Introduction
KIT is a transmembrane glycoprotein that belongs to
the type III tyrosine kinase receptor family (1). Upon binding its ligand, stem cell
factor (SCF), the KIT receptor dimerizes and initiates a signal
transduction phosphorylation cascade that results in the regulation
of cell growth. SCF is encoded by the Steel gene and is
present in both membrane-bound (mSCF) and soluble (sSCF) forms
(2). KIT that has been activated by
mSCF is considered to be stable and maintains its activity
(3). The SCF/KIT system plays a key
role in the differentiation and proliferation of the interstitial
cells of Cajal (ICCs) and hematopoietic cells (4). The SCF/KIT system is also involved in
cell proliferation in certain tumors, including mast cell leukemia,
seminoma and malignant melanoma, as well as lung, small cell,
breast, gastric, colon, cervical and ovarian cancer (5–10).
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal neoplasm of the gastrointestinal tract and they
originate from the ICCs or their precursor cells (11). GISTs are defined as tumors that are
typically immunoreactive for KIT. Unlike the SCF/KIT tumors
mentioned previously, ∼75–80% of GISTs have a gain-of-function
mutation in the KIT proto-oncogene encoding the KIT protein
(12–15). These mutations lead to constitutive
oncogenic signaling in the absence of SCF. Uncontrolled KIT
activity results in the oncogenesis and proliferation of GISTs.
However, it has been demonstrated that 94% of GIST mutations are
heterozygous, i.e., wild-type KIT remains present in the majority
of GISTs (16). It is not yet known
whether these wild-type KIT are capable of being activated by their
ligand, SCF, and are involved in the proliferation of GISTs.
Imatinib, a small molecule tyrosine kinase
inhibitor, has been demonstrated to be effective in the treatment
of recurrent or metastatic GISTs by inhibition of KIT activation.
Findings of a previous study confirmed that KIT activation is a
ubiquitous feature of GISTs, even in the absence of KIT
mutations (14). However, based on
certain clinical trials, the best response rates to imatinib have
been observed in GISTs with KIT mutations (12,17–18).
GISTs with wild-type KIT have demonstrated primary
resistance to imatinib, and a considerable proportion of GISTs with
mutant KIT have demonstrated acquired resistance at a later
stage (19). These clinical trials
revealed that imatinib was not capable of effectively inhibiting
wild-type KIT activation in GISTs lacking a KIT mutation. If
SCF is able to function as the ligand that activates wild-type KIT
in heterozygous GISTs, we hypothesize that imatinib is also likely
to fail to inhibit wild-type KIT activation.
In the present study, we examined the expression of
SCF in GISTs, analyzed the relationship between SCF expression and
the proliferative activity of GIST cells, and studied the role of
SCF in the proliferation of GIST cells. We verified our theory by
examining KIT activation in SCF-stimulated GIST cells pretreated
with imatinib. We also observed the level of SCF expression in GIST
cells following treatment with imatinib.
Materials and methods
Patient samples
Clinical samples were obtained with informed
consent. A total of 68 GIST samples were included in the study. All
cases were confirmed as GIST by at least two pathologists. The
tumor size, number of mitotic cells in 50 high-power fields (HPF)
and Ki-67 index were observed by hematoxylin and eosin (HE)
staining or immunohistochemistry. For the histological and
immunohistochemical analyses, tissue specimens were fixed in 10%
formalin and embedded in paraffin. Core tissue biopsies (2 mm in
diameter) were taken from individual paraffin-embedded tumor
tissues and arranged in new recipient paraffin blocks using a
Tissue Microarrayer (Beecher Instruments, Silver Spring, MD, USA).
For each tumor, the representative tumor areas were selected using
HE staining. For western blot analysis and mutation detection, 21
fresh tissue specimens were frozen and stored at −80°C.
