The mitochondria are double-layered membrane
organelles composed of outer and inner membranes (foldable into
cristae), an inner membrane gap, matrix and mitochondrial DNA. They
mainly participate in energy metabolism, redox processes, calcium
ion (Ca2+) concentration regulation, cell apoptosis and
other cellular processes and serve a crucial role in maintaining
cellular homeostasis (1). The
outer mitochondrial membrane (OMM) encloses the organelle, whereas
the inner mitochondrial membrane (IMM) forms intricate structures
known as cristae. The inner membrane cristae are involved in a wide
range of processes, including the electron transport chain, ATP
synthase, protein transport, metabolite exchange, protein
translation and degradation (2).
Protons and electrons can be distributed asymmetrically on both
sides of the membrane, resulting in a change in the membrane
potential and energy generation, thereby completing the cell energy
conversion. Mitochondria are highly dynamic organelles
intrinsically linked to their function. There are several levels of
mitochondrial dynamics within a single mitochondrion, among
multiple mitochondria and between mitochondria and other organelles
(3). Mitochondria interact with
other cellular structures through contact points that facilitate
the transfer of ions, lipids, proteins or metabolites and regulate
mitochondrial dynamics, quality control and mitochondrial DNA
replication (4). The formation of
mitochondrial contact points is responsive to cellular and
metabolic states, which fine-tune the mitochondrial output in
numerous aspects. Crosstalk between organelles are essential for
numerous intracellular functions, with the
mitochondrial-endoplasmic reticulum (ER) axis exemplifying a
paradigmatic inter-organelle system (5,6). The
ER and mitochondria are physically interconnected, as demonstrated
by electron tomography, which reveals that these organelles are
adjoined by tethers and electron-dense structures (7,8).
Further research has revealed a protein complex that connects these
organelles (9).
Organelle dynamics, including mitochondrial
fission/fusion and ER-mitochondrial tethering, are critical for
maintaining cellular homeostasis through regulated oxidative
phosphorylation, ATP production, calcium storage and reactive
oxygen species. Mitochondrial dysfunction disrupts oxidative
phosphorylation and calcium handling, whilst ER stress induces
protein misfolding, exacerbating oxidative stress and apoptosis.
Emerging evidence highlights that ER-mitochondrial interorganellar
communication, particularly via inositol 1,4,5-triphosphate
receptor (IP3R) type 1- voltage-dependent anion channel-1
(VDAC1)-glucose-regulated protein 75 (Grp75) complexes, directly
modulates calcium flux and apoptotic thresholds in cardiovascular
disease (CVD) pathogenesis (10–12).
Mitochondria-associated ER membranes (MAMs) are
crucial communication centers that facilitate the exchange of ions,
lipids, metabolites and signaling molecules (13). MAMs are maintained by interactions
between complementary tethering molecules located on their
surfaces. Several tethering proteins, such as VDAC1, Grp75 and
mitofusin-1/2, are crucial for the establishment and regulation of
this complex intracellular communication network and serve
essential roles in these interactions (14). Cellular physiology is coordinated
by the swift exchange of molecules between organelles at
specialized organelle-organelle contact sites (15). Numerous studies have demonstrated
that MAMs serve essential roles in calcium homeostasis, lipid
synthesis and transport and mitochondrial functions and homeostasis
(16–18). Additionally, these sites act as
signaling hubs for intracellular stress responses such as oxidative
stress, energy stress and stimulus signals (19). Moreover, MAMs are considered
pivotal sites for transmitting stress signals between the ER and
mitochondria. The cell fate also depends on these contact sites for
the decision between autophagy and apoptosis (20,21).
The interaction between organelles is a rapidly
emerging field that could allow the identification of key proteins,
help describe new regulatory pathways and clarify their
significance in CVD (22). Recent
research findings suggest that the pathogenic factors of CVD may
interfere with the interactions between mitochondria and other
organelles and the lack of specific functional proteins or
interactions involved in mitochondrial-organelle connections will
lead to several pathological changes in different tissues (23,24).
Understanding MAM proteins and their influence on cellular
physiological and pathological processes may help reveal their
diagnostic and therapeutic potential. Therefore, the present review
summarizes and discusses research progress on the interaction
between mitochondria and ER in CVD, specifically focusing on the
functional role and characteristics of MAM proteins in CVD.
Mitochondria are intimately juxtaposed to the ER and
their membrane contacts range from 10–50 nm in width, referred to
as MAMs or mitochondria-ER contacts (25). It has been observed that MAMs exist
in a wide range of species, from yeast to mammals. They facilitate
inter-organelle communication, help cells detect extracellular
signals and respond to stressful stimuli (26).
MAMs are integral to numerous cellular processes
such as lipid transport and synthesis, calcium exchange,
mitochondrial function and apoptosis/survival (26–28),
facilitated by protein complexes that exhibit both tethering
capabilities and specialized functions (29). MAM-localized proteins have been
categorized into three groups: i) MAM-specific proteins, (such as
the IP3R1-VDAC1-Grp75 complex, which directly mediates
ER-mitochondria calcium flux); ii) dual-organelle proteins [such as
phosphofurin acidic cluster sorting protein 2 (PACS-2), regulating
lipid transfer and mitophagy]; and iii) transient translocators
[such as σ-1 receptor (Sig-1R), dynamically adjusting MAM
organization under stress] (30,31).
Among these, six core protein complexes structurally organize MAMs
to mediate ER-mitochondria crosstalk: i) The IP3R1-VDAC1-Grp75
axis, critical for calcium signaling; ii) the vesicle-associated
membrane protein-associated protein B (VAPB)-protein tyrosine
phosphatase interacting protein 51 (PTPIP51) complex, integral to
the formation and stability of MAMs; iii) mitofusin 2 (Mfn2),
governing MAM structure and functional homeostasis; iv) PACS-2,
which binds to beclin 1 (BECN1) and mediates its relocation to the
MAM, facilitating MAM formation and mitophagy; v) Sig-1R, which
acts as a Ca2+-sensitive, ligand-operated chaperone; and
vi) mitochondrial contact site and cristae organizing system
(MICOS), which is evolutionarily conserved, connecting these inner
membrane domains by forming and stabilizing crista junctions
(Fig. 1).
In the context of the MAM, the most extensively
studied protein complexes related to the ER and mitochondria that
act as molecular tethers include IP3R1, VDAC1 and Grp75 (32). Structurally, IP3Rs serve as crucial
Ca2+ efflux channels on the ER membrane, facilitating
the transfer of Ca2+ from the ER lumen to the cytoplasm
(33). Voltage-dependent anion
channels (VDACs) are ion channels located on the OMM that regulate
the passage of metabolites and ions across the mitochondrial
membranes. Grp75 bridges IP3Rs and VDACs, thereby maintaining the
architecture of MAMs (34).
