Introduction
Sepsis, resulting from a dysregulated host immune
response to infection and characterized by the excessive release of
inflammatory mediators and abnormal immune cell activation, remains
a major challenge in global healthcare (1). In 2017, ~11.0 million sepsis-related
mortalities were reported worldwide, representing 19.7% of all
global mortalities. Despite a 52.8% decline in the age-standardized
sepsis mortality from 1990 to 2017, sepsis remains a leading cause
of death, prompting researchers to explore new treatment strategies
(2).
The attachment of N-acetylglucosamine (GlcNAc) via
an oxygen linkage (O-GlcNAcylation) is a post-translational
modification (PTM) of the serine (Ser) and threonine (Thr) residues
in proteins. This PTM can regulate the activation, proliferation
and apoptosis of immune cells, as well as the production of
inflammatory mediators, by influencing various metabolic pathways
and cellular processes (3,4). The regulatory role of O-GlcNAcylation
is particularly important in the context of sepsis (5), as it not only regulates the immune
response to infection but also controls autophagy, the main
mechanism by which cells clear pathogens and damaged organelles.
Therefore, O-GlcNAcylation may play a critical role in the
development of sepsis by affecting immune cell function and the
efficiency of autophagy. The present review aims to investigate the
underlying mechanisms of O-GlcNAcylation as a bridge between
infection immunity and autophagy in sepsis, and to assess its
potential as a therapeutic target.
Infection immunity in sepsis
Sepsis is a life-threatening condition characterized
by organ dysfunction resulting from dysregulation of the host
response to infection (6), and
affected ~48.9 million individuals worldwide in 2017, posing a
great threat to health (2). The
pathogens responsible for sepsis include bacteria, fungi, viruses
and parasites.
Immune cells play a key role in the pathogenesis of
sepsis. Immunological studies have shown that the host immune
response in sepsis involves several sequential or concurrent
processes that lead to excessive inflammation and immune
suppression. This immune dysfunction results in impaired innate and
adaptive immune responses (7).
During the initial phase of sepsis, the body mounts
a robust inflammatory response to infection. Immune cells such as
macrophages, dendritic cells, neutrophils and lymphocytes are
activated following the recognition of pathogen-associated and
damage-associated molecular patterns, which triggers the
inflammatory response (8). As
sepsis progresses, patients often develop immunosuppression,
characterized by diminished immune cell function, particularly
decreased activity of T cells and B cells, and increased activity
of regulatory T (Treg) cells. This immunosuppressive state
increases susceptibility to secondary infections and may lead to
clinical deterioration and even death (Fig. 1) (8,9).
In sepsis, autophagy plays a vital role in
cytoplasmic quality control, cellular metabolism, and innate and
adaptive immune responses. It involves a variety of immune cells,
including neutrophils, macrophages, T cells and B cells (10,11).
In neutrophils, autophagy facilitates the clearance of pathogenic
microorganisms, supports cellular function and prevents cell death.
Studies have shown that the autophagic activity of neutrophils
increases during sepsis, which may help to maintain their survival
and function. However, the hyperactivation or inhibition of
autophagy can impair the function of neutrophils, thereby affecting
the progression of sepsis (12,13).
In macrophages, autophagy not only facilitates the clearance of
pathogens but also regulates the inflammatory response. During
sepsis, the autophagic activity of macrophages may be suppressed,
leading to the excessive release of inflammatory mediators and
amplification of the inflammatory response. Conversely, the
activation of autophagy can attenuate macrophage-mediated
inflammation, offering protection against sepsis (14). Autophagy is also essential for the
survival and function of T cells. In sepsis, the autophagic
activity of T cells is weakened, which results in increased T-cell
apoptosis and immunosuppression. In a murine model of sepsis, mice
deficient in a T-cell-specific autophagy gene known as autophagy
related 7 exhibited a higher mortality rate and greater
immunosuppression than was observed in wild-type mice, suggesting a
protective role for T-cell autophagy in the regulation of T-cell
apoptosis and immunosuppression (10,15).
The role of B cells in sepsis has not been fully elucidated;
however, autophagy has been indicated to affect the immune response
in sepsis by regulating B cell maturation and antibody production,
with the activation of autophagy being suggested to contribute to
the maintenance of B cell function and prevent immunosuppression
(10). In summary, the effect of
autophagy on immune cells in sepsis is multifaceted, involving
various immune cells, such as neutrophils, macrophages, T cells and
B cells. Activation or inhibition of autophagy may have an
important effect on the pathogenesis of sepsis, and the regulation
of autophagy may be a novel therapeutic strategy for sepsis.
Basic biological functions of protein
O-GlcNAc modification
O-GlcNAcylation is a crucial PTM in which GlcNAc
molecules attach to Ser or Thr residues in proteins. O-GlcNAc
modifications differ from other types of glycosylation in several
important aspects (3,16–18):
i) They involve the addition of a monosaccharide, unlike other
types where complex sugar chains are involved; ii) they primarily
occur in the cytoplasm and nucleus, rather than via the Golgi or
endoplasmic reticulum pathways; iii) the modification process is
dynamic, reversible and regulated by O-GlcNAc transferase (OGT) and
O-GlcNAcase (OGA); iv) they interact with other PTMs, such as
phosphorylation, methylation and ubiquitination, to regulate
protein activity and function; and v) they play a role in the
regulation of key cellular biological functions. Due to these
characteristics, O-GlcNAcylation makes a multifaceted contribution
to cell biology and disease development.
Impact of the interaction between
O-GlcNAcylation and phosphorylation in terms of sepsis
O-GlcNAcylation and phosphorylation are two key
protein PTMs that regulate a variety of physiological and
pathological processes through dynamic competition or synergy.
O-GlcNAcylation involves the addition of a single GlcNAc group to
Ser or Thr residues, whereas phosphorylation involves the addition
of phosphate groups, often at the same or adjacent sites, resulting
in a competitive or cooperative regulatory relationship between the
two mechanisms (19). For example,
in metabolic regulation, O-GlcNAcylation inhibits the Ser9
phosphorylation of glycogen synthase kinase-3β, thereby reducing
the hyperphosphorylation of Tau protein, which may protect against
neurodegenerative diseases such as Alzheimer's disease (20,21).
Similarly, the Ser40 phosphorylation of tyrosine hydroxylase (TH)
promotes dopamine synthesis, whereas O-GlcNAcylation reduces TH
activity by inhibiting phosphorylation at this site, thereby
affecting L-DOPA levels (22). In
addition, O-GlcNAcylation can increase the activity of glycogen
phosphorylase, suggesting that synergistic effects may exist
between these two PTMs in certain contexts (23).
In the pathological process of sepsis, the balance
between O-GlcNAcylation and phosphorylation markedly influences the
regulation of inflammatory responses and cell death. Recent studies
have shown that O-GlcNAcylation can inhibit phosphorylation-driven
pro-inflammatory signaling by targeting key inflammation-related
proteins. For example, the O-GlcNAcylation of gasdermin D (GSDMD)
inhibits its phosphorylation-dependent activation, thereby
attenuating lipopolysaccharide (LPS)-induced pyroptosis, a
mechanism that may protect against vascular endothelial injury in
sepsis (24). Similarly,
homeodomain-interacting protein kinase 2 (HIPK2) inhibits
hyperinflammation by promoting the acetylation of NF-κB via the
phosphorylation of histone deacetylase 3 (HDAC3), while
O-GlcNAcylation may affect this pathway indirectly by regulating
the activity or stability of HIPK2 (25). The NF-κB acetylation promoted by
the HIPK2-mediated phosphorylation of HDAC3 has been shown to
ameliorate colitis-associated colorectal cancer and sepsis in mice.
In another study, Toll-like receptor 4 (TLR4) was shown to activate
ERK1/2 phosphorylation, leading to the downregulation of
Kruppel-like factor 4, thereby exacerbating the inflammatory
response in sepsis (26).
In sepsis, phosphorylation is often associated with
the amplification of pro-inflammatory and cell death signals. For
example, peptidylprolyl cis/trans isomerase, NIMA-interacting 1
exacerbates the inflammatory response in septic shock by promoting
the phosphorylation of p38 MAPK, which increases the activation of
the NLR family pyrin domain containing 3 (NLRP3) inflammasome.
O-GlcNAcylation can counteract this effect by competitively
modifying the same or adjacent sites of p38 MAPK, thereby
preventing its phosphorylation and blocking downstream inflammatory
signaling (27,28). Notably, O-GlcNAcylation and
phosphorylation may act synergistically in certain contexts. For
example, O-GlcNAc modification has been shown to increase
glycogenolysis by promoting the phosphorylation of glycogen
phosphorylase L during energy stress (23). In summary, the dynamic interplay
between O-GlcNAcylation and phosphorylation in sepsis reflects a
precisely regulated balance that controls inflammatory intensity,
cell death patterns and metabolic adaptation. Targeting this
interaction may offer novel strategies for the treatment of
sepsis.
Impact of the interaction between
O-GlcNAcylation and ubiquitination in terms of sepsis
There is a complex, cross-regulatory relationship
between O-GlcNAcylation and ubiquitination, both of which are
involved in cellular stress responses, metabolic regulation and
disease progression. O-GlcNAc modification affects the stability,
activity and interactions of proteins by the dynamic addition or
removal of GlcNAc groups, thereby influencing the ubiquitination
process (29). For example,
O-GlcNAcylation can increase the stability of key proteins by
inhibiting their ubiquitination; the O-GlcNAcylation of the
circadian regulators brain and muscle ARNT-like 1 and circadian
locomotor output cycles kaput prevents their ubiquitin-mediated
degradation, thereby maintaining the normal functioning of the
biological clock (30). In
addition, the synergistic or antagonistic effects of
O-GlcNAcylation and ubiquitination play critical roles in processes
such as DNA damage repair and cancer metabolism (31,32).
The activity of certain E3 ubiquitin ligases, such as members of
the cullin-RING ligase (CRL) family, can be regulated by O-GlcNAc
modifications, thereby affecting the ubiquitination levels of their
substrate proteins (33). The
ubiquitin-proteosome system itself is highly complex, with various
ubiquitin chain types, including lysine (Lys)48- and Lys63-linked
chains, and dynamic modification patterns. These features form a
multi-level interaction network with O-GlcNAcylation to coordinate
cellular responses to stresses such as oxidative stress and
nutrient deprivation (29,34).
