dRib-treated RTECs
To determine whether the ferroptosis-inhibitory
effect of BM depends on Nrf2 activation, we used the specific Nrf2
inhibitors, ML385 and brusatol. In isolated rat RTECs, ML385 and
brusatol significantly attenuated the effects of BM in restoring
cystine uptake, GSH content, and viability, which were reduced by
dRib (Fig. S1). The same results
were observed in NRK-52E cells (Fig.
2A, B and F). In addition, the ability of BM to suppress
intracellular iron, MDA, and C11-BODIPY fluorescence levels, which
were elevated by dRib, was also counteracted by ML385 and brusatol
(Figs. 2C-E and S2). These findings indicate that the
ferroptosis-inhibitory effect of BM is dependent on Nrf2
activation.
BM increases the expression of SLC7A11
and Nrf2 target genes while reducing the expression of
ferroptosis-related markers
Nrf2 protein expression was increased in a
dose-dependent manner by dRib and was further enhanced by the
addition of BM (Fig. 3A and B).
Although Nrf2 mRNA expression was also increased dose-dependently
by dRib, the addition of BM significantly suppressed the
dRib-induced increase (Fig. 4A).
Meanwhile, SLC7A11 protein, which is known to be degraded by dRib
(7), showed a dose-dependent
decrease upon dRib treatment, but this decrease was significantly
prevented by BM (Fig. 3A and
C).
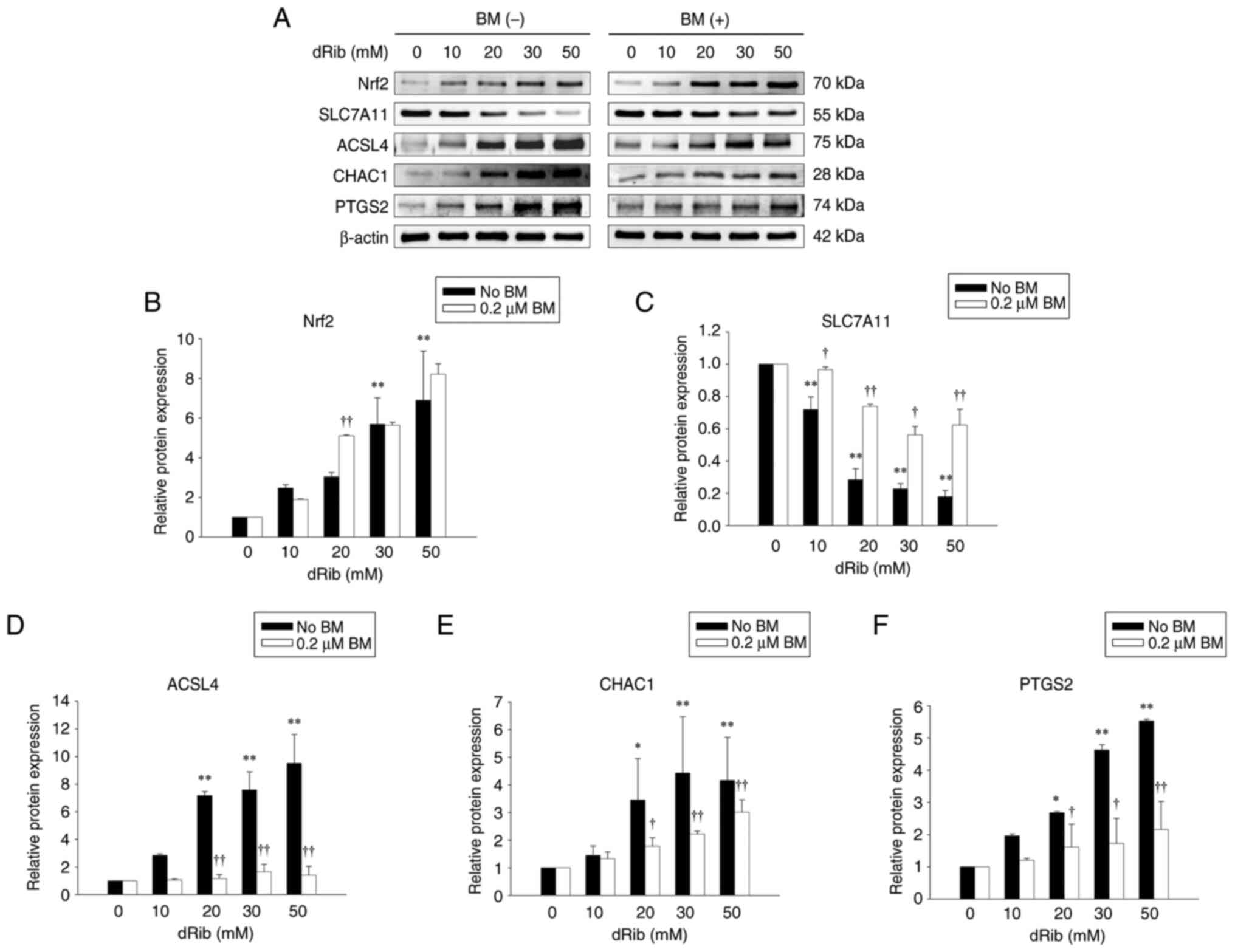 | Figure 3.Effects of BM treatment on
dRib-induced changes in Nrf2, SLC7A11, ACSL4, CHAC1 and PTGS2
protein expressions in NRK-52E cells after treatment with various