The VAPB-PTPIP51 complex is another important
component of MAMs. The membrane protein VAPB is located in the ER
membrane, whilst the membrane protein PTPIP51 is located in the
OMM. Together, VAPB and PTPIP51 are integral to the formation and
stability of MAMs (45). Emerging
evidence reveals that VAPB molecules can rapidly enter and exit
MAMs within seconds, allowing MAM to dynamically reshape according
to the intracellular and extracellular environment to precisely
regulate cellular metabolic demands. This demonstrates the critical
role of VAPB diffusion kinetics in maintaining MAMs homeostasis
(46). Oxysterol-binding
protein-related protein 5 is found at the interface and interacts
with OMM protein PTPIP51, thereby serving a role in mitochondrial
function (47). Following ischemic
stroke, the expression of VAPB-PTPIP51 is downregulated, which
damages the MAMs structure, potentially exacerbating cerebral
ischemia-reperfusion injury by inhibiting the phosphatidylinositol
3-kinase pathway and activating autophagy (48). Nucleoporin 358, a nucleoporin
resident in the annulate lamellae, can interact with the
VAPB-PTPIP51 complex, thereby suppressing the mTORC2/Akt/glycogen
synthase kinase-3β (GSK3β) signaling pathway activation and
disrupting the MAMs (49). The
VAPB-PTPIP51 tethers also serve a crucial role in the regulation of
autophagy by mediating the transfer of Ca2+ from ER
stores to the mitochondria (50).
Additionally, neuronal protein α-synuclein interacts with the VAPB,
thereby disrupting MAMs and Ca2+ homeostasis, as well as
mitochondrial ATP production (51).
Mfn2-mediated MAM structure and functional
homeostasis are crucial for coordinating vital cellular homeostatic
processes. There are three main mechanisms by which the
mitochondrial dynamics protein Mfn2 affects MAMs are as follows: i)
It directly affects the linkage of MAMs; ii) it promotes
oligomerization; and iii) it facilitates the formation of complexes
with other proteins, thereby affecting MAMs linkages (52,53).
Splicing of Mfn2 produces ER-specific variants ERMIT2 and ERMIN2.
ERMIN2 regulates the morphology of the ER, whereas ERMIT2
associates with Mfn2 and engages with mitochondrial mitofusins to
facilitate the tethering of the ER to mitochondria. This
interaction promotes enhanced mitochondrial Ca2+ uptake
and phospholipid transfer (54).
The substantial expression of dimethylarginine
dimethylaminohydrolase-1 in dopaminergic neurons of the substantia
nigra may confer neuroprotective effects by sustaining the
formation of MAMs and preserving mitochondrial function through
oligomerization of Mfn2 (55).
Mfn2 not only mediates MAM formation but also regulates
mitochondria-ER interactions by binding to Diaphanous-1, modulating
MAM proximity and ischemia-reperfusion injury susceptibility
(56). Knocking out Mfn2 reduces
the interaction between the ER and mitochondria via the
VAPB-PTPIP51 tethering complex, whilst overexpressing Mfn2
increases the interaction (57).
PDZ-domain-containing protein synaptojanin-2 binding protein
(SYNJ2BP) maintains mitochondrial Zn2+ homeostasis in
nucleus pulposus cells during intervertebral disc degeneration by
stabilizing MAM contacts via Mfn2 and facilitating the formation of
the NOD-like receptor X1-the zinc transporter solute carrier family
39 member 7 complex (58). Mfn2
regulates protein kinase RNA-like endoplasmic reticulum kinase
(PERK) and inositol-requiring enzyme 1 axis signaling and maintains
MAM integrity. Its deficiency disrupts MAM structure, inducing ER
stress, mitochondrial ROS accumulation and apoptosis in cisplatin
nephropathy (59). Exposure to
Di-(2-ethylhexyl) phthalate induces the downregulation of Mfn2,
which impairs MAMs by inhibiting Mfn2-PERK interaction (60). Conversely, skeletal muscle-specific
knockdown of the mitochondrial fusion mediator optic atrophy 1
(OPA1) upregulates Mfn2 but impairs MAM formation through
activating transcription factor 4-dependent mechanisms (61).
Rab32 is a small GTPase located in the ER and
mitochondria, where it regulates ER Ca2+ handling in the
ER and disrupts calnexin enrichment on MAM without affecting the ER
distribution of protein-disulfide isomerase or Mfn2. Furthermore,
Rab32 influences the targeting of protein kinase A to mitochondrial
and ER membranes, thereby regulating the phosphorylation of Bcl-2
agonist of cell death and dynamin-related protein 1 (Drp1)
(72). Moreover, Rab32 modulates
the positioning of the Ca2+ regulatory transmembrane
protein calnexin to MAM (72).
Thioredoxin-related transmembrane protein 1 is selectively degraded
via a Rab32-dependent process with the long isoform of reticulon-3
(RTN3L) acting as a Rab32 effector. Together, Rab32 and RTN3L
promote autophagic degradation of the mitochondrial-proximal ER
membranes (73). Proteins in the
Rab32 subfamily, including Rab32A, Rab32B, Rab29 and Rab38, serve
an evolutionarily conserved role in interacting with Drp1, which is
essential for mitochondrial dynamics and dependent on their
localization in the ER and MAM (74).
The outer mitochondria membrane links the
mitochondria to other organelles, whereas the inner membrane
consists of a boundary region and a folded crista. The MICOS
system, which is evolutionarily conserved, connects these inner
membrane domains by forming and stabilizing crista junctions.
Moreover, MICOS creates contact sites between the inner and outer
membranes through interactions with outer membrane proteins
(75). Mitochondrial contact site
and cristae organizing system complex (Mic)19 forms the sorting and
assembly machinery (Sam) 50-Mic19-Mic60 axis by interacting with
Sam50 (the outer membrane protein) and Mic60 (the inner membrane
protein), linking the S-adenosylmethionine and MICOS
complexes to the MIB super complex that connects the mitochondrial
outer and inner membranes (76).
As a key MICOS subunit, Mic19 also regulates ER-mitochondria
contacts via the EMC2/SLC25A46/Mic19 pathway, with disruptions
potentially leading to nonalcoholic steatohepatitis and liver
fibrosis (77).
ER and mitochondria are key sites for membrane
biogenesis in eukaryotes, facilitating lipid exchange through
membrane contact sites (78,79).
In yeast, this process is mediated by the endoplasmic
reticulum-mitochondria encounter structure (ERMES) (9,80), a
complex composed of at least four proteins: The mitochondrial outer
membrane proteins (Mdm10 and Mdm34), the ER membrane component
(Mmm1) and the cytoplasmic protein (Mdm12). ERMES forms ~25
bridge-like complexes at contact sites, each featuring three
synaptotagmin-like domains in a zig-zag pattern (79). These ER-mitochondrial contact sites
are essential for importing hydrophobic mitochondrial precursor
proteins into the IMM through the ER- syndrome of undifferentiated
recurrent fever pathway. ERMES, in conjunction with translocase of
outer mitochondrial membrane 70 (Tom 70), Djp1 and Lam6, form two
parallel, partially redundant pathways for ER-to-mitochondrial
transport. Disruption of these contact sites results in several
mitochondrial inner membrane protein precursors becoming trapped in
the ER membrane, leading to mitochondrial dysfunction (81). Djp1, a chaperone involved in the
mitochondrial import of ER-resident proteins, is located near the
ER exit sites (ERES)-ERMES region, suggesting a potential link
between the proximity of ERES and ERMES and mitochondrial protein
import. Besides lipid transport, ERMES facilitates protein transfer
from the ER to mitochondria, overlapping with the function of the
Tom70-Djp1/Lam6 contact site (82). To date, ERMES components have only
been identified in fungi, to the best of the authors' knowledge
(83). Further investigations are
required to determine whether these proteins are involved in lipid
transfer in other species.