The cross-regulation between O-GlcNAcylation and
ubiquitination plays a key role in modulating the intensity of the
inflammatory response and maintaining cellular homeostasis. Studies
have shown that O-GlcNAc modification can dynamically modulate
pro-inflammatory signaling pathways by targeting key components of
the ubiquitination system. For example, in macrophages,
O-GlcNAc-modified NOD-like receptor X1 (NLRX1) downregulates IL-1β
expression by interacting with inhibitor of κB kinase α (IKK-α) and
inhibiting its ubiquitination-dependent activation, thereby
potentially limiting tissue damage due to excessive inflammatory
responses in sepsis (35). In
addition, the E3 ubiquitin ligase tripartite motif containing 27
(TRIM27) exacerbates sepsis-related oxidative stress and
endothelial dysfunction by promoting the ubiquitin-mediated
degradation of peroxisome proliferator-activated receptor γ
(PPARγ), and O-GlcNAc modification may counteract the
pro-inflammatory effects of TRIM27 by stabilizing PPARγ protein
levels (30,36). Notably, the diversity of
ubiquitination systems, including different chain linkages such as
Lys48- and Lys63-linked chain types, contributes to the dual
regulatory features observed in sepsis: Lys48-linked ubiquitination
typically targets proteins for proteosomal degradation, helping to
clear damaged proteins, whereas Lys63-linked ubiquitination
amplifies inflammation by activating pathways such as the NF-κB
pathway (29,37). O-GlcNAc modification may balance
the inflammatory phenotype of macrophages by selectively regulating
the activity of specific E3 ligases, such as E3 ubiquitin-protein
ligase COP1, and affecting the ubiquitination status of
transcription factors such as CCAAT/enhancer binding protein β
(38).
Ubiquitination often serves as a central node for
pro-inflammatory and cell death signaling in sepsis, while O-GlcNAc
modification may exert a protective effect through multi-level
regulatory mechanisms. For example, microbial infection-induced
endothelial cell dysfunction is associated with the aberrant
ubiquitination of a variety of proteins, such as NADPH oxidase 4
(NOX4), whose stability is regulated by ubiquitination, and its
overexpression exacerbates oxidative stress (36,37).
O-GlcNAc modification may reduce the production of reactive oxygen
species (ROS) by directly modifying NOX4 or regulating the activity
of its E3 ligases, such as TRIM27, analogous to its modulatory role
in the CRL family ubiquitin ligases involved in DNA damage repair
(33). Small-molecule drugs
targeting the ubiquitination system, such as deubiquitinase
inhibitors, have been shown to modulate the inflammatory cascade in
sepsis (39). Similarly, the
development of OGT inhibitors or activators may provide new
therapeutic strategies by modulating the O-GlcNAc-ubiquitination
axis. Notably, there may be synergistic effects between these
pathways: For example, O-GlcNAcylation may enhance the stability of
NLRX1, promoting its binding to IKK-α and thereby indirectly
inhibiting the ubiquitin-dependent activation of the IKK complex;
this regulatory homeostasis may exhibit distinct regulatory
characteristics at different stages of sepsis (35). In conclusion, the interplay between
O-GlcNAcylation and ubiquitination constitutes a complex regulatory
network in sepsis. Further elucidation of the underlying mechanism
may lay a foundation for the development of targeted interventions
to prevent inflammatory imbalance and organ injury.
Impact of the interaction between
O-GlcNAcylation and methylation in terms of sepsis
There is extensive cross-regulation between O-GlcNAc
modification and both DNA and histone methylation, which together
contribute to the maintenance of epigenetic homeostasis and the
regulation of gene expression. Studies have shown that OGT directly
modifies DNA methyltransferase 1 through glucose-dependent
O-GlcNAcylation, thereby inhibiting its activity, reducing the
overall level of genomic methylation and affecting transposon
silencing (40,41). At the histone level, OGT is
recruited to specific chromatin regions by disruptor of telomeric
silencing 1-like, a histone methyltransferase, where it catalyzes
the O-GlcNAcylation of histone H2B, which works synergistically
with histone H3 Lys79 (H3K79) methylation to regulate gene
transcriptional activation (31).
In addition, metabolic signals, such as glucose availability, are
integrated into the epigenetic regulatory network via O-GlcNAc
modification. For example, the c-Myc oncoprotein modulates
mitochondrial metabolism and O-GlcNAc cycling, influencing the
activity and subcellular localization of DNA and RNA demethylases,
thereby linking cellular metabolic state to epigenetic
reprogramming (42).
The crosstalk between O-GlcNAcylation and
methylation regulates epigenetic reprogramming and metabolic
adaptation, which affects the inflammatory response and organ
injury. Dysregulation of this epigenetic modification is
particularly prominent in monocytes from patients with sepsis and
is strongly associated with the aberrant expression of
pro-inflammatory cytokines such as TNF-α and IL-6, as well as organ
dysfunction (43). In addition,
the synergistic interaction of histone methylation with
O-GlcNAcylation has a dual role in sepsis: During the endotoxin
tolerance stage, G9a-mediated H3K9 methylation and DNA methylation
contribute to silencing of the TNF-α gene (44), while O-GlcNAc modification
dynamically regulates the expression of pro- and anti-inflammatory
genes by regulating DOT1-like histone Lys
methyltransferase-dependent H3K79 methylation (31). This multilevel
epigenetic-metabolic-ubiquitination regulatory network provides a
new paradigm for targeting immunometabolic imbalances in
sepsis.
In conclusion, O-GlcNAcylation forms a
multi-dimensional regulatory network with other PTMs, including
phosphorylation, ubiquitination and methylation, that supports a
shift in sepsis treatment from a single anti-inflammatory mode to a
systematic regulation based on the dynamic integration of the PTM
network. This integrated perspective could provide enable the
reversal of immunometabolic imbalances in sepsis.
O-GlcNAcylation and autophagy
The PTM of proteins plays a key role in autophagy,
either by directly regulating the expression of autophagy-related
genes or by modulating key components of the autophagy signaling
pathway (45–47). Among these PTMS, O-GlcNAcylation
has been shown to be closely associated with the regulation of
autophagy; it regulates autophagy by modifying key
autophagy-related proteins, thereby affecting their activity,
stability and subcellular localization (18). Several proteins with different
roles in the autophagy pathway have been identified as potential
targets of O-GlcNAcylation (Fig. 2
and Table I) (28,48–60).
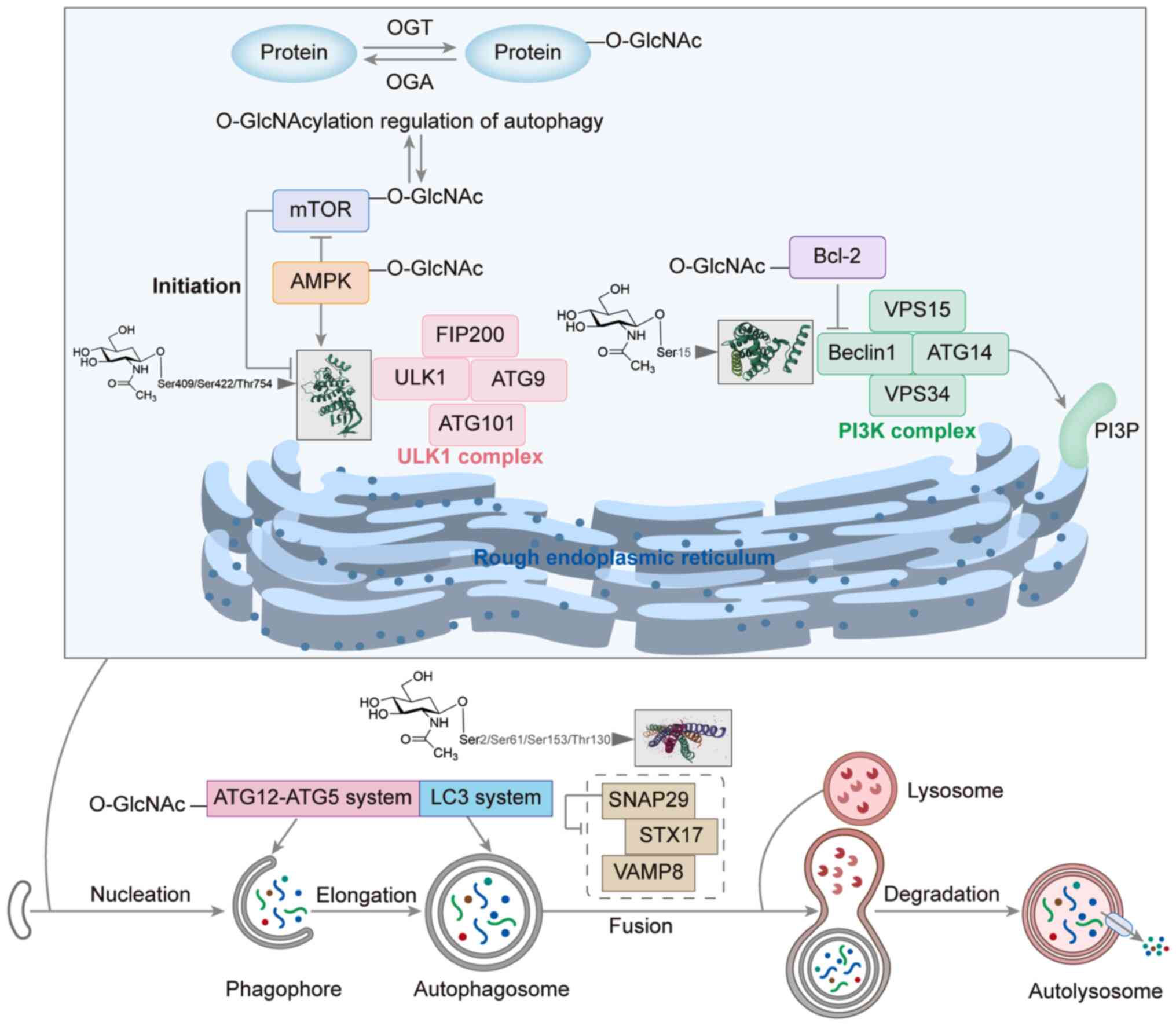 | Figure 2.Role of O-GlcNAcylation in the
initiation, nucleation, elongation and fusion stages of autophagy
and lysosomal activity. AMPK, AMP-activated protein kinase; ATG,
autophagy related; FIP200, FAK family kinase-interacting protein of
200 kDa; LC3, microtubule-associated protein 1A/1B-light chain 3;
O-GlcnAc, O-linked N-acetylglucosamine; O-GlcNAcylation, attachment
of GlcNAc via an oxygen linkage; OGA, O-GlcNAcase; OGT, O-GlcNAc
transferase; PI3K, phosphoinositide 3-kinase; PI3P,
phosphatidylinositol-3-phosphate; SNAP29, synaptosome-associated
protein 29; STX17, syntaxin 17; ULK1, unc-51 like autophagy
activating kinase 1; VAMP8, vesicle-associated membrane protein 8;
VSP, voltage-sensing phosphatase. |
 | Table I.Summary of O-GlcNAc modification
sites and their effects on protein function in autophagy
regulation. |
Table I.
