Contents
Introduction
Benign soft tissue tumors
Intermediate soft tissue tumors
Conclusions
Introduction
Soft tissue tumors are a highly heterogeneous group
of mesenchymal neoplasms that are classified according to the cell
type they resemble. Within the various histogenetic categories,
soft tissue tumors are usually divided into benign, intermediate
(locally aggressive), intermediate (rarely metastasizing) and
malignant forms (1).
In current practice, cytogenetic and molecular
cytogenetic [fluorescence in situ hybridization (FISH)]
assays can serve as a useful diagnostic adjunct for soft tissue
tumors. Many benign and intermediate soft tissue tumors are
characterized by specific cytogenetic translocations or other
rearrangements (2). This review
provides updated information on the cytogenetic and molecular
cytogenetic characteristics of benign and intermediate soft tissue
tumors as well as their clinicopathological features, including
nodular fasciitis, chondroid lipoma, collagenous fibroma
(desmoplastic fibroblastoma), giant cell tumor of tendon sheath
(GCTTS)/pigmented villonodular synovitis (PVNS), angiofibroma of
soft tissue, myxoinflammatory fibroblastic sarcoma (MIFS) and
ossifying fibromyxoid tumor (OFMT). The consistent genetic
alterations are summarized in Table
I.
 | Table IChromosomal aberrations and
associated molecular events in benign and intermediate soft tissue
tumors. |
Table I
Chromosomal aberrations and
associated molecular events in benign and intermediate soft tissue
tumors.
| Tumor type | Chromosomal
aberration | Molecular
event |
|---|
| Benign | | |
| Nodular
fasciitis | Not assigned | USP6
rearrangementa |
| 3q21
rearrangement | Not known |
| 15q22–26
rearrangement | Not known |
| Chondroid
lipoma |
t(11;16)(q13;p13) |
C11of95-MKL2 |
| Collagenous
fibroma | 11q12
rearrangementb | FOSL1
rearrangement |
| GCTTS/PVNS | 1p11–13
rearrangementc | CSF1
rearrangement |
| 16q24
rearrangement | Not known |
| Trisomy 5 and/or
trisomy 7 | Not known |
| Angiofibroma of
soft tissue |
t(5;8)(p15;q13) |
AHRR-NCOA2 |
| Intermediate
(locally metastasizing) | | |
| MIFSd |
t(1;10)(p22;q24) | TGFBR3,
MGEA5 rearrangement |
| Ossifying
fibromyxoid tumor | 6p21
rearrangemente | PHF1
rearrangement |
Benign soft tissue tumors
Nodular fasciitis is a mass-forming, self-limited
reactive process of unknown pathogenesis. It occurs in all age
groups but more often in young adults. Males and females are about
equally affected. Most nodular fasciitis arise in the subcutaneous
tissues of the upper extremities (especially the volar aspect of
the forearm), trunk and head and neck. Nodular fasciitis typically
grows rapidly and reaches its final size within a few weeks. It
usually measures ≤2 cm in its greatest dimension (1). Soreness, tenderness or slight pain may
be present. Histologically, nodular fasciitis is composed of plump,
immature-appearing fibroblasts and myofibroblasts lacking nuclear
hyperchromasia and pleomorphism. Mitotic figures are fairly common,
but atypical mitoses are virtually never evident (3). Due to its rapid growth, high
cellularity and high mitotic activity, nodular fasciitis can be
misdiagnosed as a malignant soft tissue tumor, especially
fibrosarcoma or low-grade myxofibrosarcoma, often leading to
unnecessarily aggressive therapy.
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in five cases of nodular fasciitis (4–8).
Rearrangements involving 3q21 and 15q22–26 have been identified in
a subset of nodular fasciitis. Velagaleti et al(8) suggested that FGF7 and
NTRK3 are potential candidate target genes for the 15q
rearrangement. A conventional comparative genomic hybridization
(CGH) study has revealed gains of 10p14–15 and 20q12–13.3 in only
one of five nodular fasciitis cases (9). A gene expression analysis has shown
higher expression of the SYK, LYN, EPHA4,
OAS1, TCF20, MITF, CXCL9,
CXCL10, MMP1, MMP9, MMP13, CTSC,
CTSL and PLAU genes (10). Bacac et al(10) suggested that nodular fasciitis and
desmoid-type fibromatosis may be distinguished on the basis of the
expression profile of several gene clusters.
More recently, Erickson-Johnson et
al(11) reported that
USP6 rearrangements with the formation of the fusion gene
MYH9-USP6 occur in most examples of nodular
fasciitis. USP6 is located on chromosome 17p13 and has a
limited expression in normal cells. USP6 rearrangements were
first identified in aneurysmal bone cyst (ABC) (12). The presence of USP6
rearrangements has also been reported in two cases of ABC-like
myositis ossificans (13). However,
USP6 rearrangements are absent in its histological mimics in
soft tissue, including desmoid-type fibromatosis, fibrosarcoma and
myxofibrosarcoma. Therefore, USP6 FISH is an extremely
useful adjunct in the diagnosis of nodular fasciitis.
Chondroid lipoma, despite its worrisome histological
appearance, is a benign soft tissue tumor with features of both
embryonal fat and embryonal cartilage (14). It usually occurs in the third or
fourth decade of life with a female predominance. The tumor
typically presents as a slow-growing, painless mass in the proximal
extremities and limb girdles. Most chondroid lipomas measure 2–7 cm
in their greatest dimension (1).
Chondroid lipoma is often deep seated, involving skeletal muscle or
deep fibrous connective tissues. Histologically, chondroid lipoma
has a lobular pattern and consists of sheets, nests and cords of
round cells embedded in a myxoid to hyalinized chondroid matrix,
with a variable amount of mature adipose tissue. The cells show
cytoplasmic vacuolation or eosinophilic granular cytoplasm.