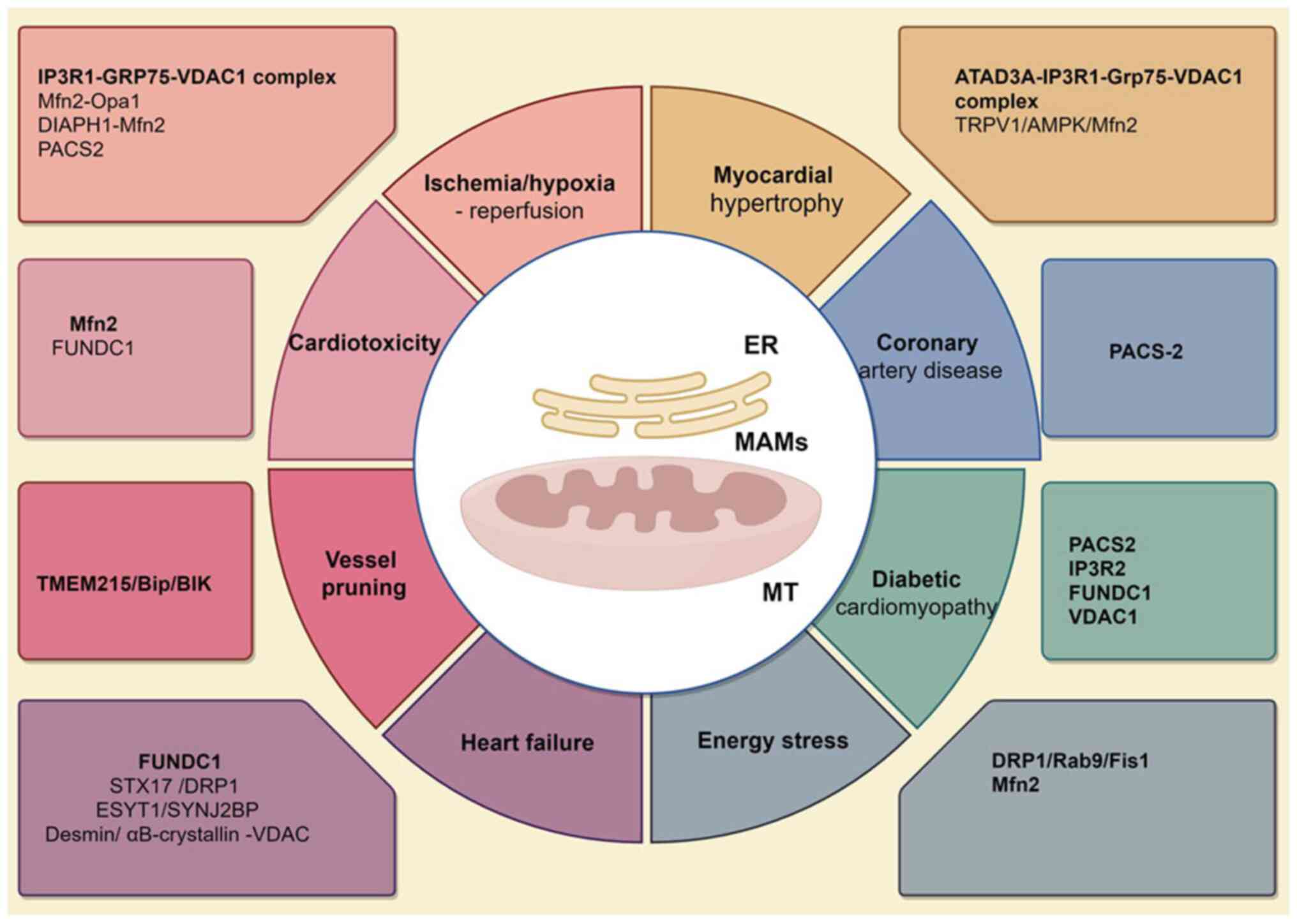
MAMs critically regulate pathological processes in
ischemic heart disease through coordinated control of calcium
homeostasis, mitochondrial dynamics and metabolic signaling. The
IP3R1-GRP75-VDAC1 complex serves as a central hub: GRP75 mediates
ER-mitochondrial tethering, enabling VDAC1-dependent calcium flux
that modulates apoptosis and glycolytic adaptation under hypoxia.
Experimental ablation of GRP75 disrupts this coupling, exacerbating
calcium overload and impairing adaptive stress responses in
cardiomyocytes (84).
GRP75-mediated ER-mitochondrial interactions via the IP3R1-VDAC1
complex are essential for Ca2+ homeostasis and ER stress
adaptation in cardiomyocyte hypoxia-ischemia. Targeting GRP75
regulates Ca2+ flux, glycolysis and cell survival
(38). In pulmonary hypertension,
downregulation of OPA1/Mfn2 impairs mitochondrial fusion,
accelerating right ventricular (RV) hypertrophy via reactive oxygen
species overproduction, whereas their overexpression preserves
mitochondrial integrity and attenuates maladaptive remodeling
(85). Diaphanous related formin 1
(DIAPH1)-Mfn2 interaction governs MAM architecture by regulating
ER-mitochondrial proximity, as synthetic linkers disrupting this
interaction negate the silencing of the DIAPH1 cardioprotective
effects during ischemia (86).
CypD-VDAC1/GRP75/IP3R1 complex dynamics further fine-tune calcium
transfer, with hypoxia-reoxygenation enhancing CypD binding to
amplify mitochondrial calcium overload, making this complex a
therapeutic target for reperfusion injury (87). Finally, Pacs2 deficiency disrupts
MAM-mediated mitophagy and energy metabolism, exacerbating RV
dysfunction in hypobaric hypoxia, whilst Pacs2 overexpression
restores calcium flux and mitophagic flux, highlighting its role as
a metabolic checkpoint (88).
ATPase family AAA-domain containing protein 3A
(ATAD3A), which is located in the MAM, maintains ER-mitochondrial
contact homeostasis, prevents mitochondrial Ca2+
overload and protects the mitochondrial bioenergetics from ER
stress. It is a substrate of sirtuin 3 and its acetylation at K134
disrupts its oligomerization. ATAD3A monomer interacts with the
IP3R1-GRP75-VDAC1 complex, leading to mitochondrial Ca2+
overload and dysfunction in myocardial hypertrophy (29). A nonselective cation channel,
transient receptor potential vanilloid type 1, enhances the
formation of MAMs and maintains mitochondrial function through the
AMP/APK/Mfn2 pathway, reducing myocardial hypertrophy caused by
pressure overload in cardiomyocytes (Fig. 2) (89).