Summary of O-GlcNAc modification
sites and their effects on protein function in autophagy
regulation.
| Protein | O-GlcNAc
modification sites | O-GlcNAcylation
regulatory mechanism | Autophagy
phase | (Refs.) |
|---|
| Beclin1 | Ser15 | O-GlcNAcylation
enhances beclin1 binding to Bcl-2, blocks its independent activity,
inhibits autophagosome formation, and reduces basal autophagy
levels | Initiation | (48,49) |
| Bcl-2 | Unclear | O-GlcNAcylation
upregulates Bcl-2 expression, enhances binding stability with
beclin1, inhibits autophagy, promotes apoptosis, and balances
cellular stress responses | Initiation | (49,50) |
| AMPK | Unclear | O-GlcNAc
modification inhibits AMPK activity, suppressing ULK1 activation
and autophagy | Initiation | (28) |
| mTOR | Unclear | Inhibiting OGT
enhances autophagy in rat cortical neurons; this effect is further
amplified when treated with the mTOR inhibitor rapamycin | Initiation | (51) |
| ULK1 | Thr754, Ser409 and
Ser422 | O-GlcNAcylation at
Thr754 is critical for VPS34 activation via ATG14L, promoting
autophagy; Ser409 modification inhibits phosphorylation at Ser423,
stabilizing ULK1; maresin-2 activates GFAT1, enhances O-GlcNAc
modification and autophagosome formation via TAK1-TAB1 inhibition,
but ULK1 Ser409 and Ser422 mutations counteract this effect | Initiation | (52–54) |
| PINK1 | Unclear | O-GlcNAcylation
inhibits PINK1 stability, blocks PARKIN recruitment during
mitochondrial depolarization, suppresses mitophagy, and leads to
the accumulation of damaged mitochondria | Elongation | (55) |
| SNAP29 | Ser2, Ser61, Thr130
and Ser153 | O-GlcNAcylation
blocks SNARE complex assembly with STX17-VAMP8, thereby interfering
with membrane fusion and inhibiting autophagy | Fusion | (56–60) |
The activation of unc-51 like autophagy activating
kinase 1 (ULK1) by stressors such as nutrient deprivation (for
example, amino acid starvation) and energy stress (for example, ATP
depletion/AMP elevation) triggers the autophagic process. ULK1 and
ULK2 form protein complexes with various mitosome-associated
proteins, including 200 kDa FAK family kinase-interacting protein,
autophagy related (ATG)13 and ATG101, which jointly regulate the
initiation phase of autophagy (61). These ULK protein complexes play a
key regulatory role in the formation of autophagic vesicles and are
responsive to signals from multiple signaling pathways. The
activity of the ULK1 complex is regulated by both mTOR complex 1
and AMP-activated protein kinase (AMPK) (62,63).
Specifically, activation of mTOR via Akt and MAPK signaling
inhibits autophagy, while the inhibition of mTOR by AMPK or p53
signaling promotes autophagy. The ULK complex promotes the
localized synthesis of phosphatidylinositol-3-phosphate by
phosphorylating components of the class III phosphatidylinositol
3-kinase complex, which includes beclin1, vacuolar protein sorting
(VPS)15, VPS34 and ATG14 (64).
Beclin1 has been identified as a direct target of O-GlcNAcylation,
and the O-GlcNAcylation and phosphorylation of beclin1 at the Ser15
site has been shown to regulate autophagy in HTR-8/SVneo cells
(48). Bcl-2 inhibits autophagy by
interacting with beclin1, and Bcl-2 is also a target of
O-GlcNAcylation (49). The
O-GlcNAcylation of beclin1 enhances its binding to Bcl-2, thereby
suppressing autophagic activity, whereas the O-GlcNAcylation of
Bcl-2 regulates autophagy and apoptosis by influencing its
expression levels (50,65).
The elongation of phagophores is promoted by two
ubiquitin-like conjugation systems: The ATG12-ATG5 system and the
microtubule-associated protein 1A/1B-light chain 3 (LC3) system
(66). Wang and Hanover (67) demonstrated that the deletion of OGT
in Drosophila resulted in a reduction in O-GlcNAc levels and
a significant increase in the accumulation of GFP-LGG-1 (an
ATG8/LC3 homolog) and its phosphatidylethanolamine-modified form
under starvation conditions, indicating enhanced autophagosome
formation. Furthermore, OGT knockdown upregulated the mRNA
expression of ATG1 and ATG5, whereas OGT overexpression inhibited
the transcription of ATG5 and ATG8 (68). These findings suggest that
O-GlcNAcylation limits autophagic activity by suppressing the
expression of ATG genes. This mechanism may operate synergistically
with the stabilization of the beclin1/Bcl-2 complex by
O-GlcNAcylation in mammals to maintain the basal autophagy in a
low-activity state under normal conditions.
During autophagic fusion, intact autophagosomes fuse
with lysosomes to form autolysosomes, which enables the degradation
of substances via the action of lysosomal enzymes. O-GlcNAcylation
has been shown to regulate the autophagosome-lysosome fusion phase
of autophagy in an arsenic-exposed environment by targeting
synaptosome-associated protein 29 (SNAP29) (29). In SNAP29-mediated vesicle fusion,
autophagosome membranes containing syntaxin 17 (STX17) are linked
to lysosomes containing vesicle-associated membrane protein 8
(VAMP8) (69). The O-GlcNAcylation
of SNAP29 blocks the assembly of the SNARE complex from STX17,
SNAP29 and VAMP8, thereby impairing normal autophagy (70).
Infection and immune regulation by O-GlcNAc
modification
O-GlcNAc modification affects cellular metabolism,
autotransduction, translation and signal transduction as a nutrient
and stress sensor. The maintenance of proper O-GlcNAcylation is
crucial for health, as imbalances in O-GlcNAcylation can result in
pathological conditions such as tumors, diabetes and
neurodegenerative diseases (71–73).
A number of studies have focused on the role of O-GlcNAcylation in
the regulation of the immune response and maintenance of
homeostasis during infection. The present review offers insights
into the role of O-GlcNAcylation in the modulation of immune
responses against viral, bacterial, fungal and parasitic
infections, and explores its potential as a therapeutic target in
sepsis (Fig. 3).
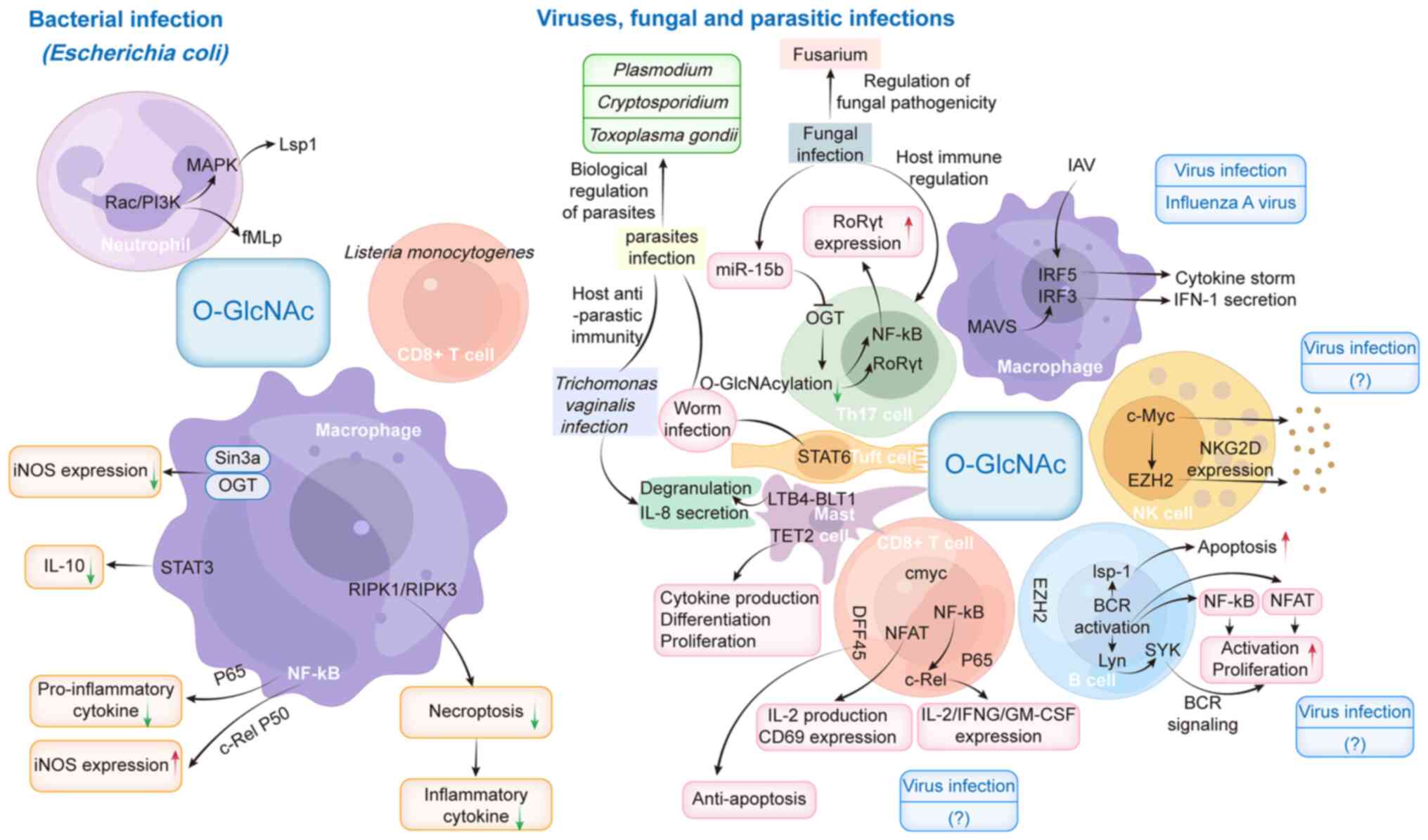 | Figure 3.Role of O-GlcNAcylation in the immune
response to infection with different pathogens. BCR, B-cell
receptor; BLT1, LTB4 receptor 1; c-Rel, cellular-Rel; DFF45, DNA
fragmentation factor 45; EZH2, enhancer of zeste 2; fMLP,
N-formyl-methionyl-leucyl-phenylalanine; GM-CSF, granulocyte
macrophage-colony-stimulating factor; IAV, influenza A virus; IFN,
interferon; IFNG, IFN-g; iNOS, inducible nitric oxide synthase;
IRF, IFN regulatory factor 3; Lsp1, lymphocyte-specific protein 1;
LTB4, leukotriene B4; MAVS, mitochondrial antiviral signaling
protein; miR-15b, microRNA-15b; NFAT, nuclear factor of activated
T-cells; NK, natural killer; NKG2D, NK group 2D; O-GlcNAc, O-linked
N-acetylglucosamine; OGT, O-GlcNAc transferase; PI3K,
phosphoinositide 3-kinase; RIPK, receptor-interacting protein
kinase; RoRyt, retinoic acid receptor-related orphan receptor gt;
TET2, Tet methylcytosine dioxygenase 2; Th17, T helper 17. |
Function of O-GlcNAcylation in immune
cell recognition of bacterial infections
Neutrophils are key cells of the innate immune
system that rapidly migrate to sites of bacterial infection and
remove pathogens via phagocytosis, the release of ROS and necrosis.
Neutrophils are the initial responders to injury and infection, and
the prognosis of sepsis is poor when neutrophil migration is
impaired (74). In both animal
models and patients with advanced sepsis, neutrophil function is
notably compromised, as evidenced by diminished bacterial
clearance, reduced reactivity, decreased ROS production and a
significant reduction in neutrophil numbers within infected tissues
(75).