concentrations of dRib. The cells were stimulated with 0, 10, 20,
30 or 50 mM dRib with or without 0.2 µM BM for 6 h in DMEM
containing 10% FBS. The protein expression was analyzed by western
blotting. β-Actin was used as loading control. (A) Representative
blots of two independent experiments are shown. Densitometric
quantification of (B) Nrf2, (C) SLC7A11, (D) ACSL4, (E) CHAC1 and
(F) PTGS2 protein levels normalized to β-actin. Data are presented
as the mean ± SD. *P<0.05 and **P<0.01 vs. 0 mM dRib group;
†P<0.05 and ††P<0.01 vs. dRib alone, as
determined by one-way analysis of variance and Tukey's post-hoc
test or Welch's ANOVA followed by Dunnett's T3 post hoc test,
depending on the result of Levene's test. SLC7A11, solute carrier
family 7 member 11; ACSL4, acyl-CoA synthetase long chain family
member 4; CHAC1, ChaC glutathione-specific
gamma-glutamylcyclotransferase 1; PTGS2, prostaglandin-endoperoxide
synthase 2; BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose. |
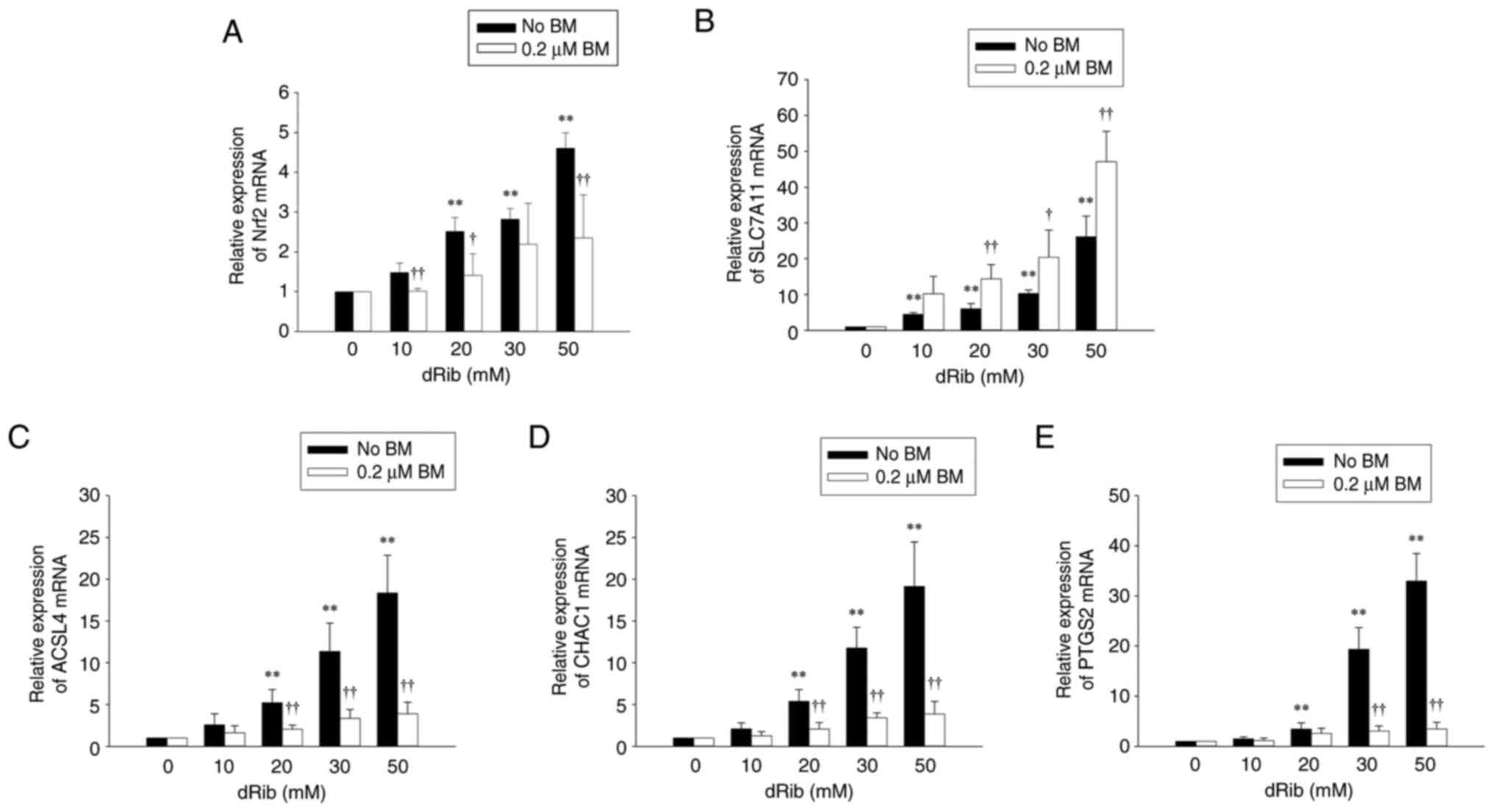 | Figure 4.Effects of BM treatment on
dRib-induced changes in (A) Nrf2, (B) SLC7A11, (C) ACSL4, (D) CHAC1
and (E) PTGS2mRNA expressions. NRK-52E cells were stimulated with
0, 10, 20, 30 or 50 mM dRib for 6 h in DMEM containing 10% FBS. The
mRNA levels were analyzed by reverse transcription-quantitative
polymerase chain reaction. Relative expression of target genes was
calculated using the 2−ΔΔCq method. This experiment was
performed thrice, in triplicate. Data are presented as the mean ±
SD. **P<0.01 vs. 0 mM dRib group; †P<0.05 and