Sorafenib (SOR), a first-line drug for the treatment
of advanced hepatocellular carcinoma, induces cardiac dysfunction
via mitochondrial Ca2+ overload, activating
calcium/calmodulin dependent protein kinase II δ (CaMKIIδ) and the
receptor interacting protein 3 (RIP3)/mixed lineage kinase
domain-like protein (MLKL) cascade, with excessive MAM formation
and tight ER-mitochondria contact serving as the key pathogenic
mechanisms (90). SOR also
downregulates Mfn2 expression. Lowering Mfn2 enhances SOR-induced
MAM biosynthesis and mitochondrial MAM binding in cardiomyocytes.
Furthermore, SOR inactivates mTOR, activated transcription factor
EB and promotes mitochondrial phagocytosis and Mfn2 degradation.
Sorafenib triggers necroptosis through the
Mfn2-MAM-Ca2+-CaMKIIδ pathway, but Mfn2 overexpression
prevents cardiac dysfunction and necroptosis caused by sorafenib by
blocking the MAM-CaMKIIδ-RIP3/MLKL pathway (90). Upon tunicamycin treatment, the
oxidoreductase ER oxidoreductase 1 α (ERO1α) has been reported to
form a covalent bond with the protein kinase PERK, which requires
the C-terminal active site of ERO1α and cysteine 216 of PERK. This
interaction oxidizes MAMs and regulates mitochondrial dynamics,
enhancing ER-mitochondria Ca2+ flux to maintain
bioenergetics and reduce oxidative stress (91). MAMs initiate autophagy and form
autophagosomes, whilst FUN14 domain containing 1 (FUNDC1) acts as a
tethering protein. Overexpression of FUNDC1 is reported to restore
autophagosome biogenesis by maintaining the MAM structure and
aiding in the formation of the autophagy-related
(ATG)5-ATG12/ATG16L1 complex without affecting mitophagy (92). FUNDC1 also reduces doxorubicin
(DOX)-induced oxidative stress and cardiomyocyte death through
autophagy. Therefore, FUNDC1-mediated MAMs provide
cardio-protection against DOX-induced cardiotoxicity by restoring
autophagosome biogenesis (Fig.
2).
In patients with coronary heart disease, the
mitochondria exhibit increased oxygen consumption, higher ATP
production and tighter connections with the ER, thereby forming
MAMs. This Ca2+ transfer through MAMs sustains
mitochondrial hyperactivity and is dependent on the inactivation of
GSK3β (93). Moulis et al
(66) reported that PACS-2
accumulates at MAM sites in VSMCs exposed to oxidized low-density
lipoprotein. PACS-2 enhances MAM contacts and its deletion disrupts
these connections, leading to impaired mitophagosome formation and
enhanced VSMC apoptosis. Further research by Assis et al
(94) reported that Pravastatin
reduces ER-mitochondrial interactions, leading to increased
mitochondrial branching. Moreover, Pravastatin upregulated the
expression of the mitochondrial dynamics regulators fission 1
protein (Fis1) and Mfn2 in bone marrow-derived macrophage from
Ldlr−/− mice. Mendelian randomization-transcriptomic
analysis further identified MAM-related KLRC1/SOCS2 as protective
AS resistance genes with reduced expression in atherosclerotic
plaques (Fig. 2) (95).
Under high glucose conditions, the formation of MAMs
is markedly increased through the involvement of PACS2, IP3R2,
FUNDC1 and VDAC1 in H9c2 cardiomyoblasts. This increase in MAMs
coincides with decreased mitochondrial biogenesis, fusion and
oxidative phosphorylation. However, ferulic acid has been reported
to effectively counteract changes in MAM formation and the
associated cellular dysfunction (102). IP3R1-GRP75-VDAC1 complex mediates
ER-mitochondrial calcium dysregulation, driving atrial remodeling
and atrial fibrillation in type 2 diabetes mellitus via exacerbated
oxidative stress. GRP75 ablation attenuates these pathological
processes in diabetic rat and cell models, identifying this complex
as a therapeutic target for diabetes-associated atrial fibrillation
(103).
Transmembrane protein 215 (TMEM215) is a two-pass
protein located in the ER. Knockdown of TMEM215 in endothelial
cells triggers apoptosis. TMEM215 interacts with chaperone-BiP and
facilitates its interaction with the pro-apoptotic protein BCL-2
interacting killer (BIK). Reducing TMEM215 levels increases the
number and proximity of mitochondria-associated ER membranes,
leading to enhanced mitochondrial Ca2+ influx. It also
reduces the distance between MAMs, leading to a higher
mitochondrial Ca2+ influx. TMEM215, induced by shear
stress through enhancer of zeste homolog 2 downregulation,
safeguards endothelial cells from BIK-induced mitochondrial
apoptosis via Ca2+ influx during vessel pruning
(Fig. 2) (104).
During the chronic phase of HFD consumption, DRP1 is
phosphorylated at Ser616, localizes to MAMs and interacts with Rab9
and Fis1. By contrast, during the acute phase, DRP1 regulates
mitophagy independently of MAMs (105). Under energy stress, AMPK
translocated from the cytosol to MAMs and mitochondria during
mitochondrial fission, where it directly interacts with Mfn2.
Mfn2−/− mouse embryonic fibroblasts (MEFs) have
been reported to exhibit notably reduced autophagic ability under
energy stress compared with wildtype MEFs, but re-expressing Mfn2
restored their autophagy. Furthermore, Mfn2−/−
cells were reported to have a markedly lower abundance of MAMs
compared to control (17).
The emerging evidence emphasizes that the MAM is a
key hub for cellular homeostasis, integrating lipid metabolism,
calcium signaling (IP3R-VDAC1-Grp75 complex) and mitochondrial
dynamics (MFN2-mediated binding and Drp1-regulated fission)
(16). The existing research
results indicate that MAM dysfunction serves a key role in the
pathogenesis of CVD, particularly through mechanisms involving ER
mitochondrial calcium overload and lipid toxicity induced membrane
remodeling (107,108).
There are still three limitations of the present
review: i) The complexity of MAM regulatory networks: Current
models inadequately capture the multi-layered regulation of MAM
integrity. Whilst canonical pathways (such as Sig-1R-mediated ER
stress modulation) have been characterized, systemic
interconnections between MAM signaling nodes remain poorly defined;
ii) Causal inference in MAM-CVD relationships: Despite robust
associative data linking MAM disruption with CVD phenotypes, causal
validation remains elusive. The need for cell-type-specific MAM
editing tools (such as CRISPRa/i in human induced cardiomyocytes)
to dissect tissue-specific patho-mechanisms; and iii) Translational
barriers in MAM-targeted therapeutics: The MAM simultaneously
participates in pathways such as calcium signaling
(IP3R-VDAC1-GRP75), lipid metabolism (Sig-1R-ER stress) and
mitochondrial dynamics (MFN2-mediated mitochondrial anchoring).
Targeting a single node may trigger compensatory feedback.
The future directions of MAMs in CVD include: i)
Dynamic MAM profiling, with implementation of live-cell
super-resolution imaging (such as lattice light-sheet microscopy)
to resolve MAM remodeling kinetics during CVDs; ii) multi-omic
integration, with the combining of spatial lipidomics with
proximity-dependent biotinylation (BioID2) to construct
disease-specific MAM interaction networks; and iii) organoid
models, where patient-derived cardiac organoids are developed with
engineered MAM architectures to test genotype-specific
therapies.