O-GlcNAcylation has been found to significantly
influence neutrophil chemotaxis and cell migration. For example,
treatment with the chemoattractant
N-formylmethionyl-leucyl-phenylalanine significantly elevates
O-GlcNAcylation levels, thereby promoting neutrophil chemotaxis and
migration (76). In addition,
glucosamine (GlcN) promotes O-GlcNAcylation by increasing the
availability of UDP-GlcNAc, which is the donor substrate for OGT;
GlcN enters the hexosamine biosynthetic pathway (HBP) downstream of
the rate-limiting enzyme glutamine-fructose-6-phosphate
transaminase 1, effectively bypassing it and boosting UDP-GlcNAc
production (77). Mechanistically,
enhanced O-GlcNAcylation promotes Rac- and phosphoinositide
3-kinase-dependent chemotaxis. Rac is an key small GTPase that
regulates neutrophil mobilization by activating downstream MAPK
signaling and promoting the phosphorylation of lymphocyte specific
protein 1 at Ser243 (78–80). These signaling pathways are
critical for neutrophil migration and effector functions.
O-GlcNAcylation is inducible in neutrophils and modulates their
dynamic responses, potentially providing a novel therapeutic target
for the treatment of sepsis.
Macrophages play a key role in resistance to
bacterial infections by eliminating pathogens through various
mechanisms, including the activation of inducible nitric oxide
synthase (iNOS) to produce nitric oxide (NO). NO is a gaseous
signaling molecule with antibacterial properties, capable of
inhibiting bacterial metabolic enzymes, thereby impairing bacterial
viability. In response to bacterial infection or inflammatory
stimuli, macrophages increase iNOS expression, thereby increasing
NO production (81). NF-κB is a
key regulator of the inflammatory response in macrophages and can
be modified via O-GlcNAcylation. In RAW264.7 macrophage-like cells,
the interaction of OGT with murine Sin3A disrupts NF-κB activation
and downregulates iNOS gene expression upon LPS stimulation
(82). In microglia,
O-GlcNAcylation of the NF-κB subunit cellular-Rel (c-Rel) promotes
the formation of the c-Rel-p50 heterodimer and increases iNOS
production (83). Notably, the
response of the NF-κB subunit p65 to O-GlcNAcylation differs
markedly from that of c-Rel. Thiamet-G, a potent and selective
inhibitor of OGA, exerts anti-inflammatory effects by promoting the
O-GlcNAcylation of p65 in microglia, which subsequently modulates
its nuclear localization and reduces the expression of inflammatory
genes (84).
Inflammatory diseases such as sepsis are
characterized by elevated glucose metabolism in immune cells.
Despite macrophages exhibiting increased glycolytic activity and
pentose phosphate pathway activation during inflammation, endotoxin
exposure inhibits HBP activity, resulting in decreased protein
O-GlcNAcylation. Loss of OGT promotes the innate immune response
and exacerbates inflammation in sepsis. Specifically, OGT-mediated
O-GlcNAcylation of receptor-interacting protein kinase (RIPK)3 at
Thr467 disrupts its interaction with RIPK1 and RIPK3 proteins,
thereby inhibiting downstream innate immune responses, necroptosis
and antibacterial activity (85).
CD8+ T cells are essential for
cell-mediated immunity. Upon acute infection, naive CD8+
T cells differentiate into effector cells following antigen
stimulation. These effector cells eliminate bacteria by releasing
granules containing perforins and granzymes, and secreting
cytokines such as interferon (IFN)-γ and TNF-α (86,87).
This immune response involves the phosphorylation of various
intracellular signaling proteins. Protein O-GlcNAcylation often
acts synergistically with phosphorylation (16). Lopez Aguilar et al (88) demonstrated that O-GlcNAcylation was
dynamically elevated in murine CD8+ T cells during
Listeria infection and in vitro differentiation, with
activated effector cells exhibiting increased global levels in
vivo, and memory-like cells exhibiting even higher levels
(including distinct high-molecular weight modifications) in
vitro. Proteomics revealed that O-GlcNAc in effector-like cells
primarily targets transcription/translation machinery (such as
ribosomal proteins) to enable rapid proliferation, while in
memory-like cells it modifies transcriptional regulators, mRNA
processing factors and tRNA synthetases, suggesting roles in
establishing a poised state. Notably, O-GlcNAc contributes to the
‘histone code’ in both subsets, implicating it in epigenetic
regulation of T cell function.
O-GlcNAcylation plays a key regulatory role in the
immune response triggered by bacterial infection by dynamically
modifying inflammation-related proteins. Pathogen-associated
molecular patterns, such as bacterial LPS, can increase
O-GlcNAcylation levels in endothelial cells, thereby exacerbating
inflammation via the activation of inflammatory signaling
pathways (89). At the
molecular level, the OGT-mediated glycosylation of NLRX1
facilitates its interaction with IKK-α and promotes the expression
of IL-1β in M1 macrophages (35). In parallel, O-GlcNAcylation of the
NLRP3 inflammasome not only directly regulates pyroptosis but also
promotes the release of inflammatory cytokines by enhancing the
activity of the NIMA related kinase 7/NLRP3 axis (90,91).
Furthermore, this PTM can create a positive feedback loop, whereby
the O-GlcNAcylation of STAT3 further amplifies pro-inflammatory
signaling by enhancing JAK2/STAT3 pathway signaling
activity (92,93). As an upstream regulator, the phase
separation process of TNF receptor-associated factor 6 may also
contribute to this regulatory network. Notably, inhibition of
O-GlcNAcylation has been shown to effectively block STAT3- and
forkhead box protein O1-mediated oxidative stress-induced
apoptosis (93–95), suggesting that targeting this
modification may be a promising strategy for the alleviation of
hyperinflammation during bacterial infections.
These findings suggest that targeting O-GlcNAc
modifications may represent a novel therapeutic strategy for the
treatment of sepsis by optimizing immune cell function and reducing
the tissue damage caused by excessive inflammation.
Influence of O-GlcNAcylation on immune
cell detection of fungal infections
O-GlcNAcylation has been implicated in the
differentiation of T helper (Th)17 cells during fungal infections.
Naive CD4+ T cells can differentiate into Th1, Th2, Th17
and Treg cells (96). The
downregulation of OGT through microRNA-15b has been shown to
inhibit Th17 cell differentiation, thereby contributing to the
pathogenesis of multiple sclerosis (97). In addition, elevated O-GlcNAc
levels have been shown to promote the secretion of IL-17A by Th17
cells and upregulate the production of lipid ligands for the
transcription factor RAR-related orphan receptor γt in diet-induced
obese mice (98). Furthermore, in
Treg cells, O-GlcNAc-mediated post-translational modifications of
the T-cell receptor (TCR) have been demonstrated to increase the
stability of forkhead box P3 and activate STAT5, effects that act
synergistically to control Treg cell homeostasis and function
(96).
Notably, fungal O-GlcNAc metabolism itself directly
affects pathogenicity. As a key signaling molecule, GlcNAc
regulates morphological transitions, the expression of virulence
factors and environmental adaptation in a variety of fungi such as
Fusarium. For example, deletion of the OGT homologous gene
FpOGT in Fusarium proliferatum inhibits the expression of
glucose metabolism-related genes, such as glucokinase, resulting in
restricted fungal growth, a downregulated stress response and
markedly reduced virulence (99).
In addition, GlcNAc has been demonstrated to be a key metabolic
target for fungal pathogenesis since it regulates cell wall
synthesis, biofilm formation and host tissue invasion pathways
(100,101). From a therapeutic perspective,
interventions targeting GlcNAc sensing and metabolism, such as
GlcNAc analogs or metabolic engineering, have been shown to
effectively reduce the virulence of pathogenic fungi (102).
Therefore, in sepsis complicated by fungal
infection, the combined modulation of host O-GlcNAc levels to
enhance Th17-mediated antifungal immunity while inhibiting fungal
GlcNAc-dependent virulence pathways may offer a synergistic
therapeutic strategy. Future studies are necessary to further
elucidate the molecular mechanisms of the host-fungus O-GlcNAc
interaction network and to develop dual regulatory strategies to
optimize immunometabolic balance.
Role of O-GlcNAcylation in immune cell
recognition of viral infections
Macrophages play a crucial role in the innate immune
system, particularly for the early detection of and response to
viral infections. Research indicates that O-GlcNAcylation in
macrophages is essential for the recognition of RNA viruses. These
viruses are detected by cytosolic retinoic acid-inducible gene
I-like receptors (RLRs), particularly retinoic acid-inducible gene
I and melanoma differentiation-associated gene 5. Upon viral
recognition, these receptors engage mitochondrial antiviral
signaling protein (MAVS), which activates IFN regulatory factor
(IRF)3 and promotes the production of type I IFN (103). In mouse bone marrow-derived
macrophages, the absence of OGT disrupts MAVS-dependent RLR
signaling and reduces inflammatory cytokine expression (104). Research has further shown that
O-GlcNAcylation is crucial for modulating immune responses and
increasing infection resistance in macrophages. During influenza A
virus infection, O-GlcNAcylation regulates the inflammatory
response by modulating the activity of specific transcription
factors.
Specifically, O-GlcNAcylation at the Ser430 residue
of IRF5 is necessary for its Lys63-linked ubiquitination, which
promotes IRF5 activation by facilitating nuclear translocation and
interaction with the E3 ubiquitin ligase TRAF6. This modification
drives downstream proinflammatory cytokine production during
influenza infection (105). Thus,
both IRF3 and IRF5 undergo O-GlcNAcylation and ubiquitination,
which are crucial for the regulation of macrophage-mediated
inflammatory responses following influenza infection. Balancing
these modifications is essential to ensure the appropriate
intensity of the immune response while preventing excessive
inflammation.
Natural killer (NK) cells are crucial components of
the innate immune system, responsible for targeting and eliminating
virus-infected cells. O-GlcNAcylation has been shown to modulate
the function of NK cells as a PTM; specifically, the elevated
O-GlcNAcylation of NK cells attenuates their cytotoxicity. Notably,
certain molecules, such as the glutathione S-transferase-soluble
HLA-G1α chain fusion protein, inhibit O-GlcNAcylation in NK cells,
thereby enhancing their cytotoxic function (106).
NK group 2D (NKG2D) is an activating receptor on NK
cells that facilitates the recognition and elimination of
virus-infected cells presenting stress-induced ligands associated
with major histocompatibility complex class I molecules (107). The cytotoxic function of
NKG2D-expressing NK cells is regulated by the transcription factor
enhancer of zeste 2 (EZH2), a core subunit of polycomb repressive
complex 2. Inhibition of EZH2 has been shown to accelerate the
maturation of NK cells and improve their killing ability. Notably,
EZH2 contains five O-GlcNAcylation sites, which are pivotal for its
stabilization and function (108–110).