Significant nuclear pleomorphism or mitotic activity is not
evident. Chondroid lipoma may be mistaken for a malignant soft
tissue tumor, especially myxoid liposarcoma or extraskeletal myxoid
chondrosarcoma (15,16).
An identical reciprocal translocation,
t(11;16)(q13;p13), has been detected by cytogenetic analysis in six
cases of chondroid lipoma (17–20).
Recurrent involvement of 11q13 has also been described in
hibernoma, but not in association with 16p13 (15).
In their study, Huang et al(20) reported that the t(11;16) (q13;p13)
translocation results in a fusion of C11orf95 and
MKL2. These gene rearrangements have not been identified in
other soft tissue tumors thus far, and appear to be characteristic
for chondroid lipoma. FISH assay for C11orf95 or MKL2
rearrangements is therefore useful for the differential diagnosis
of chondroid lipoma and its histological mimics, particularly when
evaluating small samples (20).
Collagenous fibroma, also known as desmoplastic
fibroblastoma, is a benign fibrous soft tissue tumor first
described by Evans (21) in 1995.
This tumor primarily occurs in the subcutaneous tissues or skeletal
muscle of upper extremities and has a peak incidence in the fifth
to seventh decades of life with a male predominance. Few cases have
been encountered in children (22,23).
Collagenous fibroma typically presents as a firm, slow-growing,
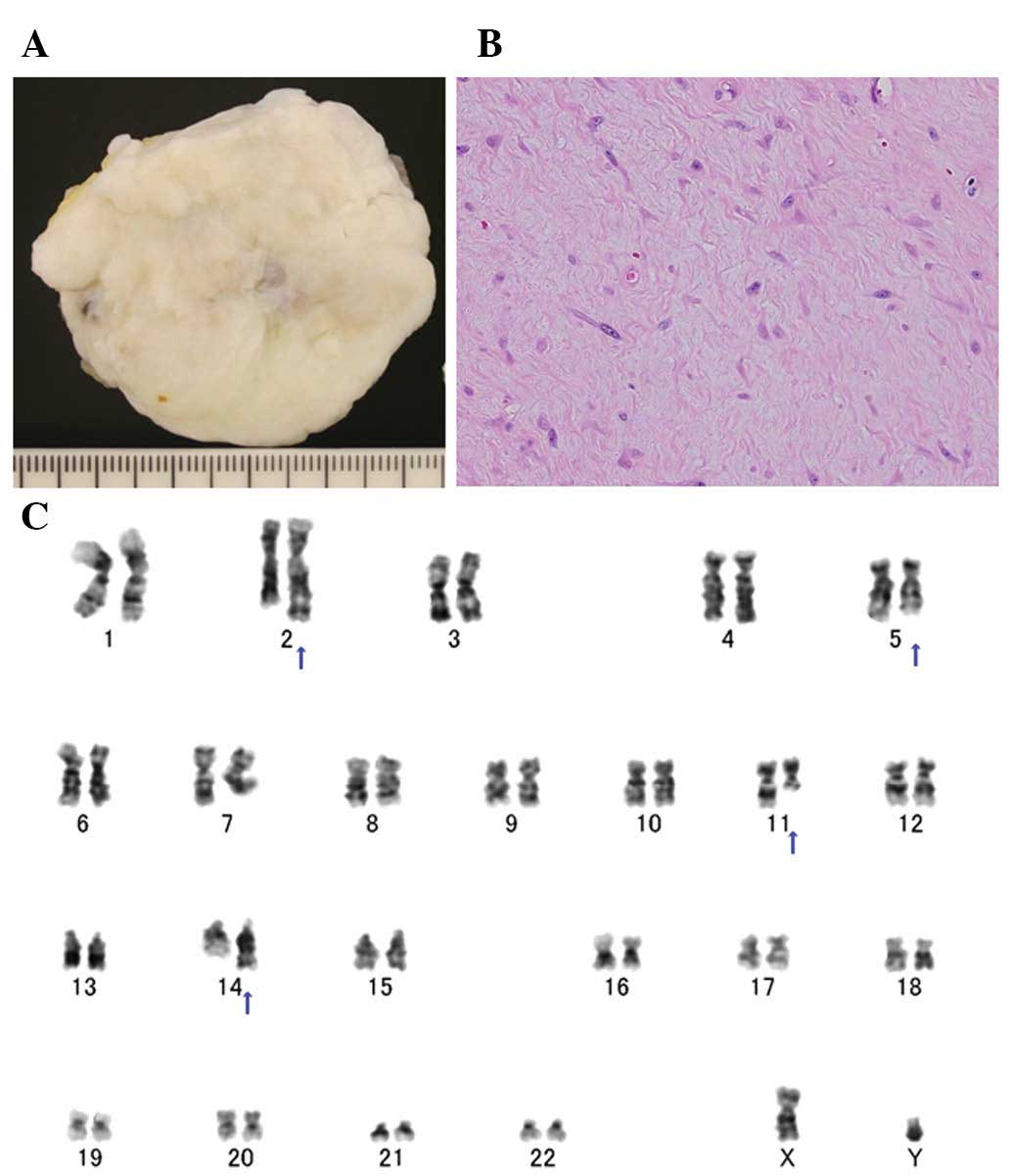
painless mass, measuring ≤4 cm in its greatest dimension (3). Grossly, collagenous fibroma appears as
a well-circumscribed mass with a white to gray cut surface
(Fig. 1A). Histologically, the
lesion is hypocellular and consists of spindle- to stellate-shaped
cells embedded in a collagenous or myxocollagenous stroma (Fig. 1B). Mitotic figures are rare or
absent and necrosis is not present. Collagenous fibroma may be
mistaken for several other benign and low-grade malignant soft
tissue tumors, including desmoid-type fibromatosis and low-grade
fibromyxoid sarcoma.