In conclusion, while the present MAM research
provides potential for CVD treatment, bridging molecular mechanisms
to clinical applications requires addressing methodological
heterogeneity, refining causal inference frameworks and innovating
organelle-specific delivery platforms. The present review
highlighted the urgency of establishing standardized MAM assessment
protocols to accelerate therapeutic discovery.
The present study was funded by the National Natural Science
Foundation of China (grant no. 82100475), Sichuan Science and
Technology Program (grant no. 2023NSFSC0617) and Chengdu Women's
and Children's Central Hospital Talent Program (grant nos.
YC2021003 and YC2022002).
Not applicable.
SX and YL contributed to conceptualization. SX, YL
and XG contributed to the methodology. The formal analysis,
investigation and preparation of the original draft was performed
by YL and XG. The review and editing of the manuscript, acquisition
of funding and supervision was performed by SX. All authors have
read and approved the final manuscript. Data authentication is not
applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Suomalainen A and Nunnari J: Mitochondria
at the crossroads of health and disease. Cell. 187:2601–2627. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Caron C and Bertolin G: Cristae shaping
and dynamics in mitochondrial function. J Cell Sci.
137:jcs2609862024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kondadi AK and Reichert AS: Mitochondrial
dynamics at different levels: From cristae dynamics to
interorganellar cross talk. Annu Rev Biophys. 53:147–168. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Wu Y, Zhang M, Li Z, Liu B, Liu
H, Hao J and Li X: Synergistic mechanism between the endoplasmic
reticulum and mitochondria and their crosstalk with other
organelles. Cell Death Discov. 9:512023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu H, Chen W, Chen Z, Li X and Wang M:
Novel tumor therapy strategies targeting endoplasmic
reticulum-mitochondria signal pathways. Ageing Res Rev.
88:1019512023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daw CC, Ramachandran K, Enslow BT, Maity
S, Bursic B, Novello MJ, Rubannelsonkumar CS, Mashal AH,
Ravichandran J, Bakewell TM, et al: Lactate elicits
ER-mitochondrial Mg(2+) dynamics to integrate cellular metabolism.
Cell. 183:474–489. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mannella CA, Buttle K, Rath BK and Marko
M: Electron microscopic tomography of rat-liver mitochondria and
their interaction with the endoplasmic reticulum. Biofactors.
8:225–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Csordas G, Renken C, Varnai P, Walter L,
Weaver D, Buttle KF, Balla T, Mannella CA and Hajnóczky G:
Structural and functional features and significance of the physical
linkage between ER and mitochondria. J Cell Biol. 174:915–921.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kornmann B, Currie E, Collins SR,
Schuldiner M, Nunnari J, Weissman JS and Walter P: An
ER-mitochondria tethering complex revealed by a synthetic biology
screen. Science. 325:477–481. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giacomello M, Pyakurel A, Glytsou C and
Scorrano L: The cell biology of mitochondrial membrane dynamics.
Nat Rev Mol Cell Biol. 21:204–224. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ali MA, Gioscia-Ryan R, Yang D, Sutton NR
and Tyrrell DJ: Cardiovascular aging: Spotlight on mitochondria. Am
J Physiol Heart Circ Physiol. 326:H317–H333. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Z and Zhang SL: Endoplasmic reticulum
stress: A key regulator of cardiovascular disease. DNA Cell Biol.
42:322–335. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hernandez-Alvarez MI, Sebastian D, Vives
S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR,
Hernández V, Vasconcelos N, et al: Deficient endoplasmic
reticulum-mitochondrial phosphatidylserine transfer causes liver
disease. Cell. 177:881–895. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mao H, Chen W, Chen L and Li L: Potential
role of mitochondria-associated endoplasmic reticulum membrane
proteins in diseases. Biochem Pharmacol. 199:1150112022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salvador-Gallego R, Hoyer MJ and Voeltz
GK: SnapShot: Functions of endoplasmic reticulum membrane contact
sites. Cell. 171:1224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao WB and Sheng R: The correlation
between mitochondria-associated endoplasmic reticulum membranes
(MAMs) and Ca(2+) transport in the pathogenesis of diseases. Acta
Pharmacol Sin. 46:271–291. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang N, Wang C, Zhao H, He Y, Lan B, Sun L
and Gao Y: The MAMs structure and its role in cell death. Cells.
10:6572021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barbuti PA, Guardia-Laguarta C, Yun T,
Chatila ZK, Flowers X, Santos BF, Larsen SB, Hattori N, Bradshaw E,
Dettmer U, et al: The role of alpha-synuclein in synucleinopathy:
Impact on lipid regulation at mitochondria-ER membranes. NPJ
Parkinsons Dis. 11:1032025. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Elwakiel A, Mathew A and Isermann B: The
role of endoplasmic reticulum-mitochondria-associated membranes in
diabetic kidney disease. Cardiovasc Res. 119:2875–2883. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang
H, Fan H, Meng T, He Z, Huang H, et al: The AMPK-MFN2 axis
regulates MAM dynamics and autophagy induced by energy stresses.
Autophagy. 17:1142–1156. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao Y, Chen Z, Hu J, Feng J, Zhu Z, Fan Y,
Lin Q and Ding G: Mfn2 regulates high glucose-induced MAMs
dysfunction and apoptosis in podocytes via PERK pathway. Front Cell
Dev Biol. 9:7692132021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao H, Xie Y, Li C, Liu W and Yi G:
Mitochondria-Associated organelle crosstalk in myocardial
ischemia/reperfusion injury. J Cardiovasc Transl Res. 17:1106–1118.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang M, Ding Y, Hu Y and Li Z, Luo W, Liu
P and Li Z: SIRT3 improved peroxisomes-mitochondria interplay and
prevented cardiac hypertrophy via preserving PEX5 expression. Redox
Biol. 62:1026522023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu L, Tang D, Qi B, Guo D, Wang Y, Geng J,
Zhang X, Song L, Chang P, Chen W, et al: Mfn2/Hsc70 complex
mediates the formation of mitochondria-lipid droplets membrane
contact and regulates myocardial lipid metabolism. Adv Sci (Weinh).
11:e23077492024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vance JE: MAM (mitochondria-associated
membranes) in mammalian cells: Lipids and beyond. Biochim Biophys
Acta. 1841:595–609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lang A, Peter AT and Kornmann B:
ER-mitochondria contact sites in yeast: Beyond the myths of ERMES.
Curr Opin Cell Biol. 35:7–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Naon D and Scorrano L: At the right
distance: ER-mitochondria juxtaposition in cell life and death.
Biochim Biophys Acta. 1843:2184–2194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prudent J and McBride HM: The
mitochondria-endoplasmic reticulum contact sites: A signalling
platform for cell death. Curr Opin Cell Biol. 47:52–63. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barazzuol L, Giamogante F and Cali T:
Mitochondria associated membranes (MAMs): Architecture and
physiopathological role. Cell Calcium. 94:1023432021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Poston CN, Krishnan SC and Bazemore-Walker
CR: In-depth proteomic analysis of mammalian
mitochondria-associated membranes (MAM). J Proteomics. 79:219–230.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X, Wen Y, Dong J, Cao C and Yuan S:
Systematic in-depth proteomic analysis of mitochondria-associated
endoplasmic reticulum membranes in mouse and human testes.