Another transcription factor that induces
cytotoxicity by regulating the O-GlcNAcylation of NK cells is
c-Myc. It upregulates the expression of granzyme B, thereby
increasing the cytotoxic activity of NK cells against
virus-infected cells. O-GlcNAcylation helps to maintain the
stability and function of c-Myc. The substrate for this
modification, UDP-GlcNAc, is generated through glycolysis and the
glutamine metabolic pathway. Glutamine depletion reduces UDP-GlcNAc
levels in NK cells, subsequently impacting c-Myc function and
diminishing cytotoxic activity (111). Thus, O-GlcNAc modification
potentially boosts NK cell immune responses, aiding in the
elimination of infected cells and protecting against the
progression of sepsis.
B cells are activated via the B-cell receptor (BCR)
on their surface, a process that is regulated by O-GlcNAcylation.
This PTM affects the YN proto-oncogene, an Src family tyrosine
kinase, and its interaction with spleen tyrosine kinase to initiate
BCR signaling (112). In
addition, the O-GlcNAcylation of nuclear factor of activated
T-cells, cytoplasmic 1 (NFATc1) and NF-κB further amplifies B cell
activation (113). Following BCR
crosslinking, the interaction between O-GlcNAc-modified
lymphocyte-specific protein 1 (Lsp1) and protein kinase C-β1 is
crucial for the regulation of apoptosis in activated B cells. The
O-GlcNAcylation of Lsp1 at Ser209 facilitates its phosphorylation
at Ser243, thereby modulating Lsp1 function (114). Also, the dysregulation of EZH2
activity or aberrant O-GlcNAcylation may impair antibody production
in B cells, indicating the importance of O-GlcNAcylation in the
modulation of B cell function and antibody responses (115).
CD8+ cytotoxic T cells identify and
destroy infected cells during viral infections. NF-κB and c-Myc are
essential transcription factors in the activation of
CD8+ T cells, and TCR-mediated stimulation induces their
O-GlcNAcylation. The O-GlcNAcylation of c-Rel enhances its binding
to the CD28 response element, resulting in the production of
proinflammatory cytokines, including IL-2, IFN-γ and granulocyte
macrophage-colony-stimulating factor. In addition, site-specific
O-GlcNAcylation stabilizes c-Myc, while the inhibition of OGT
decreases c-Myc levels (116,117). Furthermore, NFATc1 is mainly
O-GlcNAcylated in the cytosol, and translocates to the nucleus
after TCR activation. The O-GlcNAc modification of NFAT has been
demonstrated to be crucial for its nuclear translocation and
function, as the inhibition of OGT decreases TCR-induced IL-2
production and CD69 expression (113). These findings highlight the
importance of O-GlcNAcylation in T cells. The inhibition of OGT has
also been shown to affect the transcriptional activity of NF-κB in
T cells, with the p65 subunit of NF-κB undergoing O-GlcNAcylation
(113,118). In addition, the O-GlcNAcylation
of DNA fragmentation factor 45 shields proteins from
caspase-mediated cleavage during DNA damage-induced T-cell
apoptosis, indicating its protective role against apoptotic cell
death in T cells (119).
Modulating O-GlcNAc glycosylation levels or its
downstream signaling pathways may help to address immune imbalances
in patients with sepsis, suppress excessive inflammation, prevent
organ dysfunction and ultimately improve clinical outcomes.
Role of O-GlcNAcylation in immune cell
recognition of parasitic infections
The dual role of O-GlcNAcylation in host
anti-parasitic immunity and parasite self-regulation has become
increasingly evident. This involves an interaction between the
regulation of host immune cell function and the metabolic
dependence of the parasite on O-GlcNAc pathways. In the
anti-parasitic immunity of the host, O-GlcNAc contributes to key
pathways that regulate type 2 immune responses. For example, the
O-GlcNAcylation of STAT6 in intestinal epithelial cells forms the
first line of defense against helminth infection by driving tuft
cell proliferation and the release of gasdermin C-dependent
alarmins (120). Restoration of
STAT6 activation partially rescues the impaired immune responses to
helminth infection in OGT-deficient mice, which exhibit defective
STAT6 signaling and intestinal villus dysplasia (121). In addition, mast cell function is
closely associated with O-GlcNAc modification: In Trichomonas
vaginalis infection, leukotriene B4 (LTB4) secreted by the
parasite binds to LTB4 receptor 1 on mast cells, elevates O-GlcNAc
levels and promotes NADPH oxidase-dependent migration,
degranulation and IL-8 secretion (122). However, Tet methylcytosine
dioxygenase 2 (TET2)-mediated O-GlcNAcylation affects mast cell
differentiation and cytokine production by regulating chromatin
accessibility (123). These
findings suggest that O-GlcNAc contributes to the anti-parasitic
response through the multifaceted regulation of host immune cell
functions, including Th2 polarization, epithelial barrier defense
and mast cell effector functions.
At the biological level, glycosylation
modifications, including O-GlcNAc-related pathways, are essential
for the survival, development and pathogenicity of parasites. For
example, Plasmodium falciparum relies on the HBP to produce
glycosylphosphatidylinositol-anchored proteins, which are critical
for the invasion of red blood cells and immune evasion. Therefore,
blocking this pathway can lead to the growth arrest of
Plasmodium at the schizont stage (124). Although Cryptosporidium
lacks certain glycosylases, it compensates by hijacking host
glycosylation resources to perform its protein modifications
(125). In addition,
glycosylation in various parasites, such as Plasmodium and
Toxoplasma gondii, affects host immune recognition by
altering the function of surface proteins (126,127). Notably, there is an interaction
between host and parasite O-GlcNAc metabolism: Parasites may
disrupt the glycosylation homeostasis of host immune cells by
secreting metabolites such as GlcNAc, while host-targeted drugs
against key enzymes involved in parasite glycosylation, such as
glutamine:fructose-6-phosphate transaminase (GFAT) in the HBP, can
simultaneously inhibit parasite growth and enhance immune clearance
(124,127).
In summary, O-GlcNAcylation affects infection
outcomes through host-parasite bidirectional regulation: It
regulates host type 2 immune responses and effector cell functions
through targets such as STAT6 and TET2, while parasites rely on
glycosylation pathways to complete their developmental cycles and
evade host immunity. Future studies should aim to further elucidate
the parasite-specific O-GlcNAc modification network and explore
dual intervention strategies, such as the development of GFAT
inhibitors combined with immunomodulators, to synergistically
enhance anti-parasitic immunity and block pathogen
metabolism-dependent virulence mechanisms.
Discussion
O-GlcNAcylation plays diverse roles in different
types of infection by modulating host defense mechanisms and
affecting immune cell function. Specifically, macrophage responses
to infection reveal a key functional divergence of O-GlcNAcylation:
It promotes antiviral defenses but suppresses antibacterial
responses. Given the dual role of O-GlcNAcylation in different
types of infection, the modulation of O-GlcNAcylation activity may
serve as a novel therapeutic strategy. For example, enhancing
O-GlcNAcylation may strengthen the host antiviral response, while
in bacterial infections, moderate inhibition of O-GlcNAcylation may
help to limit inflammatory damage and promote tissue repair.
Therefore, targeting O-GlcNAcylation may represent a potential
approach for the treatment of infectious diseases such as
sepsis.
O-GlcNAc glycosylation is a crucial PTM that
influences protein function by interacting with other PTMs, such as
phosphorylation and ubiquitination (16). For example, O-GlcNAcylation has
been demonstrated to promote protein phosphorylation, including
that of Lsp1 in B cells, where it facilitates interaction with
F-actin (114). Also,
O-GlcNAcylation can modulate the function of ubiquitinated
proteins, including macrophage proteins MAVS and IRF5, which depend
on downstream ubiquitination signaling for proper immune activation
(103–105). Understanding the interplay
between O-GlcNAcylation and other PTMs, such as phosphorylation and
ubiquitination, is crucial for elucidating the role of
O-GlcNAcylation in the regulation of immune cell function in both
homeostatic and infectious states.
Intervention strategies targeting O-GlcNAc
homeostasis have demonstrated multidimensional potential in the
treatment of sepsis. For example, GlcN significantly inhibits
LPS-induced activation of the MAPK and NF-κB inflammatory pathways
by increasing the O-GlcNAc modification of nucleoplasmic proteins,
thereby attenuating lung tissue damage and improving survival in
mouse and zebrafish models (128). In addition, OGT-mediated
modification of O-GlcNAc at RIPK3 Thr467 has been shown to block
the formation of necroptotic complexes and inhibit the inflammatory
storm in sepsis (129). Also, an
acute increase in overall O-GlcNAc levels, such as that induced by
the OGA inhibitor N-butyryl-glucosamine-1,5-lactone
O-(phenylcarbamoyl)oxime, significantly reduces mortality in septic
shock models by inhibiting the cleavage and activation of GSDMD, a
key pyroptosis protein (130,131). At the metabolic level,
O-GlcNAcylation regulates metabolic enzymes such as ATP citrate
lyase in rat models of juvenile sepsis and helps to modulate
systemic inflammatory responses through non-transcriptional
pathways, suggesting its multi-target regulatory advantage
(132,133). However, the clinical application
of OGT/OGA inhibitors faces complex challenges. Specifically, the
inhibition of OGT may have off-target effects that interfere with
protective mechanisms in sepsis. For example, inhibition of OGT may
inadvertently weaken its inhibitory effect on GSDMD-mediated
pyroptosis or impair the glutamine-maintained gut mucus barrier by
interfering with mucin-type glycosylation, potentially through the
cross-inhibition of enzymes such as UDP-GalNAc:polypeptide
N-acetylgalactosaminyltransferase (5,24,134). In addition, the compensatory
stimulation of OGA or fluctuations in the UDP-GlcNAc metabolic pool
triggered by OGT inhibition may disrupt the N-glycosylation of
immune receptors such as TLR4, thereby exacerbating inflammatory
dysregulation (134–136). However, the spatiotemporal
heterogeneity of O-GlcNAcylation, such as the contrasting roles
observed between endothelial cells and macrophages, and the lack of
cell-specific regulatory tools make it challenging to accurately
target the dynamic pathological processes of sepsis (137,138). Future strategies combining
nano-targeted delivery and dynamic biomarker monitoring are
necessary for the development of spatiotemporally accurate
modification approaches that optimize therapeutic benefits while
minimizing risks.
In summary, as a key PTM, O-GlcNAcylation acts as a
critical link between the immune response to infection and the
progression of sepsis. Future studies should aim to fully elucidate
the specific role of O-GlcNAcylation in the pathogenesis of sepsis,
including the mechanisms by which it activates immune cells,
regulates the inflammatory response and controls the autophagic
process. In addition, the identification of specific
O-GlcNAcylation markers as potential biomarkers may improve the
early diagnosis and prognostic assessment of sepsis, providing
deeper mechanistic insights. Intervention strategies targeting
O-GlcNAcylation may enable the regulation of autophagy and immune
responses, thereby providing new therapeutic options for patients
with sepsis. Such approaches may include the development of
small-molecule drugs to modulate the activity of OGT and OGA, or
the design of interventions targeting specific O-GlcNAcylation
sites.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the National
Natural Science Foundation of China (grant no. 82260135).
Availability of data and materials
Not applicable.
Authors' contributions
ZH and XL wrote and revised this manuscript. PL
designed the subject of review. LZ, YL and XM reviewed and revised
the manuscript. Data authentication is not applicable. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nedeva C: Inflammation and cell death of
the innate and adaptive immune system during sepsis. Biomolecules.