††P<0.01 vs. dRib alone, as determined by one-way
analysis of variance and Tukey's post-hoc test or Welch's ANOVA
followed by Dunnett's T3 post hoc test, depending on the result of
Levene's test. SLC7A11, solute carrier family 7 member 11; ACSL4,
acyl-CoA synthetase long chain family member 4; CHAC1, ChaC
glutathione-specific gamma-glutamylcyclotransferase 1; PTGS2,
prostaglandin-endoperoxide synthase 2; BM, bardoxolone methyl;
dRib, 2-deoxy-d-ribose. |
We further examined the expression of Nrf2 target
genes, including SLC7A11, HO-1, NQO1, the catalytic subunit of
glutamate-cysteine ligase (GCLC), and the modifier subunit of
glutamate-cysteine ligase (GCLM). These genes were upregulated by
dRib and showed a further increase upon BM treatment. The
BM-induced increase in these Nrf2 target genes was significantly
attenuated by ML385 and brusatol (Figs. 4B and 5).
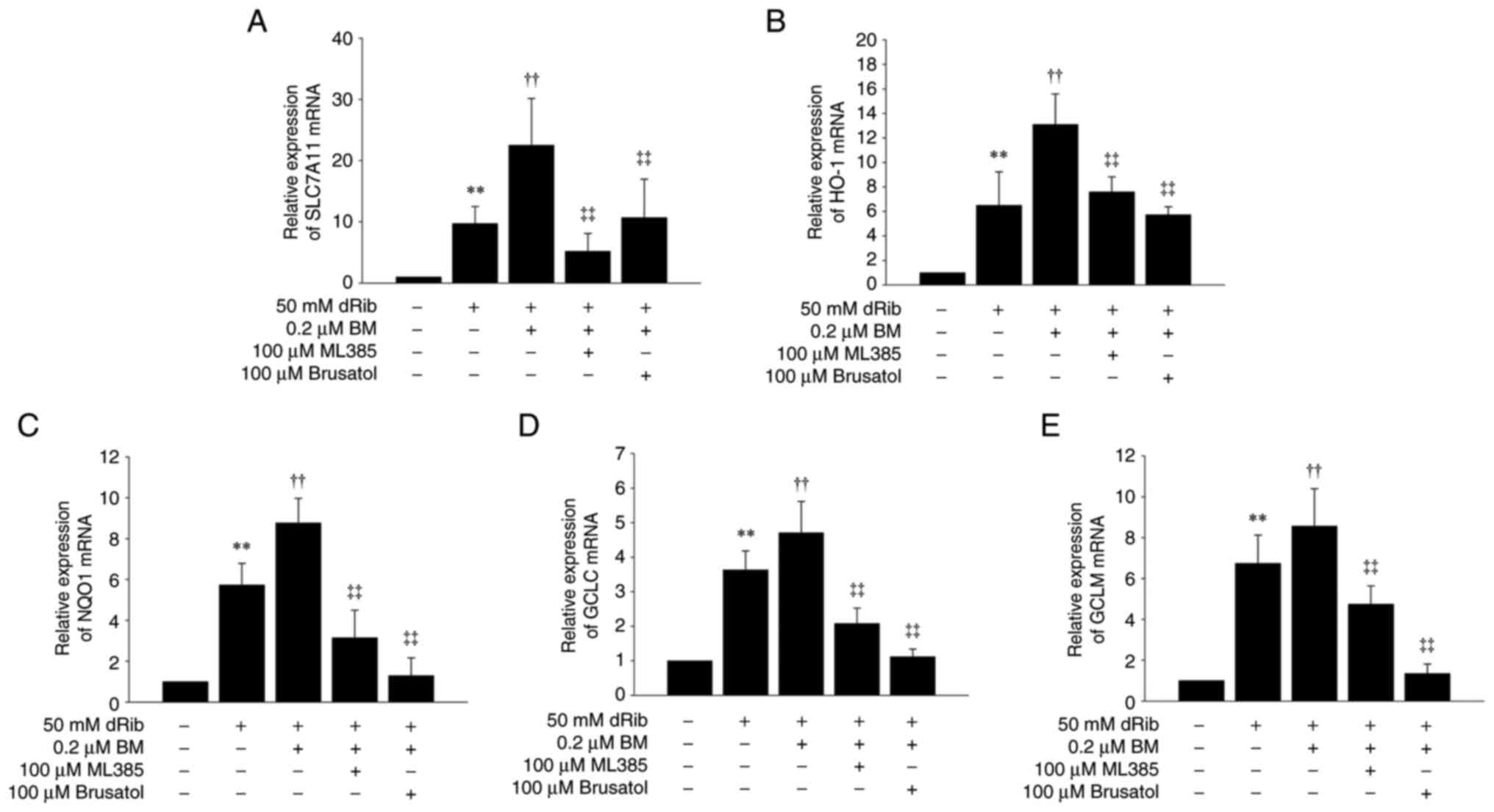 | Figure 5.Effects of BM and ML385 or brusatol
treatments on dRib-induced changes in SLC7A11 and Nrf2-antioxidant
response element-dependent genes. NRK-52E cells were co-stimulated
with 0.2 µM BM, 100 µM ML385, or 100 µM brusatol and 50 mM dRib for
6 h in DMEM containing 10% FBS. The mRNA levels of (A) SLC7A11, (B)
HO-1, (C) NQO1, (D) GCLC and (E) GCLM were analyzed by reverse
transcription-quantitative polymerase chain reaction. Relative
expression of target genes was calculated using the
2−ΔΔCq method. This experiment was performed thrice, in
triplicate. Data are presented as the mean ± SD. **P<0.01 vs.