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in 10 cases of collagenous fibroma (24–29).
The long arm of chromosome 11, in particular 11q12, is involved in
all cases except one (Fig. 1C).
Several chromosomal segments have been recognized as translocation
partners to 11q12 and the most preferred rearrangement is
t(2;11)(q31;q12). Notably, the same translocation has also been
observed in a case of fibroma of tendon sheath (30), suggesting a pathogenetic link
between these two entities.
Macchia et al(29) recently reported that FOSL1
(formerly known as Fra-1) is a candidate target gene
for 11q12 rearrangements in collagenous fibroma. This gene is a
member of the Fos family and encodes leucine zipper proteins.
FOSL1 has been reported to be activated in multiple human
carcinomas, including lung, colon, breast, prostate, brain and head
and neck cancers (31). However,
FOSL1 rearrangements have not been detected in other soft
tissue tumors thus far. FOSL1 FISH may therefore be
important in the diagnosis of collagenous fibroma.
Tenosynovial giant cell tumor is classified into two
types, with different clinical features and biological behavior,
although their histological finding is similar (1). The localized type, also known as
GCTTS, may occur at any age but has a peak incidence in the third
to fourth decades of life with a female predominance. GCTTS usually
presents as a painless, slow-growing mass in the extremities,
especially the fingers. Grossly, GCTTS is well circumscribed and
encapsulated. Recurrences are non-destructive and are easily
controlled by re-excision. The diffuse type, also known as PVNS,
tends to occur in younger patients and is slightly more common in
females. PVNS usually presents as a painful, longstanding mass with
hemarthrosis in large joints, especially the knee. Grossly, PVNS is
poorly circumscribed and has a prominent villonodular growth
pattern. This potentially aggressive lesion frequently recurs,
occasionally necessitating radical surgery (1). Histologically, the lesion is composed
of sheets of round or polygonal cells admixed with multinucleated
giant cells and xanthoma cells with hemosiderin granules. Compared
with GCTTS, cleft-like or pseudoglandular spaces are more prominent
in PVNS. Mitotic activity is usually present, but atypical mitoses
and nuclear atypia are not evident.
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in 45 cases of GCTTS/PVNS (32–45).
GCTTS/PVNS exhibits mostly simple karyotypes characterized by one
or a few chromosomal rearrangements or numerical aberrations. The
short arm of chromosome 1, in particular 1p11–13, is frequently
involved in GCTTS/PVNS. Several chromosomal segments have been
recognized as translocation partners to 1p11–13 and the most
preferred rearrangement is t(1;2)(p11–13;q35–37) resulting in a
COL6A3-CSF1 fusion gene (46,47).
In addition, another possible cytogenetic subgroup might be
characterized by translocations involving 16q24. Moreover, trisomy
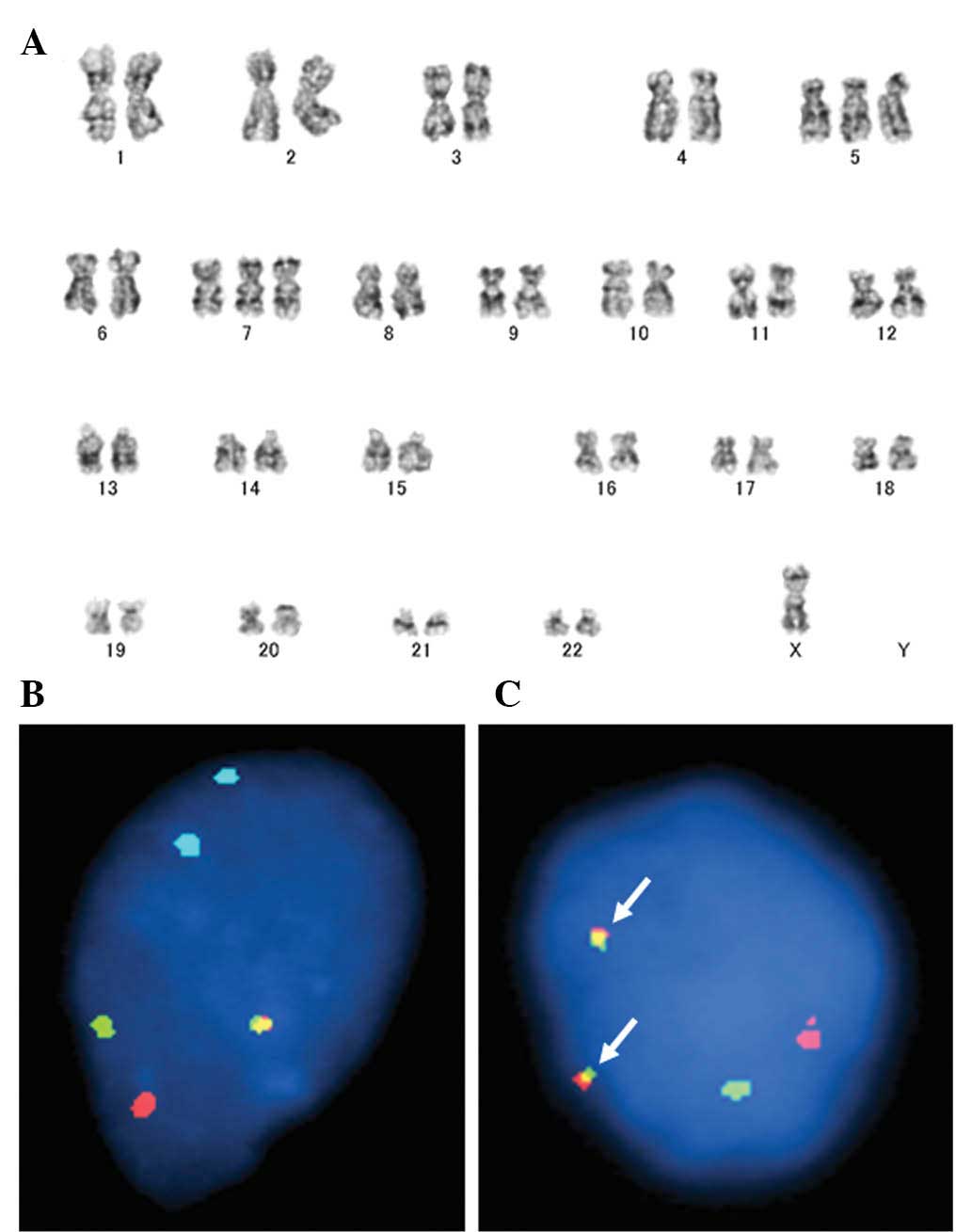
for chromosomes 5 and/or 7, usually as the sole anomaly, has been
observed only in PVNS (Fig.