Proteomics. 18:e17004782018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Z, Hu O, Xu S, Lin C, Yu W, Ma D, Lu J
and Liu P: The SIRT3-ATAD3A axis regulates MAM dynamics and
mitochondrial calcium homeostasis in cardiac hypertrophy. Int J
Biol Sci. 20:831–847. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Woll KA and Van Petegem F: Calcium-release
channels: Structure and function of IP(3) receptors and ryanodine
receptors. Physiol Rev. 102:209–268. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Massa R, Marliera LN, Martorana A, Cicconi
S, Pierucci D, Giacomini P, De Pinto V and Castellani L:
Intracellular localization and isoform expression of the
voltage-dependent anion channel (VDAC) in normal and dystrophic
skeletal muscle. J Muscle Res Cell Motil. 21:433–442. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu F, Courjaret R, Assaf L, Elmi A, Hammad
A, Fisher M, Terasaki M and Machaca K: Mitochondria-ER contact
sites expand during mitosis. iScience. 27:1093792024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Narayanan D, Adebiyi A and Jaggar JH:
Inositol trisphosphate receptors in smooth muscle cells. Am J
Physiol Heart Circ Physiol. 302:H2190–H2210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garcia MI and Boehning D: Cardiac inositol
1,4,5-trisphosphate receptors. Biochim Biophys Acta Mol Cell Res.
1864:907–914. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim JC, Son MJ, Subedi KP, Li Y, Ahn JR
and Woo SH: Atrial local Ca2+ signaling and inositol
1,4,5-trisphosphate receptors. Prog Biophys Mol Biol. 103:59–70.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nandwani A, Rathore S and Datta M: LncRNA
H19 inhibition impairs endoplasmic reticulum-mitochondria contact
in hepatic cells and augments gluconeogenesis by increasing VDAC1
levels. Redox Biol. 69:1029892024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Honrath B, Metz I, Bendridi N, Rieusset J,
Culmsee C and Dolga AM: Glucose-regulated protein 75 determines
ER-mitochondrial coupling and sensitivity to oxidative stress in
neuronal cells. Cell Death Discov. 3:170762017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang C, Liu B, Sheng J, Wang J, Zhu W,
Xie C, Zhou X, Zhang Y, Meng Q and Li Y: Potential targets for the
treatment of MI: GRP75-mediated Ca(2+) transfer in MAM. Eur J
Pharmacol. 971:1765302024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Y, Zhu L, Cai MX, Wang ZL, Zhuang M,
Tan CY, Xie TH, Yao Y and Wei TT: TGR5 supresses cGAS/STING pathway
by inhibiting GRP75-mediated endoplasmic reticulum-mitochondrial
coupling in diabetic retinopathy. Cell Death Dis. 14:5832023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Basso V, Marchesan E and Ziviani E: A trio
has turned into a quartet: DJ-1 interacts with the IP3R-Grp75-VDAC
complex to control ER-mitochondria interaction. Cell Calcium.
87:1021862020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jiang T, Ruan N, Luo P, Wang Q, Wei X, Li
Y, Dai Y, Lin L, Lv J, Liu Y and Zhang C: Modulation of
ER-mitochondria tethering complex VAPB-PTPIP51: Novel therapeutic
targets for aging-associated diseases. Ageing Res Rev.
98:1023202024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Obara CJ, Nixon-Abell J, Moore AS, Riccio
F, Hoffman DP, Shtengel G, Xu CS, Schaefer K, Pasolli HA, Masson
JB, et al: Motion of VAPB molecules reveals ER-mitochondria contact
site subdomains. Nature. 626:169–176. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Galmes R, Houcine A, van Vliet AR,
Agostinis P, Jackson CL and Giordano F: ORP5/ORP8 localize to
endoplasmic reticulum-mitochondria contacts and are involved in
mitochondrial function. EMBO Rep. 17:800–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li M, Zhang Y, Yu G, Gu L, Zhu H, Feng S,
Xiong X and Jian Z: Mitochondria-associated endoplasmic reticulum
membranes tethering protein VAPB-PTPIP51 protects against ischemic
stroke through inhibiting the activation of autophagy. CNS Neurosci
Ther. 30:e147072024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kalarikkal M, Saikia R, Oliveira L,
Bhorkar Y, Lonare A, Varshney P, Dhamale P, Majumdar A and Joseph
J: Nup358 restricts ER-mitochondria connectivity by modulating
mTORC2/Akt/GSK3β signalling. EMBO Rep. 25:4226–4251. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gomez-Suaga P, Paillusson S, Stoica R,
Noble W, Hanger DP and Miller CCJ: The ER-mitochondria tethering
complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 27:371–385.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paillusson S, Gomez-Suaga P, Stoica R,
Little D, Gissen P, Devine MJ, Noble W, Hanger DP and Miller CCJ:
α-Synuclein binds to the ER-mitochondria tethering protein VAPB to
disrupt Ca2+ homeostasis and mitochondrial ATP
production. Acta Neuropathol. 134:129–149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang T, Zhu Q, Cao B, Cai Y, Wen S, Bian
J, Zou H, Song R, Gu J, Liu X, et al: Ca2+ transfer via
the ER-mitochondria tethering complex in neuronal cells contribute
to cadmium-induced autophagy. Cell Biol Toxicol. 38:469–485. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Naon D, Hernandez-Alvarez MI, Shinjo S,
Wieczor M, Ivanova S, de Brito OM, Quintana A, Hidalgo J, Palacín
M, Aparicio P, et al: Splice variants of mitofusin 2 shape the
endoplasmic reticulum and tether it to mitochondria. Science.
380:eadh93512023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao Y, Shen W, Zhang M, Guo M, Dou Y, Han
S, Yu J, Cui M and Zhao Y: DDAH-1 maintains endoplasmic
reticulum-mitochondria contacts and protects dopaminergic neurons
in Parkinson's disease. Cell Death Dis. 15:3992024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kirshenbaum LA, Dhingra R, Bravo-Sagua R
and Lavandero S: DIAPH1-MFN2 interaction decreases the endoplasmic
reticulum-mitochondrial distance and promotes cardiac injury
following myocardial ischemia. Nat Commun. 15:14692024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Han S, Zhao F, Hsia J, Ma X, Liu Y, Torres
S, Fujioka H and Zhu X: The role of Mfn2 in the structure and
function of endoplasmic reticulum-mitochondrial tethering in vivo.
J Cell Sci. 134:jcs2534432021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Song Y, Geng W, Zhu D, Liang H, Du Z, Tong
B, Wang K, Li S, Gao Y, Feng X, et al: SYNJ2BP ameliorates
intervertebral disc degeneration by facilitating
mitochondria-associated endoplasmic reticulum membrane formation
and mitochondrial Zn(2+) homeostasis. Free Radic Biol Med.