control; ††P<0.01 vs. 50 mM dRib-alone group;
‡‡P<0.01 vs. 50 mM dRib plus 0.2 µM BM group, as
determined by one way analysis of variance and Tukey's post hoc
test. BM, bardoxolone methyl; dRib, 2-deoxy-d-ribose; SLC7A11,
solute carrier family 7 member 11; GCLC, glutamate-cysteine ligase
catalytic subunit; GCLM, glutamate-cysteine ligase modifier
subunit; HO-1, heme oxygenase-1; NQO1, NADPH quinone oxidoreductase
I. |
Next, we investigated the effect of BM on the
expression of ferroptosis biomarkers. The mRNA and protein levels
of ACSL4, ChaC glutathione-specific gamma-glutamylcyclotransferase
1 (CHAC1), and prostaglandin-endoperoxide synthase 2 (PTGS2) were
all significantly increased by dRib in a dose-dependent manner.
However, treatment with BM markedly reduced the expression of these
ferroptosis-related markers (Figs.
3 and 4C-E). These regulatory
effects on SLC7A11, ACSL4, CHAC1, and PTGS2 gene expression were
also consistently observed in primary cultured rat RTECs (Fig. S3). These findings further support
the notion that BM prevents ferroptosis by activating Nrf2
pathway.
BM disrupts Nrf2-Keap1 interaction and
promotes the nuclear translocation of Nrf2
To elucidate the mechanism through which BM
activates the Nrf2 pathway, we investigated the interaction between
Nrf2 and Keap1 and their intracellular localization.
In the co-IP assay assessing their interaction,
Keap1 was pulled down by Nrf2 in both the control and dRib-treated
groups, indicating a physical association. However, when BM was
added, the pull-down of Keap1 by Nrf2 was reduced (Fig. 6A and B). This indicates that
although the physical interaction between Nrf2 and Keap1 remained
intact under dRib treatment alone, the addition of BM weakened
their association, implying that BM disrupts the Nrf2-Keap1
interaction.
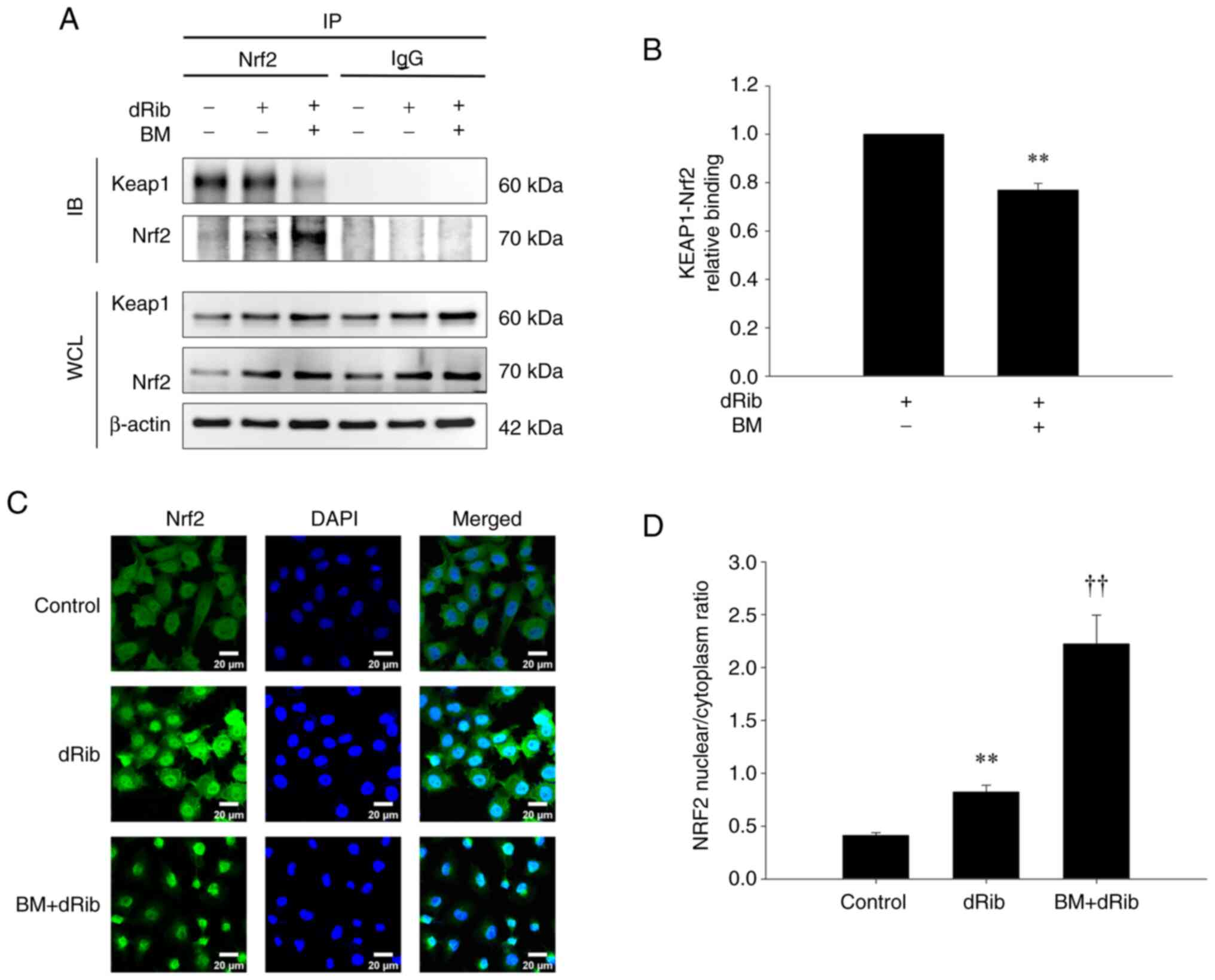 | Figure 6.Investigation of Nrf2 and Keap1
interaction and intracellular localization using (A and B) co-IP
and (C and D) immunofluorescence confocal microscopy. (A) NRK-52E
cells were stimulated with 50 mM dRib with or without 0.2 µM BM for
6 h in DMEM media containing 10% FBS. The cell lysates were
immunoprecipitated using an anti-Nrf2 antibody or control
immunoglobulin (IgG), followed by IB with anti-Keap1 and anti-Nrf2
antibodies. The lower panels show IB of WCL. (B) Densitometric
quantification of the immunoprecipitated Keap1 and Nrf2 protein
bands from (A), normalized to IgG controls. Data represent the mean
± SD from three independent experiments. **P<0.01 vs. dRib