2A).
In a previous study, West et al(46) demonstrated that CSF1 at 1p13
is often rearranged in GCTTS/PVNS. The authors also detected high
levels of its receptor (CSF1R) expression in most of the cells.
However, only a minority of tumor cells (2–16%) carry the
translocation and express CSF1. These findings suggest that
aberrant CSF1 signaling is crucial in the pathogenesis of
GCTTS/PVNS. CSF1 rearrangements can be detected by FISH
(Fig. 2B and C), although probes
are not yet commercially available.
Blay et al(48) initially reported a case of recurrent
PVNS showing a complete response after imatinib therapy. More
recently, Cassier et al(49)
reported that the overall response rate of a daily dose of imatinib
(400 mg) in patients with locally advanced/metastatic PVNS was 19%.
Moreover, the overall symptomatic response rate was 73%. These
results suggest that the use of targeted inhibitors of CSF1R, such
as imatinib, are a good therapeutic option in the treatment
strategy of locally advanced/metastatic or recurrent PVNS.
Angiofibroma of soft tissue is a distinctive benign
fibrovascular soft tissue tumor first described by Mariño-Enríquez
and Fletcher (50) in 2012. It
usually occurs in middle-aged adults, with a female predominance.
Angiofibroma of soft tissue typically presents as a slow-growing,
painless, well-defined mass in the extremities, often in
relationship to joints or fibrotendinous structures. The lesions
range from 1.2 to 12 cm in their greatest dimension, with a median
size of 3.5 cm (50).
Histologically, the tumor consists of a proliferation of relatively
uniform, bland spindle cells set in a variably myxoid-to
collagenous stroma with a prominent and complex vascular pattern.
Mitotic activity is occasionally observed, but cytologic atypia and
nuclear hyperchromasia are absent. Immunohistochemically, the
neoplastic cells are focally positive for epithelial membrane
antigen. Occasional cases may show scattered cells that stain CD34,
smooth muscle actin and desmin. Angiofibroma of soft tissue may be
mistaken for a number of benign and low-grade malignant soft tissue
tumors, including cellular angiofibroma, solitary fibrous tumor,
low-grade fibromyxoid sarcoma, low-grade myxofibrosarcoma and
myxoid liposarcoma (50).
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in seven cases of soft tissue angiofibroma
(50–52). A reciprocal translocation,
t(5;8)(p15;q13), has been identified in five cases. A three-way
t(5;8;8)(p15;q13;p11) translocation has also been described in one
case (50). A gene expression
analysis has shown higher expression of CYP1A1 in
angiofibroma of soft tissue compared with myxofibrosarcoma
(51).
In their study, Jin et al(51) reported that the t(5;8)(p15;q13)
translocation results in a fusion of AHRR and NCOA2.
AHRR is located on chromosome 5p15 and is not involved in gene
fusions. NCOA2 is located on chromosome 8q13 and has been
identified as the 3′-partner in fusions with PAX3 and
HEY1 in soft tissue sarcomas (53,54).
However, these gene rearrangements have not been detected in its
histological mimics. FISH assay for AHRR or NCOA2
rearrangements can therefore be applied as a reliable laboratory
test in the diagnosis of soft tissue angiofibroma.
Intermediate soft tissue tumors
MIFS is an intermediate (rarely metastasizing) soft
tissue tumor first described by Montgomery et al(55) in 1998. It primarily occurs in the
subcutaneous tissues of distal extremities and has a peak incidence
in the fourth and fifth decades of life with no gender
predilection. MIFS typically presents as a slow-growing, painless,
poorly defined mass. The preoperative diagnosis in most cases is
benign and may include tenosynovitis, ganglion cyst and GCTTS/PVNS
(1). Histologically, MIFS is
multinodular, poorly circumscribed and characterized by a prominent
myxoid matrix containing numerous inflammatory cells, including
lymphocytes, plasma cells, neutrophils and eosinophils (56). Germinal centers are occasionally
encountered. Neoplastic cells include spindle-shaped and
epithelioid cells with mild to moderate nuclear atypia, large
polygonal and bizarre ganglion-like cells, Reed-Sternberg-like
cells with huge inclusion-like nucleoli and multivacuolated
lipoblast-like cells. Hemosiderin deposition may be evident.
Mitotic activity is usually low and necrosis is rarely present.
Immunohistochemically, the neoplastic cells are diffusely positive
for vimentin and focally for CD68 and CD34. Occasional cases may
show scattered cells that stain for cytokeratin or smooth muscle
actin. More importantly, immunostains for leukocyte common
antigens, CD15 and CD30, are negative (16). MIFS can be histologically mistaken
for a number of benign and malignant soft tissue tumors, including
GCTTS/PVNS, inflammatory myofibroblastic tumor and
myxofibrosarcoma.