11:10112021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lockridge A and Hanover JA: A nexus of
lipid and O-Glcnac metabolism in physiology and disease. Front
Endocrinol (Lausanne). 13:9435762022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai H, Xiong W, Zhu H, Wang Q, Liu S and
Lu Z: Protein O-GlcNAcylation in multiple immune cells and its
therapeutic potential. Front Immunol. 14:12099702023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu D, Su S, Zha X, Wei Y, Yang G, Huang Q,
Yang Y, Xia L, Fan S and Peng X: Glutamine promotes O-GlcNAcylation
of G6PD and inhibits AGR2 S-glutathionylation to maintain the
intestinal mucus barrier in burned septic mice. Redox Biol.
59:1025812023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Srzić I, Nesek Adam V and Tunjić Pejak D:
Sepsis definition: What's new in the treatment guidelines. Acta
Clin Croat. 61 (Suppl 1):S67–S72. 2022.PubMed/NCBI
|
|
7
|
Wiersinga WJ and van der Poll T:
Immunopathophysiology of human sepsis. EBioMedicine. 86:1043632022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Torres LK, Pickkers P and van der Poll T:
Sepsis-induced immunosuppression. Annu Rev Physiol. 84:157–181.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu D, Huang SY, Sun JH, Zhang HC, Cai QL,
Gao C, Li L, Cao J, Xu F, Zhou Y, et al: Sepsis-induced
immunosuppression: Mechanisms, diagnosis and current treatment
options. Mil Med Res. 9:562022.PubMed/NCBI
|
|
10
|
Wen X, Xie B, Yuan S and Zhang J: The
‘Self-sacrifice’ of ImmuneCells in sepsis. Front Immunol.
13:8334792022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang L, Zhou L, Li F, Chen X, Li T, Zou Z,
Zhi Y and He Z: Diagnostic and prognostic value of
Autophagy-related key genes in sepsis and potential correlation
with immune cell signatures. Front Cell Dev Biol. 11:12183792023.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reglero-Real N, Pérez-Gutiérrez L,
Yoshimura A, Rolas L, Garrido-Mesa J, Barkaway A, Pickworth C,
Saleeb RS, Gonzalez-Nuñez M, Austin-Williams SN, et al: Autophagy
modulates endothelial junctions to restrain neutrophil diapedesis
during inflammation. Immunity. 54:1989–2004.e9. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu CL, Wang Y, Liu Q, Li HR, Yu CM, Li P,
Deng XM and Wang JF: Dysregulation of neutrophil death in sepsis.
Front Immunol. 13:9639552022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin Y, Li W, Liu J, Wang F, Zhou W, Xiao
L, Zhou P, Wu F, Chen X, Xu S, et al: Andrographolide ameliorates
sepsis-induced acute lung injury by promoting autophagy in alveolar
macrophages via the RAGE/PI3K/AKT/mTOR pathway. Int
Immunopharmacol. 139:1127192024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Bai G, Chen J, Han W, Guo R and
Cui N: mTOR deletion ameliorates CD4 + T cell apoptosis during
sepsis by improving autophagosome-lysosome fusion. Apoptosis.
27:401–408. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ye L, Ding W, Xiao D, Jia Y, Zhao Z, Ao X
and Wang J: O-GlcNAcylation: Cellular physiology and therapeutic
target for human diseases. MedComm (2020). 4:e4562023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gonzalez-Rellan MJ, Fondevila MF, Dieguez
C and Nogueiras R: O-GlcNAcylation: A sweet hub in the regulation
of glucose metabolism in health and disease. Front Endocrinol
(Lausanne). 13:8735132022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chatham JC, Zhang J and Wende AR: Role of
O-Linked N-acetylglucosamine protein modification in cellular
(patho)physiology. Physiol Rev. 101:427–493. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Q, Zhou S, Lou W, Qian H and Xu Z:
Crosstalk between O-GlcNAcylation and phosphorylation in
metabolism: Regulation and mechanism. Cell Death Differ.
32:1181–1199. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
El Hajjar L, Page A, Bridot C, Cantrelle
FX, Landrieu I and Smet-Nocca C: Regulation of Glycogen Synthase
Kinase-3β by Phosphorylation and O-β-Linked
N-Acetylglucosaminylation: Implications on tau protein
phosphorylation. Biochemistry. 63:1513–1533. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cardozo CF, Vera A, Quintana-Peña V,
Arango-Davila CA and Rengifo J: Regulation of Tau protein
phosphorylation by glucosamine-induced O-GlcNAcylation as a
neuroprotective mechanism in a brain ischemia-reperfusion model.
Int J Neurosci. 133:194–200. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
da Costa Rodrigues B, Dos Santos Lucena
MC, Costa A, de Araújo Oliveira I, Thaumaturgo M, Paes-Colli Y,
Beckman D, Ferreira ST, de Mello FG, de Melo Reis RA, et al:
O-GlcNAcylation regulates tyrosine hydroxylase serine 40
phosphorylation and l-DOPA levels. Am J Physiol Cell Physiol.
328:C825–C835. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YF, Zhu JJ, Li J and Ye XS:
O-GlcNAcylation increases PYGL activity by promoting
phosphorylation. Glycobiology. 32:101–109. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu F, Zhang Z, Leng Y and Chen AF:
O-GlcNAc modification of GSDMD attenuates LPS-induced endothelial
cells pyroptosis. Inflamm Res. 73:5–17. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Correction for Zhang et al. HIPK2
phosphorylates HDAC3 for NF-κB acetylation to ameliorate
colitis-associated colorectal carcinoma and sepsis. Proc Natl Acad
Sci USA. 122:e25060471222025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li C, Yu L, Mai C, Mu T and Zeng Y: KLF4
down-regulation resulting from TLR4 promotion of ERK1/2
phosphorylation underpins inflammatory response in sepsis. J Cell
Mol Med. 25:2013–2024. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dong R, Xue Z, Fan G, Zhang N, Wang C, Li
G and Da Y: Pin1 Promotes NLRP3 inflammasome activation by
phosphorylation of p38 MAPK Pathway in Septic Shock. Front Immunol.
12:6202382021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin L, Yuan F, Dai G, Yao Q, Xiang H, Wang
L, Xue B, Shan Y and Liu X: Blockage of O-linked GlcNAcylation
induces AMPK-dependent autophagy in bladder cancer cells. Cell Mol
Biol Lett. 25:172020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sheng X, Xia Z, Yang H and Hu R: The
ubiquitin codes in cellular stress responses. Protein Cell.
15:157–190. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li MD, Ruan HB, Hughes ME, Lee JS, Singh
JP, Jones SP, Nitabach MN and Yang X: O-GlcNAc signaling entrains
the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell
Metab. 17:303–310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu B, Zhang C, Jiang A, Zhang X, Liang F,
Wang X, Li D, Liu C, Liu X, Xia J, et al: Histone methyltransferase
Dot1L recruits O-GlcNAc transferase to target chromatin sites to
regulate histone O-GlcNAcylation. J Biol Chem. 298:1021152022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Q, Wei D, Li C, Yang X, Su K, Wang T,
Zou R, Wang L, Cun D, Tang B, et al: O-GlcNAcylation with
ubiquitination stabilizes METTL3 to promoting HMGB1 degradation to
inhibit ferroptosis and enhance gemcitabine resistance in
pancreatic cancer. Mol Med. 31:2282025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koo SY, Park EJ, Noh HJ, Jo SM, Ko BK,
Shin HJ and Lee CW: Ubiquitination links DNA damage and repair
signaling to cancer metabolism. Int J Mol Sci. 24:84412023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dikic I and Schulman BA: An expanded
lexicon for the ubiquitin code. Nat Rev Mol Cell Biol. 24:273–287.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Li Y, Zeng S, Duan S, Huang Z and
Liang Y: The interaction of O-GlcNAc-modified NLRX1 and IKK-α
modulates IL-1β expression in M1 macrophages. In Vitro Cell Dev
Biol Anim. 58:408–418. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ning M, Liu Y, Wang D, Wei J, Hu G and
Xing P: Knockdown of TRIM27 alleviated sepsis-induced inflammation,
apoptosis, and oxidative stress via suppressing ubiquitination of
PPARγ and reducing NOX4 expression. Inflamm Res. 71:1315–1325.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, He Y and Zhou D: The role of
ubiquitination in microbial infection induced endothelial
dysfunction: Potential therapeutic targets for sepsis. Expert Opin
Ther Targets. 27:827–839. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu Y, Fu Q and Li J, Zen X and Li J: E3
ubiquitin ligase COP1-mediated CEBPB ubiquitination regulates the
inflammatory response of macrophages in sepsis-induced myocardial
injury. Mamm Genome. 35:56–67. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li ZQ, Chen X and Wang Y: Small molecules
targeting ubiquitination to control inflammatory diseases. Drug
Discov Today. 26:2414–2422. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
O-GlcNAc transferase is a key regulator of
DNA methylation and transposon silencing. Nat Struct Mol Biol.
32:1137–1138. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shin H, Leung A, Costello KR, Senapati P,
Kato H, Moore RE, Lee M, Lin D, Tang X, Pirrotte P, et al:
Inhibition of DNMT1 methyltransferase activity via
glucose-regulated O-GlcNAcylation alters the epigenome. Elife.
12:e855952023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin AP, Qiu Z, Ethiraj P, Sasi B, Jaafar
C, Rakheja D and Aguiar R: MYC, mitochondrial metabolism and
O-GlcNAcylation converge to modulate the activity and subcellular
localization of DNA and RNA demethylases. Leukemia. 36:1150–1159.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lorente-Sorolla C, Garcia-Gomez A,
Català-Moll F, Toledano V, Ciudad L, Avendaño-Ortiz J, Maroun-Eid
C, Martín-Quirós A, Martínez-Gallo M, Ruiz-Sanmartín A, et al:
Inflammatory cytokines and organ dysfunction associate with the
aberrant DNA methylome of monocytes in sepsis. Genome Med.
11:662019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
El Gazzar M, Yoza BK, Chen X, Hu J,
Hawkins GA and McCall CE: G9a and HP1 couple histone and DNA
methylation to TNFalpha transcription silencing during endotoxin
tolerance. J Biol Chem. 283:32198–32208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhu Z, Ren W, Li S, Gao L and Zhi K:
Functional significance of O-linked N-acetylglucosamine protein
modification in regulating autophagy. Pharmacol Res.