alone, as determined by unpaired Student's t-test. (C) Cells were
treated with dRib (50 mM) alone or in combination with BM (0.2 µM)
for 6 h. Representative images from three independent experiments
show Nrf2 (green), Keap1 (red), and DAPI (blue) staining, with
merged images illustrating nuclear translocation of Nrf2 under
different conditions. Scale bar=20 µm. (D) The nuclear/cytoplasmic
ratio of Nrf2 expression was measured using the ImageJ program.
Data are presented as the mean ± SD. **P<0.01 vs. control group;
††P<0.01 vs. dRib alone, as determined by one-way
analysis of variance and Tukey's post-hoc test. BM, bardoxolone
methyl; dRib, 2-deoxy-d-ribose; IB, immunoblotting; IP,
immunoprecipitation; WCL, whole cell lysates. |
To examine changes in the intracellular localization
of Nrf2 and Keap1 upon dRib and BM treatment, we performed IF
staining and observed the cells using confocal microscopy. In the
control group, Nrf2 was evenly distributed between the cytoplasm
and nucleus, whereas in the dRib-treated group, nuclear
translocation of Nrf2 was increased. Notably, in the BM-treated
group, nuclear translocation of Nrf2 was markedly enhanced, with
minimal Nrf2 observed in the cytoplasm (Fig. 6C and D). However, Keap1
localization remained unchanged, as it was consistently retained in
the cytoplasm regardless of dRib or BM treatment (Fig. S4). Results from co-IP and confocal
microscopy indicate that BM disrupts the interaction between
Nrf2-Keap1, thereby facilitating the nuclear translocation of
Nrf2.
Discussion
In this study, we showed that BM inhibits
ferroptosis in RTECs by activating the Nrf2 signaling pathway and
upregulating SLC7A11 expression. These findings provide further
insight into the renal protective mechanisms of BM in DKD and show
a novel strategy to overcome the clinical limitations of current
DKD treatments.
As ferroptosis is an iron-dependent form of
oxidative cell death caused by lipid peroxidation, our finding that
BM lowers intracellular iron levels and lipid peroxidation markers
supports its role in preventing ferroptosis. This is further
confirmed by BM's ability to reduce the expression of ferroptosis
markers such as ACSL4, CHAC1, and PTGS2. ACSL4 promotes lipid
peroxidation by catalyzing the esterification of polyunsaturated
fatty acids (30). CHAC1 increases
during ferroptosis triggered by system χc- inhibitors (31), and PTGS2 is a recognized marker of
ferroptosis (32). BM inhibits
ferroptosis mainly by increasing the expression of SLC7A11, which
is essential for cystine transport through system χc−.