212:220–233. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu YT, Zhang H, Duan SB, Wang JW, Chen H,
Zhan M, Zhang W, Li AM, Liu Y, Yang Y and Yang S: Mitofusin2
ameliorated endoplasmic reticulum stress and mitochondrial reactive
oxygen species through maintaining mitochondria-associated
endoplasmic reticulum membrane integrity in cisplatin-induced acute
kidney injury. Antioxid Redox Signal. 40:16–39. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhao Y, Chang YH, Ren HR, Lou M, Jiang FW,
Wang JX, Chen MS, Liu S, Shi YS, Zhu HM and Li JL: Phthalates
induce neurotoxicity by disrupting the Mfn2-PERK axis-mediated
endoplasmic reticulum-mitochondria interaction. J Agric Food Chem.
72:7411–7422. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hinton A Jr, Katti P, Mungai M, Hall DD,
Koval O, Shao J, Vue Z, Lopez EG, Rostami R, Neikirk K, et al:
ATF4-dependent increase in mitochondrial-endoplasmic reticulum
tethering following OPA1 deletion in skeletal muscle. J Cell
Physiol. 239:e312042024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
van Vliet AR, Verfaillie T and Agostinis
P: New functions of mitochondria associated membranes in cellular
signaling. Biochim Biophys Acta. 1843:2253–2262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Xue M, Fang T, Sun H, Cheng Y, Li T, Xu C,
Tang C, Liu X, Sun B and Chen L: PACS-2 attenuates diabetic kidney
disease via the enhancement of mitochondria-associated endoplasmic
reticulum membrane formation. Cell Death Dis. 12:11072021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li C, Li L, Yang M, Yang J, Zhao C, Han Y,
Zhao H, Jiang N, Wei L, Xiao Y, et al: PACS-2 Ameliorates tubular
injury by facilitating endoplasmic reticulum-mitochondria contact
and mitophagy in diabetic nephropathy. Diabetes. 71:1034–1050.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu S, Han S, Wang C, Chen H, Xu Q, Feng
S, Wang Y, Yao J, Zhou Q, Tang X, et al: MAPK1 mediates MAM
disruption and mitochondrial dysfunction in diabetic kidney disease
via the PACS-2-dependent mechanism. Int J Biol Sci. 20:569–584.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Moulis M, Grousset E, Faccini J, Richetin
K, Thomas G and Vindis C: The multifunctional sorting protein
PACS-2 controls mitophagosome formation in human vascular smooth
muscle cells through mitochondria-ER contact sites. Cells.
8:6382019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hayashi T and Su TP: Sigma-1 receptor
chaperones at the ER-mitochondrion interface regulate Ca(2+)
signaling and cell survival. Cell. 131:596–610. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Leonard A, Grose V, Paton AW, Paton JC,
Yule DI, Rahman A and Fazal F: Selective inactivation of
intracellular BiP/GRP78 attenuates endothelial inflammation and
permeability in acute lung injury. Sci Rep. 9:20962019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hayashi T, Lewis A, Hayashi E, Betenbaugh
MJ and Su TP: Antigen retrieval to improve the immunocytochemistry
detection of sigma-1 receptors and ER chaperones. Histochem Cell
Biol. 135:627–637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mahamed Z, Shadab M, Najar RA, Millar MW,
Bal J, Pressley T and Fazal F: The protective role of
mitochondria-associated endoplasmic reticulum membrane (MAM)
protein sigma-1 receptor in regulating endothelial inflammation and
permeability associated with acute lung injury. Cells. 13:52023.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang Z, Zhou H, Gu W, Wei Y, Mou S, Wang
Y, Zhang J and Zhong Q: CGI1746 targets σ1R to modulate
ferroptosis through mitochondria-associated membranes. Nat Chem
Biol. 20:699–709. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bui M, Gilady SY, Fitzsimmons RE, Benson
MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD and Simmen T:
Rab32 modulates apoptosis onset and mitochondria-associated
membrane (MAM) properties. J Biol Chem. 285:31590–31602. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Herrera-Cruz MS, Yap MC, Tahbaz N,
Phillips K, Thomas L, Thomas G and Simmen T: Rab32 uses its
effector reticulon 3L to trigger autophagic degradation of
mitochondria-associated membrane (MAM) proteins. Biol Direct.
16:222021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ortiz-Sandoval CG, Hughes SC, Dacks JB and
Simmen T: Interaction with the effector dynamin-related protein 1
(Drp1) is an ancient function of Rab32 subfamily proteins. Cell
Logist. 4:e9863992014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rampelt H, Zerbes RM, van der Laan M and
Pfanner N: Role of the mitochondrial contact site and cristae
organizing system in membrane architecture and dynamics. Biochim
Biophys Acta Mol Cell Res. 1864:737–746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tang J, Zhang K, Dong J, Yan C, Hu C, Ji
H, Chen L, Chen S, Zhao H and Song Z: Sam50-Mic19-Mic60 axis
determines mitochondrial cristae architecture by mediating
mitochondrial outer and inner membrane contact. Cell Death Differ.
27:146–160. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dong J, Chen L, Ye F, Tang J, Liu B, Lin
J, Zhou PH, Lu B, Wu M, Lu JH, et al: Mic19 depletion impairs
endoplasmic reticulum-mitochondrial contacts and mitochondrial
lipid metabolism and triggers liver disease. Nat Commun.
15:1682024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Vance JE: Phospholipid synthesis in a
membrane fraction associated with mitochondria. J Biol Chem.
265:7248–7256. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wozny MR, Di Luca A, Morado DR, Picco A,
Khaddaj R, Campomanes P, Ivanović L, Hoffmann PC, Miller EA, Vanni
S and Kukulski W: In situ architecture of the ER-mitochondria
encounter structure. Nature. 618:188–192. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Peter AT, Petrungaro C, Peter M and
Kornmann B: METALIC reveals interorganelle lipid flux in live cells
by enzymatic mass tagging. Nat Cell Biol. 24:996–1004. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Koch C, Lenhard S, Raschle M,
Prescianotto-Baschong C, Spang A and Herrmann JM: The ER-SURF
pathway uses ER-mitochondria contact sites for protein targeting to
mitochondria. EMBO Rep. 25:2071–2096. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chakraborty N, Jain BK, Shembekar S and
Bhattacharyya D: ER exit sites (ERES) and ER-mitochondria encounter
structures (ERMES) often localize proximally. FEBS Lett.
597:320–336. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cheema JY, He J, Wei W and Fu C: The
endoplasmic reticulum-mitochondria encounter structure and its
regulatory proteins. Contact (Thousand Oaks).
4:251525642110644912021.PubMed/NCBI
|
|
84
|
Szabadkai G, Bianchi K, Varnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Luo F, Fu M, Wang T, Qi Y, Zhong X, Li D
and Liu B: Down-regulation of the mitochondrial fusion protein
Opa1/Mfn2 promotes cardiomyocyte hypertrophy in
Su5416/hypoxia-induced pulmonary hypertension rats. Arch Biochem
Biophys. 747:1097432023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yepuri G, Ramirez LM, Theophall GG,
Reverdatto SV, Quadri N, Hasan SN, Bu L, Thiagarajan D, Wilson R,
Díez RL, et al: DIAPH1-MFN2 interaction regulates
mitochondria-SR/ER contact and modulates ischemic/hypoxic stress.