202:1071202024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li S, Ren W, Zheng J, Li S, Zhi K and Gao
L: Role of O-linked N-acetylglucosamine protein modification in
oxidative stress-induced autophagy: A novel target for bone
remodeling. Cell Commun Signal. 22:3582024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jeon M, Park J, Yang E, Baek HJ and Kim H:
Regulation of autophagy by protein methylation and acetylation in
cancer. J Cell Physiol. 237:13–28. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Q, Na Q and Song W: Moderate
mammalian target of rapamycin inhibition induces autophagy in
HTR8/SVneo cells via O-linked β-N-acetylglucosamine signaling. J
Obstet Gynaecol Res. 43:1585–1596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qiu Z, Cui J, Huang Q, Qi B and Xia Z:
Roles of O-GlcNAcylation in mitochondrial homeostasis and
cardiovascular diseases. Antioxidants (Basel). 13:5712024.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Marsh SA, Powell PC, Dell'italia LJ and
Chatham JC: Cardiac O-GlcNAcylation blunts autophagic signaling in
the diabetic heart. Life Sci. 92:648–656. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rahman MA, Cho Y, Hwang H and Rhim H:
Pharmacological Inhibition of O-GlcNAc transferase promotes
mTOR-Dependent autophagy in rat cortical neurons. Brain Sci.
10:9582020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pyo KE, Kim CR, Lee M, Kim JS, Kim KI and
Baek SH: ULK1 O-GlcNAcylation is crucial for activating VPS34 via
ATG14L during Autophagy initiation. Cell Rep. 25:2878–2890.e4.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shi Y, Yan S, Shao GC, Wang J, Jian YP,
Liu B, Yuan Y, Qin K, Nai S, Huang X, et al: O-GlcNAcylation
stabilizes the autophagy-initiating kinase ULK1 by inhibiting
chaperone-mediated autophagy upon HPV infection. J Biol Chem.
298:1023412022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang J, Li C, Shuai W, Chen T, Gong Y, Hu
H, Wei Y, Kong B and Huang H: Maresin2 fine-tunes ULK1
O-GlcNAcylation to improve post myocardial infarction remodeling.
Eur J Pharmacol. 962:1762232024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Alghusen IM, Carman MS, Wilkins HM, Strope
TA, Gimore C, Fedosyuk H, Shawa J, Ephrame SJ, Denson AR, Wang X,
et al: O-GlcNAc impacts mitophagy via the PINK1-dependent pathway.
Front Aging Neurosci. 16:13879312024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P,
Ma Y, Zhao B, Kovács AL, Zhang Z, et al: O-GlcNAc-modification of
SNAP-29 regulates autophagosome maturation. Nat Cell Biol.
16:1215–1226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dodson M, Liu P, Jiang T, Ambrose AJ, Luo
G, Rojo de la Vega M, Cholanians AB, Wong PK, Chapman E and Zhang
DD: Increased O-GlcNAcylation of SNAP29 drives Arsenic-induced
autophagic dysfunction. Mol Cell Biol. 38:e00595–e00517. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pellegrini FR, De Martino S, Fianco G,
Ventura I, Valente D, Fiore M, Trisciuoglio D and Degrassi F:
Blockage of autophagosome-lysosome fusion through SNAP29
O-GlcNAcylation promotes apoptosis via ROS production. Autophagy.
19:2078–2093. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Huang L, Yuan P, Yu P, Kong Q, Xu Z, Yan
X, Shen Y, Yang J, Wan R, Hong K, et al: O-GlcNAc-modified SNAP29
inhibits autophagy-mediated degradation via the disturbed
SNAP29-STX17-VAMP8 complex and exacerbates myocardial injury in
type I diabetic rats. Int J Mol Med. 42:3278–3290. 2018.PubMed/NCBI
|
|
60
|
Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An
R, Lv X, Li J and Chen B: Down-regulation of OGT promotes cisplatin
resistance by inducing autophagy in ovarian cancer. Theranostics.
8:5200–5212. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Patel A and Faesen AC: Metamorphosis by
ATG13 and ATG101 in human autophagy initiation. Autophagy.
20:968–969. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang X and Jia J: Magnolol improves
Alzheimer's disease-like pathologies and cognitive decline by
promoting autophagy through activation of the AMPK/mTOR/ULK1
pathway. Biomed Pharmacother. 161:1144732023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ge Y, Zhou M, Chen C, Wu X and Wang X:
Role of AMPK mediated pathways in autophagy and aging. Biochimie.
195:100–113. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pizzimenti C, Fiorentino V, Ruggeri C,
Franchina M, Ercoli A, Tuccari G and Ieni A: Autophagy involvement
in Non-neoplastic and neoplastic endometrial pathology: The state
of the art with a focus on carcinoma. Int J Mol Sci. 25:121182024.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang TF, Feng ZQ, Sun YW, Zhao SJ, Zou HY,
Hao HS, Du WH, Zhao XM, Zhu HB and Pang YW: Disruption of
O-GlcNAcylation homeostasis induced ovarian granulosa cell injury
in bovine. Int J Mol Sci. 23:78152022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xu J, Gu J, Pei W, Zhang Y, Wang L and Gao
J: The role of lysosomal membrane proteins in autophagy and related
diseases. FEBS J. 291:3762–3785. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang P and Hanover JA: Nutrient-driven
O-GlcNAc cycling influences autophagic flux and neurodegenerative
proteotoxicity. Autophagy. 9:604–606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Park S, Lee Y, Pak JW, Kim H, Choi H, Kim
JW, Roth J and Cho JW: O-GlcNAc modification is essential for the
regulation of autophagy in Drosophila melanogaster. Cell Mol Life
Sci. 72:3173–3183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zheng D, Tong M, Zhang S, Pan Y, Zhao Y,
Zhong Q and Liu X: Human YKT6 forms priming complex with STX17 and
SNAP29 to facilitate autophagosome-lysosome fusion. Cell Rep.
43:1137602024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ben Ahmed A, Lemaire Q, Scache J, Mariller
C, Lefebvre T and Vercoutter-Edouart AS: O-GlcNAc dynamics: The
sweet side of protein trafficking regulation in mammalian cells.
Cells. 12:13962023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Du P, Zhang X, Lian X, Hölscher C and Xue
G: O-GlcNAcylation and its roles in neurodegenerative diseases. J
Alzheimers Dis. 97:1051–1068. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
He XF, Hu X, Wen GJ, Wang Z and Lin WJ:
O-GlcNAcylation in cancer development and immunotherapy. Cancer
Lett. 566:2162582023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu C, Dong W, Li J, Kong Y and Ren X:
O-GlcNAc modification and its role in diabetic retinopathy.
Metabolites. 12:7252022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bruserud Ø, Mosevoll KA, Bruserud Ø,
Reikvam H and Wendelbo Ø: The regulation of neutrophil migration in
patients with sepsis: The complexity of the molecular mechanisms
and their modulation in sepsis and the heterogeneity of sepsis
patients. Cells. 12:10032023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhou YY and Sun BW: Recent advances in
neutrophil chemotaxis abnormalities during sepsis. Chin J
Traumatol. 25:317–324. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kneass ZT and Marchase RB: Neutrophils
exhibit rapid Agonist-induced increases in protein-associated
O-GlcNAc. J Biol Chem. 279:45759–45765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cong R, Sun L, Yang J, Cui H, Ji X, Zhu J,
Gu JH and He B: Protein O-GlcNAcylation alleviates small intestinal
injury induced by ischemia-reperfusion and oxygen-glucose
deprivation. Biomed Pharmacother. 138:1114772021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hossain M, Qadri SM, Xu N, Su Y, Cayabyab
FS, Heit B and Liu L: Endothelial LSP1 modulates extravascular
neutrophil chemotaxis by regulating nonhematopoietic vascular
PECAM-1 Expression. J Immunol. 195:2408–2416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Machin PA, Johnsson AE, Massey EJ,
Pantarelli C, Chetwynd SA, Chu JY, Okkenhaug H, Segonds-Pichon A,
Walker S, Malliri A, et al: Dock2 generates characteristic
spatiotemporal patterns of Rac activity to regulate neutrophil
polarisation, migration and phagocytosis. Front Immunol.
14:11808862023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kneass ZT and Marchase RB: Protein
O-GlcNAc modulates motility-associated signaling intermediates in
neutrophils. J Biol Chem. 280:14579–14585. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu KK, Xu X, Wu M, Li X, Hoque M, Li G,
Lian Q, Long K, Zhou T, Piao H, et al: MDM2 induces
pro-inflammatory and glycolytic responses in M1 macrophages by
integrating iNOS-nitric oxide and HIF-1α pathways in mice. Nat
Commun. 15:86242024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hwang SY, Hwang JS, Kim SY and Han IO:
O-GlcNAc transferase inhibits LPS-mediated expression of inducible
nitric oxide synthase through an increased interaction with mSin3A
in RAW264.7 cells. Am J Physiol Cell Physiol. 305:C601–C608. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hwang SY, Hwang JS, Kim SY and Han IO:
O-GlcNAcylation and p50/p105 binding of c-Rel are dynamically
regulated by LPS and glucosamine in BV2 microglia cells. Br J
Pharmacol. 169:1551–1560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
He Y, Ma X, Li D and Hao J: Thiamet G
mediates neuroprotection in experimental stroke by modulating
microglia/macrophage polarization and inhibiting NF-κB p65
signaling. J Cereb Blood Flow Metab. 37:2938–2951. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Seo J, Kim Y, Ji S, Kim HB, Jung H, Yi EC,
Lee YH, Shin I, Yang WH and Cho JW: O-GlcNAcylation of RIPK1
rescues red blood cells from necroptosis. Front Immunol.
14:11604902023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Janas ML, Groves P, Kienzle N and Kelso A:
IL-2 regulates perforin and granzyme gene expression in CD8+ T
cells independently of its effects on survival and proliferation. J
Immunol. 175:8003–8010. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Reina-Campos M, Scharping NE and Goldrath
AW: CD8+ T cell metabolism in infection and cancer. Nat Rev
Immunol. 21:718–738. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lopez Aguilar A, Gao Y, Hou X, Lauvau G,
Yates JR and Wu P: Profiling of protein O-GlcNAcylation in murine
CD8+ effector- and Memory-like T cells. ACS Chem Biol.
12:3031–3038. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen H, Shi Y, Ying J, Dong Z, Wang Y,
Zheng Y and Ruan P: O-linked N-acetylglucosamine modification
induced by lipopolysaccharide is involved in inflammatory signaling
pathway in endothelial cells. Zhonghua Wei Zhong Bing Ji Jiu Yi
Xue. 35:164–169. 2023.(In Chinese). PubMed/NCBI
|
|
90
|
Zeng Z, Liao X and Zhao X: O-GlcNAc
transferase mediates O-GlcNAcylation of NLRP3 regulates pyroptosis
in spinal cord injury. Brain Res Bull. 222:1112332025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
He C, Wu Q, Zeng Z, Yang Y, He H, Hu M and
Liu S: OGT-induced O-GlcNAcylation of NEK7 protein aggravates
osteoarthritis progression by enhancing NEK7/NLRP3 axis.
Autoimmunity. 57:23192022024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yaqin Z, Kehan W, Yi Z, Naijian W, Wei Q
and Fei M: Resveratrol alleviates inflammatory bowel disease by
inhibiting JAK2/STAT3 pathway activity via the reduction of
O-GlcNAcylation of STAT3 in intestinal epithelial cells. Toxicol
Appl Pharmacol. 484:1168822024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang CC, Li Y, Jiang CY, Le QM, Liu X, Ma
L and Wang FF: O-GlcNAcylation mediates H2O2-induced apoptosis
through regulation of STAT3 and FOXO1. Acta Pharmacol Sin.