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in 10 cases of MIFS and hybrid
MIFS/hemosiderotic fibrolipomatous tumor (HFLT) (57–63).
Cytogenetic and molecular cytogenetic studies have identified the
frequent presence of a balanced or unbalanced t(1;10) (p22;q24)
translocation and ring or marker chromosomes secondary to 3p11–12
amplifications. A reciprocal t(2;6) (q31;p21.3) translocation has
also been described as the sole anomaly in a single case (59). It is of interest that the t(1;10)
translocation has also been identified in three cases of pure HFLT
(61,63,64).
These findings suggest that MIFS and HFLT likely represent
different morphologic variants of the same entity.
Conventional and array CGH studies have shown the
amplification of 3p11–12 (61,65).
Notably, Hallor et al(61)
demonstrated that 3p11–12 amplification is associated with an
increased expression of VGLL3 and CHMP2B. Moreover,
Antonescu et al(63)
confirmed the presence of high level VGLL3 amplification by
FISH in MIFS, as well as in HFLT and cases with hybrid morphology.
VGLL3 has been shown to be amplified and overexpressed in
undifferentiated pleomorphic sarcoma and dedifferentiated
liposarcoma (66). These findings
suggest that VGLL3 is the main target of 3p12 amplification
and this genetic event may be crucial in the development and
progression of certain subsets of soft tissue sarcomas. A gene
expression analysis has shown overexpression of NPM3 and
particularly FGF8(61).
These two genes have been mapped to 10q24. FGF8 is a member
of the fibroblast growth factor family. FGF8 overexpression has
been shown to increase tumor growth and angiogenesis (67).
Antonescu et al(63) reported that FISH analysis for
TGFBR3 at 1p22 and MGEA5 at 10q24 rearrangements can
be applied as a reliable molecular test in the diagnosis of
MIFS.
According to the current World Health Organization
classification, OFMT is an intermediate (rarely metastasizing) soft
tissue tumor of uncertain lineage (1). The biological behavior of OFMT varies.
OFMT primarily occurs in the extremities, trunk and head and neck
of adults with a median age of approximately 50 years (68–70).
Males are affected more frequently than females. OFMT typically
presents as a slow-growing, painless, well-defined, subcutaneous
mass. The lesions range from 1 to 20 cm in their greatest
dimension, with a median size of 3–5 cm (68–70).
Histologically, OFMT is composed of uniform round, ovoid, or
spindle-shaped cells arranged in nests and cords and deposited in a
variably fibromyxoid stroma. In approximately 80% of cases, there
is an incomplete shell of lamellar bone found at the periphery of
the nodules (3). Small foci of
calcification and metaplastic cartilage are also evident. Mitotic
activity is usually less than 1/10 high power fields. Although a
majority of OFMTs are histologically benign and show
correspondingly benign clinical behavior, it has been recognized
that a subset of OFMTs have atypical histological findings, such as
high cellularity and/or increased mitotic activity and show
correspondingly more aggressive clinical behavior (68,70).
Immunohistochemically, the neoplastic cells are typically positive
for vimentin and S-100 protein. The cells may also express desmin,
CD10, Leu-7, neuron-specific enolase and glial fibrillary acidic
protein. OFMT may be mistaken for a number of benign and malignant
soft tissue tumors, including chondroid syringoma, low-grade
fibromyxoid sarcoma and extraskeletal osteosarcoma.
Clonal chromosomal aberrations have been detected by
cytogenetic analysis in seven cases of OFMT (68,71–74).
The short arm of chromosome 6, in particular 6p21, is frequently
involved in OFMT. Notably, a balanced or unbalanced t(6;12)
(p21;q24) translocation appears to be characteristic for OFMT. A
gene expression analysis has shown overexpression of EAAT4
and underexpression of PMP22 in OFMT compared with nerve
sheath myxoma and schwannoma (70).
Moreover, Graham et al(70)
identified loss of INI-1 at 22q11.2 in five of seven
cases by interphase FISH.
Gebre-Medhin et al(74) recently reported that PHF1 at
6p21 is frequently rearranged in OFMT, including atypical and
malignant variants. Moreover, PHF1 was fused to EP400
at 12q24 in one atypical case with the t(6;12) translocation. OFMT
is the second neoplasm, in addition to endometrial stromal tumor,
in which PHF1 is involved in fusions with ectopic sequences.
These findings suggest that FISH assay for PHF1
rearrangements serves as an excellent diagnostic tool in ambiguous
cases.
Conclusions
Traditional laboratory techniques, such as
cytogenetic analysis and FISH, have a pivotal role in the diagnosis
of soft tissue tumors. Over the past few years, some novel gene
rearrangements have been described in benign and intermediate soft
tissue tumors. Notably, the t(5;8)(p15;q13) translocation,
resulting in the AHRR-NCOA2 fusion gene, was
identified in angiofibroma of soft tissue recently proposed as a
new clinicopathologic entity (51).
FISH can be used to distinguish between entities with similar
histological appearances, although probes are not yet commercially
available. Understanding the basis of these ancillary techniques
and their application is critical to provide accurate diagnoses of
soft tissue tumors.
Acknowledgements
This study was supported in part by
Fukuoka Cancer Society, Ogata Foundation and the Foundation for the
Promotion of Medical Science.
References
|
1
|
Fletcher CDM, Unni KK and Mertens F: World
Health Organization Classification of Tumours: Pathology and
Genetics of Tumours of Soft Tissue and Bone. IARC Press; Lyon,
France: 2002
|
|
2
|
Bridge JA and Cushman-Vokoun AM: Molecular
diagnostics of soft tissue tumors. Arch Pathol Lab Med.