Nat Commun. 14:69002023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Paillard M, Tubbs E, Thiebaut PA, Gomez L,
Fauconnier J, Da Silva CC, Teixeira G, Mewton N, Belaidi E, Durand
A, et al: Depressing mitochondria-reticulum interactions protects
cardiomyocytes from lethal hypoxia-reoxygenation injury.
Circulation. 128:1555–1565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yang J, Sun M, Chen R, Ye X, Wu B, Liu Z,
Zhang J, Gao X, Cheng R, He C, et al: Mitochondria-associated
membrane protein PACS2 maintains right cardiac function in
hypobaric hypoxia. iScience. 26:1063282023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wang Y, Li X, Xu X, Qu X and Yang Y:
Transient receptor potential vanilloid type 1 protects against
pressure overload-induced cardiac hypertrophy by promoting
mitochondria-associated endoplasmic reticulum membranes. J
Cardiovasc Pharmacol. 80:430–441. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Song Z, Song H, Liu D, Yan B, Wang D,
Zhang Y, Zhao X, Tian X, Yan C and Han Y: Overexpression of MFN2
alleviates sorafenib-induced cardiomyocyte necroptosis via the
MAM-CaMKIIdelta pathway in vitro and in vivo. Theranostics.
12:1267–1285. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bassot A, Chen J, Takahashi-Yamashiro K,
Yap MC, Gibhardt CS, Le GNT, Hario S, Nasu Y, Moore J, Gutiérrez T,
et al: The endoplasmic reticulum kinase PERK interacts with the
oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep.
42:1118992023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
He W, Sun Z, Tong G, Zeng L, He W, Chen X,
Zhen C, Chen P, Tan N and He P: FUNDC1 alleviates
doxorubicin-induced cardiotoxicity by restoring
mitochondrial-endoplasmic reticulum contacts and blocked autophagic
flux. Theranostics. 14:3719–3738. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zeisbrich M, Yanes RE, Zhang H, Watanabe
R, Li Y, Brosig L, Hong J, Wallis BB, Giacomini JC and Assimes TL:
Hypermetabolic macrophages in rheumatoid arthritis and coronary
artery disease due to glycogen synthase kinase 3b inactivation. Ann
Rheum Dis. 77:1053–1062. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Assis LHP, Dorighello GG, Rentz T, de
Souza JC, Vercesi AE and de Oliveira HCF: In vivo pravastatin
treatment reverses hypercholesterolemia induced
mitochondria-associated membranes contact sites, foam cell
formation, and phagocytosis in macrophages. Front Mol Biosci.
9:8394282022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang LR, Zhang CX, Tian LB, Huang J, Jia
LJ, Tao H, Yu NW and Li BH: Identification and validation of
mitochondrial endoplasmic reticulum membrane-related genes in
atherosclerosis. Mamm Genome. 36:665–682. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X,
Huang K, Xie Z and Zou MH: Binding of FUN14 domain containing 1
with inositol 1,4,5-trisphosphate receptor in
mitochondria-associated endoplasmic reticulum membranes maintains
mitochondrial dynamics and function in hearts in vivo. Circulation.
136:2248–2266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xu H, Yu W, Sun M, Bi Y, Wu NN, Zhou Y,
Yang Q, Zhang M, Ge J, Zhang Y and Ren J: Syntaxin17 contributes to
obesity cardiomyopathy through promoting mitochondrial
Ca2+ overload in a Parkin-MCUb-dependent manner.
Metabolism. 143:1555512023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xu H, Wang X, Yu W, Sun S, Wu NN, Ge J,
Ren J and Zhang Y: Syntaxin 17 protects against heart failure
through recruitment of CDK1 to promote DRP1-dependent mitophagy.
JACC Basic Transl Sci. 8:1215–1239. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Janer A, Morris JL, Krols M, Antonicka H,
Aaltonen MJ, Lin ZY, Anand H, Gingras AC, Prudent J and Shoubridge
EA: ESYT1 tethers the ER to mitochondria and is required for
mitochondrial lipid and calcium homeostasis. Life Sci Alliance.
7:e2023023352024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Liu IF, Lin TC, Wang SC, Yen CH, Li CY,
Kuo HF, Hsieh CC, Chang CY, Chang CR, Chen YH, et al: Long-term
administration of Western diet induced metabolic syndrome in mice
and causes cardiac microvascular dysfunction, cardiomyocyte
mitochondrial damage, and cardiac remodeling involving caveolae and
caveolin-1 expression. Biol Direct. 18:92023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Diokmetzidou A, Soumaka E, Kloukina I,
Tsikitis M, Makridakis M, Varela A, Davos CH, Georgopoulos S,
Anesti V, Vlahou A and Capetanaki Y: Desmin and αB-crystallin
interplay in the maintenance of mitochondrial homeostasis and
cardiomyocyte survival. J Cell Sci. 129:3705–3720. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Raj PS, Nair A, Rani MR, Rajankutty K,
Ranjith S and Raghu KG: Ferulic acid attenuates high
glucose-induced MAM alterations via PACS2/IP3R2/FUNDC1/VDAC1
pathway activating proapoptotic proteins and ameliorates
cardiomyopathy in diabetic rats. Int J Cardiol. 372:101–109. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yuan M, Gong M, He J, Xie B, Zhang Z, Meng
L, Tse G, Zhao Y, Bao Q, Zhang Y, et al: IP3R1/GRP75/VDAC1 complex
mediates endoplasmic reticulum stress-mitochondrial oxidative
stress in diabetic atrial remodeling. Redox Biol. 52:1022892022.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang P, Yan X, Zhang X, Liu Y, Feng X,
Yang Z, Zhang J, Xu X, Zheng Q, Liang L and Han H: TMEM215 prevents
endothelial cell apoptosis in vessel regression by blunting
BIK-regulated ER-to-mitochondrial ca influx. Circ Res. 133:739–757.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tong M, Mukai R, Mareedu S, Zhai P, Oka
SI, Huang CY, Hsu CP, Yousufzai FAK, Fritzky L, Mizushima W, et al:
Distinct roles of DRP1 in conventional and alternative mitophagy in
obesity cardiomyopathy. Circ Res. 133:6–21. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lu X, Gong Y, Hu W, Mao Y, Wang T, Sun Z,
Su X, Fu G, Wang Y and Lai D: Ultrastructural and proteomic
profiling of mitochondria-associated endoplasmic reticulum
membranes reveal aging signatures in striated muscle. Cell Death
Dis. 13:2962022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Li YE, Sowers JR, Hetz C and Ren J: Cell
death regulation by MAMs: From molecular mechanisms to therapeutic
implications in cardiovascular diseases. Cell Death Dis.
13:5042022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Chen X, Yang Y, Zhou Z, Yu H, Zhang S,
Huang S, Wei Z, Ren K and Jin Y: Unraveling the complex interplay
between mitochondria-associated membranes (MAMs) and cardiovascular
inflammation: Molecular mechanisms and therapeutic implications.
Int Immunopharmacol. 141:1129302024. View Article : Google Scholar : PubMed/NCBI
|