45:714–727. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li Y, Peng J, Xia Y, Pan C, Li Y, Gu W,
Wang J, Wang C, Wang Y, Song J, et al: Sufu limits sepsis-induced
lung inflammation via regulating phase separation of TRAF6.
Theranostics. 13:3761–3780. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Matsui S, Ri C, Bolanos LC, Choi K,
Shibamiya A, Ishii A, Takaishi K, Oshima-Hasegawa N, Tsukamoto S,
Takeda Y, et al: Metabolic reprogramming regulated by TRAF6
contributes to the leukemia progression. Leukemia. 38:1032–1045.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Qiang A, Slawson C and Fields PE: The Role
of O-GlcNAcylation in immune cell activation. Front Endocrinol
(Lausanne). 12:5966172021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao
J, Jiang W, Zhang F, Li D, Han B, et al: MicroRNA-15b Suppresses
Th17 differentiation and is associated with pathogenesis of
multiple sclerosis by targeting O-GlcNAc transferase. J Immunol.
198:2626–2639. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Machacek M, Saunders H, Zhang Z, Tan EP,
Li J, Li T, Villar MT, Artigues A, Lydic T, Cork G, et al: Elevated
O-GlcNAcylation enhances pro-inflammatory Th17 function by altering
the intracellular lipid microenvironment. J Biol Chem.
294:8973–8990. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Thomaidis T, Maderer A, Formentini A,
Bauer S, Trautmann M, Schwarz M, Neumann W, Kittner JM, Schad A,
Link KH, et al: Proteins of the VEGFR and EGFR pathway as
predictive markers for adjuvant treatment in patients with stage
II/III colorectal cancer: Results of the FOGT-4 trial. J Exp Clin
Cancer Res. 33:832014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Gao YZ, Wang YT, He S, Li H, Wang Y and Wu
Z: FpOGT is required for fungal growth, stress response, and
virulence of Fusarium proliferatum by affecting the expression of
glucokinase and other glucose metabolism-related genes. Phytopathol
Res. 6:22024. View Article : Google Scholar
|
|
101
|
Naseem S and Konopka JB:
N-acetylglucosamine regulates virulence properties in microbial
pathogens. PLoS Pathog. 11:e10049472015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ansari S, Kumar V, Bhatt DN, Irfan M and
Datta A: N-acetylglucosamine sensing and metabolic engineering for
attenuating human and plant pathogens. Bioengineering (Basel).
9:642022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Seo J, Park YS, Kweon TH, Kang J, Son S,
Kim HB, Seo YR, Kang MJ, Yi EC, Lee YH, et al: O-Linked
N-acetylglucosamine modification of mitochondrial antiviral
signaling protein regulates antiviral signaling by modulating its
activity. Front Immunol. 11:5892592020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Song N, Qi Q, Cao R, Qin B, Wang B, Wang
Y, Zhao L, Li W, Du X, Liu F, et al: MAVS O-GlcNAcylation is
essential for host antiviral immunity against lethal RNA viruses.
Cell Rep. 28:2386–2396.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang Q, Fang P, He R, Li M, Yu H, Zhou L,
Yi Y, Wang F, Rong Y, Zhang Y, et al: O-GlcNAc transferase promotes
influenza A virus-induced cytokine storm by targeting interferon
regulatory factor-5. Sci Adv. 6:eaaz70862020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yao AY, Tang HY, Wang Y, Feng MF and Zhou
RL: Inhibition of the activating signals in NK92 cells by
recombinant GST-sHLA-G1a chain. Cell Res. 14:155–160. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Mariuzza RA, Singh P, Karade SS, Shahid S
and Sharma VK: Recognition of self and viral ligands by NK cell
receptors. Immunol Rev. 329:e134352025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yu M, Su Z, Huang X, Zhou Y, Zhang X, Wang
B, Wang Z, Liu Y, Xing N, Xia M, et al: Histone methyltransferase
Ezh2 negatively regulates NK cell terminal maturation and function.
J Leukoc Biol. 110:1033–1045. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lo PW, Shie JJ, Chen CH, Wu CY, Hsu TL and
Wong CH: O-GlcNAcylation regulates the stability and enzymatic
activity of the histone methyltransferase EZH2. Proc Natl Acad Sci
USA. 115:7302–7307. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Feinberg D, Ramakrishnan P, Wong DP,
Asthana A and Parameswaran R: Inhibition of O-GlcNAcylation
decreases the cytotoxic function of natural killer cells. Front
Immunol. 13:8412992022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Loftus RM, Assmann N, Kedia-Mehta N,
O'Brien KL, Garcia A, Gillespie C, Hukelmann JL, Oefner PJ, Lamond
AI, Gardiner CM, et al: Amino acid-dependent cMyc expression is
essential for NK cell metabolic and functional responses in mice.
Nat Commun. 9:23412018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ramakrishnan P: O-GlcNAcylation and immune
cell signaling: A review of known and a preview of unknown. J Biol
Chem. 300:1073492024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chang YH, Weng CL and Lin KI:
O-GlcNAcylation and its role in the immune system. J Biomed Sci.
27:572020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wu JL, Wu HY, Tsai DY, Chiang MF, Chen YJ,
Gao S, Lin CC, Lin CH, Khoo KH, Chen YJ and Lin KI: Temporal
regulation of Lsp1 O-GlcNAcylation and phosphorylation during
apoptosis of activated B cells. Nat Commun. 7:125262016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Guo M, Price MJ, Patterson DG, Barwick BG,
Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM and Scharer
CD: EZH2 represses the B cell transcriptional program and regulates
Antibody-secreting cell metabolism and antibody production. J
Immunol. 200:1039–1052. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ramakrishnan P, Clark PM, Mason DE, Peters
EC, Hsieh-Wilson LC and Baltimore D: Activation of the
transcriptional function of the NF-κB protein c-Rel by O-GlcNAc
glycosylation. Sci Signal. 6:ra752013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Swamy M, Pathak S, Grzes KM, Damerow S,
Sinclair LV, van Aalten DM and Cantrell DA: Glucose and glutamine
fuel protein O-GlcNAcylation to control T cell self-renewal and
malignancy. Nat Immunol. 17:712–720. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu AR and Ramakrishnan P: Regulation of
nuclear Factor-kappaB Function by O-GlcNAcylation in inflammation
and cancer. Front Cell Dev Biol. 9:7517612021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Saha A and Fernández-Tejada A: Chemical
biology tools to interrogate the roles of O-GlcNAc in immunity.
Front Immunol. 13:10898242022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhao M, Ren K, Xiong X, Xin Y, Zou Y,
Maynard JC, Kim A, Battist AP, Koneripalli N, Wang Y, et al:
Epithelial STAT6 O-GlcNAcylation drives a concerted anti-helminth
alarmin response dependent on tuft cell hyperplasia and Gasdermin
C. Immunity. 55:13272022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Xiong X, Huang R, Li Z, Yang C, Wang Q,
Ruan HB and Xu L: Intestinal epithelial STAT6 activation rescues
the defective Anti-helminth responses caused by ogt deletion. Int J
Mol Sci. 23:111372022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Min A, Lee YA, Kim KA and Shin MH:
BLT1-mediated O-GlcNAcylation is required for NOX2-dependent
migration, exocytotic degranulation and IL-8 release of human mast
cell induced by Trichomonas vaginalis-secreted LTB4.
Microbes Infect. 20:376–384. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Montagner S, Leoni C, Emming S, Della
Chiara G, Balestrieri C, Barozzi I, Piccolo V, Togher S, Ko M, Rao
A, et al: TET2 regulates mast cell differentiation and
proliferation through catalytic and Non-catalytic activities. Cell
Rep. 15:1566–1579. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Alberione MP, Avalos-Padilla Y, Rangel GW,
Ramírez M, Romero-Uruñuela T, Fenollar A, Crispim M, Smith TK,
Llinás M and Izquierdo L: Hexosamine biosynthesis disruption
impairs GPI production and arrests Plasmodium falciparum
growth at schizont stages. PLoS Pathog. 21:e10128322025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang D, Wang C and Zhu G: Genomic
reconstruction and features of glycosylation pathways in the
apicomplexan Cryptosporidium parasites. Front Mol Biosci.
9:10510722022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Gowda DC and Miller LH: Glycosylation in
malaria parasites: What do we know. Trends Parasitol. 40:131–146.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang N, Jiang N and Chen Q: Key
regulators of parasite biology viewed through a Post-translational
modification repertoire. Proteomics. December 17–2024.(Epub ahead
of print). View Article : Google Scholar
|
|
128
|
Hwang JS, Kim KH, Park J, Kim SM, Cho H,
Lee Y and Han IO: Glucosamine improves survival in a mouse model of
sepsis and attenuates sepsis-induced lung injury and inflammation.
J Biol Chem. 294:608–622. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li X, Gong W, Wang H, Li T, Attri KS,
Lewis RE, Kalil AC, Bhinderwala F, Powers R, Yin G, et al: O-GlcNAc
transferase suppresses inflammation and necroptosis by targeting
Receptor-Interacting Serine/Threonine-protein kinase 3. Immunity.
50:576–590.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Lang Y, Li J and Zhang L: O-GlcNAcylation
dictates pyroptosis. Front Immunol. 15:15135422024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ferron M, Cadiet J, Persello A, Prat V,
Denis M, Erraud A, Aillerie V, Mevel M, Bigot E, Chatham JC, et al:
O-GlcNAc stimulation: A new metabolic approach to treat septic
shock. Sci Rep. 9:187512019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Denis M, Dupas T, Persello A, Dontaine J,
Bultot L, Betus C, Pelé T, Dhot J, Erraud A, Maillard A, et al: An
O-GlcNAcylomic approach reveals ACLY as a potential target in
sepsis in the young rat. Int J Mol Sci. 22:92362021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Dupas T, Persello A, Blangy-Letheule A,
Denis M, Erraud A, Aillerie V, Leroux AA, Rivière M, Lebreton J,
Tessier A, et al: Beneficial effects of O-GlcNAc stimulation in a
young rat model of sepsis: Beyond modulation of gene expression.
Int J Mol Sci. 23:64302022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Trapannone R, Rafie K and van Aalten DM:
O-GlcNAc transferase inhibitors: Current tools and future
challenges. Biochem Soc Trans. 44:88–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Elbatrawy AA, Kim EJ and Nam G:
O-GlcNAcase: Emerging mechanism, substrate recognition and
Small-Molecule inhibitors. ChemMedChem. 15:1244–1257. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman
M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ and Walker S: A
small molecule that inhibits OGT activity in cells. ACS Chem Biol.
10:1392–1397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Fehl C and Hanover JA: Tools, tactics and
objectives to interrogate cellular roles of O-GlcNAc in disease.
Nat Chem Biol. 18:8–17. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Cheng SS, Mody AC and Woo CM:
Opportunities for therapeutic modulation of O-GlcNAc. Chem Rev.
124:12918–13019. 2024. View Article : Google Scholar : PubMed/NCBI
|