We found that BM raises both the mRNA and protein levels of
SLC7A11, and also enhances cystine uptake and intracellular GSH
levels. Notably, while dRib increased SLC7A11 mRNA expression, it
decreased the protein levels. This matches our earlier work showing
that dRib causes SLC7A11 protein degradation via the
ubiquitin-proteasome pathway (7).
This explains why SLC7A11 mRNA is upregulated as a compensatory
response to reduced cystine uptake, but protein levels drop because
of increased degradation. In contrast, BM activates Nrf2 and
restores SLC7A11 protein levels, counteracting the degradation
caused by dRib. Furthermore, the ferroptosis induced by dRib, which
was prevented by BM, was primarily caused by impaired intracellular
cystine transport (7). BM
upregulates SLC7A11 by activating the Keap1-Nrf2 pathway. Keap1
normally inhibits Nrf2 activation (33). In our study, BM increased Nrf2
protein expression and promoted its intranuclear translocation by
disrupting the interaction between Nrf2 and Keap1. Although BM does
not directly enhance the transcription of Nrf2, it stabilizes the
Nrf2 protein by interfering with its binding to Keap1, thereby
inhibiting Keap1-mediated ubiquitination and proteasomal
degradation of Nrf2 (20). As a
result, Nrf2 accumulates in the cytoplasm and is subsequently
translocated into the nucleus. Therefore, the observed increase in
Nrf2 protein levels following BM treatment is primarily attributed
to post-translational stabilization instead of increased gene
expression. Consequently, the expression of Nrf2 target genes,
including HO-1, NQO1, and GCL, was upregulated. Furthermore, the
ferroptosis-inhibitory effects of BM were abolished by the Nrf2
inhibitors ML385 and brusatol, providing strong evidence that Nrf2
activation is essential for the protective effects of BM.
Meanwhile, Nrf2 mRNA expression was increased by dRib, and this
increase was prevented by the addition of BM. It is well known that
Nrf2 mRNA expression increases in response to oxidative stress
stimuli (34,35). Since BM promotes the stabilization
of Nrf2 protein as described above, there is no need for increased
transcription of Nrf2, and thus the addition of BM is thought to
prevent the dRib-induced increase in Nrf2 mRNA expression.
BM has shown renoprotective effects in clinical
trials involving patients with DKD; however, one trial failed to
confirm these benefits. The BEACON trial, which aimed to verify the
nephroprotective effects of BM in 2,185 patients with type 2
diabetes and reduced eGFR, was terminated early after only 9 months
owing to an increased incidence of heart failure in the BM-treated
group, thus preventing definitive confirmation of its
kidney-protective effects (36).
Subsequent studies conducted in Japanese patients with DKD, such as
the TSUBAKI study (37) and the
AYAME study (27), excluded
patients with risk factors for heart failure. These studies
revealed the nephroprotective effects of BM without significant
adverse events. Therefore, heart failure remains a major obstacle
in the development of BM as a therapeutic agent for DKD. Based on
the results of our present study, a treatment strategy targeting
SLC7A11 rather than BM, which activates Nrf2, may have the
potential to prevent ferroptosis in RTECs while minimizing adverse
effects. Therefore, the development of a novel compound that
selectively upregulates SLC7A11 expression in RTECs could offer a
safer and more effective therapeutic option for DKD. In addition to
approaches targeting SLC7A11, other strategies may help reduce the
cardiovascular risks associated with BM. First, tissue-specific
drug delivery systems that selectively target the kidney can limit
systemic exposure (38), thereby
lowering the risk of off-target side effects such as heart failure.
Second, combining BM with other renoprotective agents may allow for
dose reduction, thereby improving its safety profile (39). Further research is required to
develop structural analogs of BM (21) that retain Nrf2-activating capacity
while reducing cardiotoxicity.
This study has some limitations. First, the
concentration of dRib used in our study is considerably high.
Erastin, a well-known system χc− inhibitor, is typically
used at micromolar concentrations, whereas we used dRib at
concentrations of 10–50 mM. Unlike erastin, dRib is not a small
molecule but rather a reducing sugar. Studies that have used
reducing sugars to induce oxidative stress have consistently
employed high concentrations, and some have even used
concentrations higher than those used in our study (40,41).
Second, it was conducted at the cellular level using NRK-52E cells
and primary cultured RTECs. Therefore, it does not fully replicate
the complex physiological environment of DKD, and further studies
are required to verify whether similar results are observed in
animal models of DKD. Third, instead of using erastin, a widely
used ferroptosis inducer, we induced ferroptosis in RTECs using
dRib. However, both erastin and dRib induce ferroptosis through
cysteine deprivation. Erastin inhibits system χc-, leading to a
compensatory increase in SLC7A11 gene expression. Ferroptosis
induced by erastin is known to be prevented by 2-mercaptoethanol,
which bypasses system χc- to enhance cystine uptake (3). Similarly, dRib increases SLC7A11 gene
expression in RTECs and enables dRib-induced ferroptosis to be
prevented by 2-ME (7). Therefore,
even if erastin had been used as a ferroptosis inducer, we believe
the results would have been the same as those obtained in this
study. Moreover, the reason we used dRib instead of erastin is that
dRib is more suitable for in vitro studies using RTECs.