135:588–601. 2011.PubMed/NCBI
|
|
3
|
Weiss SW and Goldblum JR: Enzinger and
Weiss’s: Soft Tissue Tumors. Mosby; Philadelphia: 2008
|
|
4
|
Sawyer JR, Sammartino G, Baker GF and Bell
JM: Clonal chromosome aberrations in a case of nodular fasciitis.
Cancer Genet Cytogenet. 76:154–156. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Birdsall SH, Shipley JM, Summersgill BM,
et al: Cytogenetic findings in a case of nodular fasciitis of the
breast. Cancer Genet Cytogenet. 81:166–168. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weibolt VM, Buresh CJ, Roberts CA, et al:
Involvement of 3q21 in nodular fasciitis. Cancer Genet Cytogenet.
106:177–179. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Donner LR, Silva T and Dobin SM: Clonal
rearrangement of 15p11.2, 16p11.2 and 16p13.3 in a case of nodular
fasciitis: additional evidence favoring nodular fasciitis as a
benign neoplasm and not a reactive tumefaction. Cancer Genet
Cytogenet. 139:138–140. 2002. View Article : Google Scholar
|
|
8
|
Velagaleti GVN, Tapper JK, Panova NE,
Miettinen M and Gatalica Z: Cytogenetic findings in a case of
nodular fasciitis of subclavicular region. Cancer Genet Cytogenet.
141:160–163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng GZ, Zhang HY, Zhang Z, Wei B and Bu
H: Myofibroblastic sarcoma vs nodular fasciitis: a comparative
study of chromosomal imbalances. Am J Clin Pathol. 131:701–709.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bacac M, Migliavacca E, Stehle JC, et al:
A gene expression signature that distinguishes desmoid tumours from
nodular fasciitis. J Pathol. 208:543–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Erickson-Johnson MR, Chou MM, Evers BR, et
al: Nodular fasciitis: a novel model of transient neoplasia induced
by MYH9-USP6 gene fusion. Lab Invest. 91:1427–1433. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oliveira AM, Hsi BL, Weremowicz S, et al:
USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res.
64:1920–1923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sukov WR, Franco MF, Erickson-Johnson M,
et al: Frequency of USP6 rearrangements in myositis ossificans,
brown tumor and cherubism: molecular cytogenetic evidence that a
subset of ‘myositis ossificans-like lesions’ are the early phases
in the formation of soft-tissue aneurysmal bone cyst. Skeletal
Radiol. 37:321–327. 2008.PubMed/NCBI
|
|
14
|
Thway K, Flora RS and Fisher C: Chondroid
lipoma: an update and review. Ann Diagn Pathol. 16:230–234. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishio J: Contributions of cytogenetics
and molecular cytogenetics to the diagnosis of adipocytic tumors. J
Biomed Biotechnol 2011. 5240672011.PubMed/NCBI
|
|
16
|
Nishio J, Iwasaki H, Nabeshima K and Naito
M: Cytogenetics and molecular genetics of myxoid soft-tissue
sarcomas. Genet Res Int. 2011:4971482011.PubMed/NCBI
|
|
17
|
Gisselsson D, Domanski HA, Höglund M, et
al: Unique cytological features and chromosome aberrations in
chondroid lipoma: a case report based on fine-needle aspiration
cytology, histopathology, electron microscopy, chromosome banding
and molecular cytogenetics. Am J Surg Pathol. 23:1300–1304. 1999.
View Article : Google Scholar
|
|
18
|
Thomson TA, Horsman D and Bainbridge TC:
Cytogenetic and cytologic features of chondroid lipoma of soft
tissue. Mod Pathol. 12:88–91. 1999.PubMed/NCBI
|
|
19
|
Ballaux F, Debiec-Rychter M, De Wever I
and Sciot R: Chondroid lipoma is characterized by
t(11;16)(q13;p12–13). Virchows Arch. 444:208–210. 2004.PubMed/NCBI
|
|
20
|
Huang D, Sumegi J, Dal Cin P, et al:
C11orf95-MKL2 is the resulting fusion oncogene of t(11;16)(q13;p13)
in chondroid lipoma. Genes Chromosomes Cancer. 49:810–818.
2010.PubMed/NCBI
|
|
21
|
Evans HL: Desmoplastic fibroblastoma: a
report of seven cases. Am J Surg Pathol. 19:1077–1081. 1995.
View Article : Google Scholar
|
|
22
|
Magro G and Venti C: Childhood
desmoplastic fibroblastoma (collagenous fibroma) with a 12-year
follow-up. Pediatr Dev Pathol. 2:62–64. 1999.PubMed/NCBI
|
|
23
|
Nishio J, Iwasaki H, Nishijima T and
Kikuchi M: Collagenous fibroma (desmoplastic fibroblastoma) of the
finger in a child. Pathol Int. 52:322–325. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sciot R, Samson I, Van Den Berghe H, Van
Damme B and Dal Cin P: Collagenous fibroma (desmoplastic
fibroblastoma): genetic link with fibroma of tendon sheath? Mod
Pathol. 12:565–568. 1999.PubMed/NCBI
|
|
25
|
Bernal K, Nelson M, Neff JR, Nielsen SM
and Bridge JA: Translocation (2;11)(q31;q12) is recurrent in
collagenous fibroma (desmoplastic fibroblastoma). Cancer Genet
Cytogenet. 149:161–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sakamoto A, Yamamoto H, Yoshida T, et al:
Desmoplastic fibroblastoma (collagenous fibroma) with a specific
breakpoint of 11q12. Histopathology. 51:859–860. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maghari A, Ma N, Aisner S, Benevenia J and
Hameed M: Collagenous fibroma (desmoplastic fibroblastoma) with a
new translocation involving 11q12: a case report. Cancer Genet
Cytogenet. 192:73–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nishio J, Akiho S, Iwasaki H and Naito M:
Translocation t(2;11) is characteristic of collagenous fibroma
(desmoplastic fibroblastoma). Cancer Genet. 204:569–571. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Macchia G, Trombetta D, Möller E, et al:
FOSL1 as a candidate target gene for 11q12 rearrangements in
desmoplastic fibroblastoma. Lab Invest. 92:735–743. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dal Cin P, Sciot R, De Smet L and Van Den
Berghe H: Translocation 2;11 in a fibroma of tendon sheath.