Erastin does not dissolve well and is difficult to handle (42). In contrast, dRib dissolves readily
in water, induces ferroptosis in RTECs in a dose-dependent manner,
and its detailed mechanisms are well understood (7). For these reasons, we chose to use
dRib in our study on ferroptosis in RTECs. Fourth, further research
on intracellular signaling pathways that may contribute to the
off-target effects of BM is required. Adverse effects of BM, such
as heart failure, remain a major obstacle to its development as a
therapeutic agent for DKD. Thus, elucidating these detailed
mechanisms is as crucial as understanding the renoprotective
mechanisms of BM, as it could contribute to the development of an
optimal DKD treatment.
This study showed that BM inhibits ferroptosis in
renal tubular epithelial cells by activating the Keap1-Nrf2 pathway
and increasing the expression of SLC7A11. BM reversed the
dRib-induced decrease in cystine uptake, GSH depletion,
intracellular iron accumulation, and lipid peroxidation, and these
effects were noted to be dependent on Nrf2 activation. Although BM
appears to be a promising therapeutic strategy for DKD, adverse
effects such as heart failure remain a significant concern. Future
research should focus on developing safer therapeutic agents that
selectively enhance SLC7A11 activity to inhibit ferroptosis.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by a research grant from Jeju National
University Hospital in 2023.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
GK, SAL and MK conceived and designed the research.
GK, JYB and SY performed experiments. GK and SY analyzed data. GK,
JYB and SY interpreted results of experiments. GK and SAL acquired
funding. GK, JYB and SY confirm the authenticity of all the raw
data. GK, JYB and SY prepared figures. GK, SY and MK drafted the
manuscript, and GK edited and revised the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Jeju National University (approval
no. 2022-0014).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu J, Xiong Y, Zhang Y, Wen J, Cai N,
Cheng K, Liang H and Zhang W: The molecular mechanisms of
regulating oxidative stress-induced ferroptosis and therapeutic
strategy in tumors. Oxid Med Cell Longev. 2020:88107852020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi
AA and Lei P: Ferroptosis: Mechanisms and links with diseases.
Signal Transduct Target Ther. 6:492021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zilka O, Shah R, Li B, Friedmann Angeli
JP, Griesser M, Conrad M and Pratt DA: On the mechanism of
cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of
lipid peroxidation in ferroptotic cell death. ACS Cent Sci.
3:232–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dringen R and Hirrlinger J: Glutathione
pathways in the brain. Biol Chem. 384:505–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim M, Bae JY, Yoo S, Kim HW, Lee SA, Kim
ET and Koh G: 2-Deoxy-d-ribose induces ferroptosis in renal tubular
epithelial cells via ubiquitin-proteasome system-mediated xCT
protein degradation. Free Radic Biol Med. 208:384–393. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao MY, Liu T, Zhang L, Wang MJ, Yang Y
and Gao J: Role of ferroptosis in neurological diseases. Neurosci
Lett. 747:1356142021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
ElSayed NA, Aleppo G, Aroda VR, Bannuru
RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D,
Johnson EL, et al: 11. chronic kidney disease and risk management:
standards of care in diabetes-2023. Diabetes Care. 46 (Suppl
1):S191–S202. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barnett AH, Bain SC, Bouter P, Karlberg B,
Madsbad S, Jervell J and Mustonen J; Diabetics Exposed to
Telmisartan and Enalapril Study Group, : Angiotensin-receptor
blockade versus converting-enzyme inhibition in type 2 diabetes and
nephropathy. N Engl J Med. 351:1952–1961. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Solomon J, Festa MC, Chatzizisis YS,
Samanta R, Suri RS and Mavrakanas TA: Sodium-glucose co-transporter
2 inhibitors in patients with chronic kidney disease. Pharmacol
Ther. 242:1083302023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang XD and Yang YY: Ferroptosis as a
novel therapeutic target for diabetes and its complications. Front
Endocrinol (Lausanne). 13:8538222022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X and Li X: Abnormal iron and lipid
metabolism mediated ferroptosis in kidney diseases and its
therapeutic potential. Metabolites. 12:582022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim S, Kang SW, Joo J, Han SH, Shin H, Nam
BY, Park J, Yoo TH, Kim G, Lee P and Park JT: Characterization of
ferroptosis in kidney tubular cell death under diabetic conditions.
Cell Death Dis. 12:1602021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mengstie MA, Seid MA, Gebeyehu NA, Adella
GA, Kassie GA, Bayih WA, Gesese MM, Anley DT, Feleke SF, Zemene MA,
et al: Ferroptosis in diabetic nephropathy: Mechanisms and
therapeutic implications. Metabol Open. 18:1002432023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Bi R, Quan F, Cao Q, Lin Y, Yue C,
Cui X, Yang H, Gao X and Zhang D: Ferroptosis involves in renal
tubular cell death in diabetic nephropathy. Eur J Pharmacol.
888:1735742020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang WJ, Jiang X, Gao CC and Chen ZW:
Salusin-β participates in high glucose-induced HK-2 cell
ferroptosis in a Nrf-2-dependent manner. Mol Med Rep. 24:6742021.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li S, Zheng L, Zhang J, Liu X and Wu Z:
Inhibition of ferroptosis by up-regulating Nrf2 delayed the
progression of diabetic nephropathy. Free Radic Biol Med.
162:435–449. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rojas-Rivera J, Ortiz A and Egido J:
Antioxidants in kidney diseases: The impact of bardoxolone methyl.
Int J Nephrol. 2012:3217142012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang YY, Yang YX, Zhe H, He ZX and Zhou
SF: Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update
on its pharmacokinetic and pharmacodynamic properties. Drug Des
Devel Ther. 8:2075–2088. 2014.PubMed/NCBI
|
|
22
|
Suzuki T and Yamamoto M: Molecular basis
of the Keap1-Nrf2 system. Free Radic Biol Med. 88((Pt B)): 93–100.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu H, Guo P, Xie X, Wang Y and Chen G:
Ferroptosis, a new form of cell death, and its relationships with
tumourous diseases. J Cell Mol Med. 21:648–657. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pergola PE, Krauth M, Huff JW, Ferguson
DA, Ruiz S, Meyer CJ and Warnock DG: Effect of bardoxolone methyl
on kidney function in patients with T2D and Stage 3b-4 CKD. Am J
Nephrol. 33:469–476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nangaku M, Takama H, Ichikawa T, Mukai K,
Kojima M, Suzuki Y, Watada H, Wada T, Ueki K, Narita I, et al:
Randomized, double-blind, placebo-controlled phase 3 study of
bardoxolone methyl in patients with diabetic kidney disease: Design
and baseline characteristics of the AYAME study. Nephrol Dial
Transplant. 38:1204–1216. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pergola PE, Raskin P, Toto RD, Meyer CJ,
Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H,
et al: Bardoxolone methyl and kidney function in CKD with type 2
diabetes. N Engl J Med. 365:327–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tadao A, Kengo Y, Tomohiro I, Kazuya M and
Masaomi N: AYAME Study: Randomized, Double-Blind,
Placebo-Controlled Phase 3 Study of Bardoxolone Methyl in Diabetic
Kidney Disease (DKD) Patients FR-OR110. JASN. 34:pB12023.
View Article : Google Scholar
|
|
28
|
Kanda H and Yamawaki K: Bardoxolone
methyl: Drug development for diabetic kidney disease. Clin Exp
Nephrol. 24:857–864. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 55:611–622.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ye L, Jin F, Kumar SK and Dai Y: The
mechanisms and therapeutic targets of ferroptosis in cancer. Expert
Opin Ther Targets. 25:965–986. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Comish PB, Tang D and Kang R:
Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol.
9:6371622021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hayes JD, Dayalan Naidu S and
Dinkova-Kostova AT: Regulating Nrf2 activity: Ubiquitin ligases and
signaling molecules in redox homeostasis. Trends Biochem Sci.
50:179–205. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taqi MO, Saeed-Zidane M, Gebremedhn S,
Salilew-Wondim D, Tholen E, Neuhoff C, Hoelker M, Schellander K and
Tesfaye D: NRF2-mediated signaling is a master regulator of
transcription factors in bovine granulosa cells under oxidative
stress condition. Cell Tissue Res. 385:769–783. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeng XP, Li XJ, Zhang QY, Liu QW, Li L,
Xiong Y, He CX, Wang YF and Ye QF: Tert-butylhydroquinone protects
liver against ischemia/reperfusion injury in rats through
Nrf2-activating anti-oxidative activity. Transplant Proc.
49:366–372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
de Zeeuw D, Akizawa T, Audhya P, Bakris
GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M,
Lambers Heerspink HJ, et al: Bardoxolone methyl in type 2 diabetes
and stage 4 chronic kidney disease. N Engl J Med. 369:2492–2503.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nangaku M, Kanda H, Takama H, Ichikawa T,
Hase H and Akizawa T: Randomized clinical trial on the effect of
bardoxolone methyl on GFR in diabetic kidney disease patients
(TSUBAKI Study). Kidney Int Rep. 5:879–890. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu Y, Aimetti AA, Langer R and Gu Z:
Bioresponsive materials. Nat Rev Mater. 2:160752017. View Article : Google Scholar
|
|
39
|
Alicic RZ, Neumiller JJ and Tuttle KR:
Combination therapy: An upcoming paradigm to improve kidney and
cardiovascular outcomes in chronic kidney disease. Nephrol Dial
Transplant. 40 (Supplement 1):i3–i17. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaneto H, Fujii J, Myint T, Miyazawa N,
Islam KN, Kawasaki Y, Suzuki K, Nakamura M, Tatsumi H, Yamasaki Y
and Taniguchi N: Reducing sugars trigger oxidative modification and
apoptosis in pancreatic beta-cells by provoking oxidative stress
through the glycation reaction. Biochem J. 320((Pt 3)): 855–863.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tanaka Y, Tran PO, Harmon J and Robertson
RP: A role for glutathione peroxidase in protecting pancreatic beta
cells against oxidative stress in a model of glucose toxicity. Proc
Natl Acad Sci USA. 99:12363–12368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang L, Chen X and Yan C: Ferroptosis: An
emerging therapeutic opportunity for cancer. Genes Dis. 9:334–346.
2020. View Article : Google Scholar : PubMed/NCBI
|