Histopathology. 32:433–435. 1998.PubMed/NCBI
|
|
31
|
Young MR and Colburn NH: Fra-1 a target
for cancer prevention or intervention. Gene. 379:1–11. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ray RA, Morton CC, Lipinski KK, Corson JM
and Fletcher JA: Cytogenetic evidence of clonality in a case of
pigmented villonodular synovitis. Cancer. 67:121–125. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fletcher JA, Henkle C, Atkins L, Rosenberg
AE and Morton CC: Trisomy 5 and trisomy 7 are nonrandom aberrations
in pigmented villonodular synovitis: confirmation of trisomy 7 in
uncultured cells. Genes Chromosomes Cancer. 4:264–266. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mertens F, Orndal C, Mandahl N, et al:
Chromosome aberrations in tenosynovial giant cell tumors and
nontumorous synovial tissue. Genes Chromosomes Cancer. 6:212–217.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rowlands CG, Roland B, Hwang WS and Sevick
RJ: Diffusevariant tenosynovial giant cell tumor: a rare and
aggressive lesion. Hum Pathol. 25:423–425. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dal Cin P, Sciot R, Samson I, et al:
Cytogenetic characterization of tenosynovial giant cell tumors
(nodular tenosynovitis). Cancer Res. 54:3986–3987. 1994.
|
|
37
|
Choong PF, Willen H, Nilbert M, et al:
Pigmented villonodular synovitis. Monoclonality and metastasis - a
case for neoplastic origin? Acta Orthop Scand. 66:64–68. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gonzalez-Campora R, Salas Herrero E,
Otal-Salaverri C, et al: Diffuse tenosynovial giant cell tumor of
soft tissues: report of a case with cytologic and cytogenetic
findings. Acta Cytol. 39:770–776. 1995.PubMed/NCBI
|
|
39
|
Dal Cin P, Sciot R, De Smet L, Van Damme B
and Van Den Berghe H: A new cytogenetic subgroup in tenosynovial
giant cell tumors (nodular tenosynovitis) is characterized by
involvement of 16q24. Cancer Genet Cytogenet. 87:85–87.
1996.PubMed/NCBI
|
|
40
|
Ohjimi Y, Iwasaki H, Ishiguro M, et al:
Short arm of chromosome 1 aberration recurrently found in pigmented
villonodular synovitis. Cancer Genet Cytogenet. 90:80–85. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sciot R, Rosai J, Dal Cin P, et al:
Analysis of 35 cases of localized and diffuse tenosynovial giant
cell tumor: a report from the chromosomes and morphology (CHAMP)
study group. Mod Pathol. 12:576–579. 1999.PubMed/NCBI
|
|
42
|
Nilsson M, Hoglund M, Panagopoulos I, et
al: Molecular cytogenetic mapping of recurrent chromosomal
breakpoints in tenosynovial giant cell tumors. Virchows Arch.
441:475–480. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ferrer J, Namiq A, Carda C, Lopez-Gines C,
Tawfik O and Llombart-Bosch A: Diffuse type of giant-cell tumor of
tendon sheath: an ultrastructural study of two cases with
cytogenetic support. Ultrastruct Pathol. 26:15–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brandal P, Bjerkehagen B and Heim S:
Molecular cytogenetic characterization of tenosynovial giant cell
tumors. Neoplasia. 6:578–583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Occhipinti E, Heinrich SD and Craver R:
Giant cell tumor of tendon sheath arising in the toe. Fetal Pediatr
Pathol. 23:171–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
West RB, Rubin BP, Miller MA, et al: A
landscape effect in tenosynovial giant-cell tumor from activation
of CSF1 expression by a translocation in a minority of tumor cells.
Proc Natl Acad Sci USA. 103:690–695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Moller E, Mandahl N, Mertens F and
Panagopoulos I: Molecular identification of COL6A3-CSF1 fusion
transcripts in tenosynovial giant cell tumors. Genes Chromosomes
Cancer. 47:21–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Blay JY, El Sayadi H, Thiesse P, Garret J
and Ray-Coquard I: Complete response to imatinib in relapsing
pigmented villonodular synovitis/tenosynovial giant cell tumor
(PVNS/TGCT). Ann Oncol. 19:821–822. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cassier PA, Gelderblom H, Stacchiotti S,
et al: Efficacy of imatinib mesylate for the treatment of locally
advanced and/or metastatic tenosynovial giant cell tumor/pigmented
villonodular synovitis. Cancer. 118:1649–1655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mariño-Enríquez A and Fletcher CDM:
Angiofibroma of soft tissue: clinicopathologic characterization of
a distinctive benign fibrovascular neoplasm in a series of 37
cases. Am J Surg Pathol. 36:500–508. 2012.PubMed/NCBI
|
|
51
|
Jin Y, Möller E, Nord KH, et al: Fusion of
the AHRR and NCOA2 genes through a recurrent translocation
t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation
of aryl hydrocarbon receptor target genes. Genes Chromosomes
Cancer. 51:510–520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schoolmeester JK, Sukov WR, Aubry MC and
Folpe AL: Angiofibroma of soft tissue: core needle biopsy
diagnosis, with cytogenetic confirmation. Am J Surg Pathol.
36:1421–1423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sumegi J, Streblow R, Frayer RW, et al:
Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma
without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear
receptor transcriptional coactivator family. Genes Chromosomes
Cancer. 49:224–236. 2010.
|
|
54
|
Wang L, Motoi T, Khanin R, et al:
Identification of a novel, recurrent HEY1-NCOA2 fusion in
mesenchymal chondrosarcoma based on a genome-wide screen of
exon-level expression data. Genes Chromosomes Cancer. 51:127–139.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Montgomery EA, Devaney KO, Giordano TJ and
Weiss SW: Inflammatory myxohyaline tumor of distal extremities with
virocyte or Reed-Sternberg-like cells: a distinctive lesion with
features simulating inflammatory conditions, Hodgkin’s disease and
various sarcomas. Mod Pathol. 11:384–391. 1998.
|
|
56
|
Meis-Kindblom JM and Kindblom LG: Acral
myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the
hands and feet. Am J Surg Pathol. 22:911–924. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lambert I, Debiec-Rychter M, Guelinckx P,
Hagemeijer A and Sciot R: Acral myxoinflammatory fibroblastic
sarcoma with unique clonal chromosomal changes. Virchows Arch.
438:509–512. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mansoor A, Fidda N, Himoe E, Payne M,
Lawce H and Magenis RE: Myxoinflammatory fibroblastic sarcoma with
complex supernumerary ring chromosomes composed of chromosome 3
segments. Cancer Genet Cytogenet. 152:61–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ida CM, Rolig KA, Hulshizer RL, et al:
Myxoinflammatory fibroblastic sarcoma showing t(2;6)(q31;p21.3) as
a sole cytogenetic abnormality. Cancer Genet Cytogenet.
177:139–142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gonzalez-Cámpora R, Ríos-Martín JJ,
Solórzano-Amoretti A, et al: Fine needle aspiration cytology of an
acral myxoinflammatory fibroblastic sarcoma: case report with
cytological and cytogenetic findings. Cytopathology. 19:118–123.
2008.PubMed/NCBI
|
|
61
|
Hallor KH, Sciot R, Staaf J, et al: Two
genetic pathways, t(1;10) and amplification of 3p11–12, in
myxoinflammatory fibroblastic sarcoma, haemosiderotic
fibrolipomatous tumour and morphologically similar lesions. J
Pathol. 217:716–727. 2009.PubMed/NCBI
|
|
62
|
Elco CP, Mariño-Enríquez A, Abraham JA,
Dal Cin P and Hornick JL: Hybrid myxoinflammatory fibroblastic
sarcoma/hemosiderotic fibrolipomatous tumor: report of a case
providing further evidence for a pathogenetic link. Am J Surg
Pathol. 34:1723–1727. 2010.
|
|
63
|
Antonescu CR, Zhang L, Nielsen GP,
Rosenberg AE, Dal Cin P and Fletcher CD: Consistent t(1;10) with
rearrangements of TGFBR3 and MGEA5 in both myxoinflammatory
fibroblastic sarcoma and hemosiderotic fibrolipomatous tumor. Genes
Chromosomes Cancer. 50:757–764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wettach GR, Boyd LJ, Lawce HJ, Magenis RE
and Mansoor A: Cytogenetic analysis of a hemosiderotic
fibrolipomatous tumor. Cancer Genet Cytogenet. 182:140–143. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baumhoer D, Glatz K, Schulten HJ, et al:
Myxoinflammatory fibroblastic sarcoma: investigations by
comparative genomic hybridization of two cases and review of the
literature. Virchows Arch. 451:923–928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hélias-Rodzewicz Z, Pérot G, Chibon F, et
al: YAP1 and VGLL3, encoding two cofactors of TEAD transcription
factors, are amplified and overexpressed in a subset of soft tissue
sarcomas. Genes Chromosomes Cancer. 49:1161–1171. 2010.PubMed/NCBI
|
|
67
|
Mattila MM and Härkönen PL: Role of
fibroblast growth factor 8 in growth and progression of hormonal
cancer. Cytokine Growth Factor Rev. 18:257–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Folpe AL and Weiss SW: Ossifying
fibromyxoid tumor of soft parts: a clinicopathologic study of 70
cases with emphasis on atypical and malignant variants. Am J Surg
Pathol. 27:421–431. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Miettinen M, Finnell V and Fetsch JF:
Ossifying fibromyxoid tumor of soft parts - a clinicopathologic and
immunohistochemical study of 104 cases with long-term follow-up and
a critical review of the literature. Am J Surg Pathol. 32:996–1005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Graham RP, Dry S, Li X, et al: Ossifying
fibromyxoid tumor of soft parts: a clinicopathologic, proteomic and
genomic study. Am J Surg Pathol. 35:1615–1625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sovani V, Velagaleti GVN, Filipowicz E,
Gatalica Z and Knisely AS: Ossifying fibromyxoid tumor of soft
parts: report of a case with novel cytogenetic findings. Cancer
Genet Cytogenet. 127:1–6. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nishio J, Iwasaki H, Ohjimi Y, et al:
Ossifying fibromyxoid tumor of soft parts: cytogenetic findings.
Cancer Genet Cytogenet. 133:124–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kawashima H, Ogose A, Umezu H, et al:
Ossifying fibromyxoid tumor of soft parts with clonal chromosomal
aberrations. Cancer Genet Cytogenet. 176:156–160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gebre-Medhin S, Nord KH, Möller E, et al:
Recurrent rearrangement of the PHF1 gene in ossifying fibromyxoid
tumors. Am J Pathol. 181:1069–1077. 2012. View Article : Google Scholar : PubMed/NCBI
|