Immunohistochemical detection
Immunohistochemical staining was performed using
anti-SCF antibody (rabbit monoclonal; 1:50 dilution; Abcam,
Cambridge, MA, USA), anti-KIT antibody (rabbit polyclonal; 1:500
dilution; DakoCytomation, Glostrup, Denmark) and anti-Ki-67
antibody (mouse monoclonal; MIB-1; 1:200 dilution; DakoCytomation).
The TMA sections (4 μm) were deparaffinized and boiled in
10% citric acid buffer solution (pH 6.0) for 20 min for antigen
retrieval. Following blocking of endogenous peroxidase activity
with 3% hydrogen peroxide, specimens were incubated with primary
antibodies at 4°C overnight. Bound antibodies were detected by a
peroxidase-labeled, polymer-conjugated secondary antibody (EnVision
HRP; DakoCytomation), and subjected to peroxidase staining using
diaminobenzidine (DAB) as a substrate. Slides were counterstained
with hematoxylin. The Ki-67 labeling index was calculated as the
percentage of Ki-67-positive cells among all tumor cells in 5
HPF.
Detection of KIT mutations
Genomic DNA of fresh or paraffin-embedded tissues
was extracted using a standard proteinase-K
digestion/phenol-chloroform procedure. KIT exons 11 and 9
were amplified using the following primer sequences and annealing
temperatures: Exon 11, forward: 5′-CCAGAGTGCTCTAATGACTG-3′ and
reverse: 5′-AGC CCCTGTTTCATACTGAC-3′ at 60°C; exon 9, forward:
5′-GCCACATCCCAAGTGTTTTATG-3′ and reverse: 5′-GAG
CCTAAACATCCCCTTAAATTG-3′ at 56°C. Sequencing analysis was performed
directly on the PCR products.
Cell isolation and primary cell
culture
Fresh GIST tissues were minced with scissors, washed
twice with phosphate-buffered saline (PBS), and then ground into
single-cell suspensions by filtering through the sieve with 200
μm mesh. After washing in cold PBS, cell pellets were
resuspended in RPMI-1640 medium supplemented with 10% fetal calf
serum (FCS; Gibco, France) and seeded onto culture dishes (Fig. 1). Cells were cultured overnight
prior to analysis. Simultaneously, all the cells that had been
isolated from GIST tissue were analyzed for KIT mutations
(Table I).
 | Table I.Details of GIST primary cultures. |
Table I.
Details of GIST primary cultures.
| GIST case no. | Age (years) | Origin | Tumor size (cm) | KIT
mutation |
|---|
| 0917833 | 53 | Stomach | 6.0 | Exon 11
DEL557–558 |
| 0919049 | 41 | Stomach | 3.0 | Exon 11
DEL555–558 |
| 0919298 | 68 | Stomach | 2.6 | Exon 11
DEL557–558 |
| 0930644 | 37 | Small intestine | 2.0 | Exon 9
INS502–503 |
| 0930830 | 58 | Small intestine | 8.0 | Exon 9
INS502–503 |
| 1002977 | 56 | Small intestine | 11.0 | Exon 9
INS502–503 |
| 1002979 | 59 | Stomach | 7.5 | Exon 11 DEL576 |
In vitro cell proliferation assay and
drug testing
GIST cells were FCS-starved for 4 h and then
cultured in the appropriate culture medium with 100 ng/ml
recombinant human SCF (rhSCF, Peprotech, Inc., Rocky Hill, NJ, USA)
for 72 h. Viable cells were measured with the Cell Counting kit 8
(Wako, Osaka, Japan) according to the manufacturer’s instructions.
GIST cells were FCS-starved for 4 h, stimulated by rhSCF for 0, 5,
15, 30, 60 or 120 min, and then harvested for KIT phosphorylation
detection by western blot analysis. SCF expression levels were
detected in GIST cells treated with or without imatinib (Novartis
Pharma, Basel, Switzerland) for 72 h by western blot analysis. KIT
phosphorylation levels were observed in GIST cells treated with
imatinib for 90 min, prior to stimulation with 100 ng/ml rhSCF for
10 min.
Western blot analysis
Frozen GIST samples were calibrated and homogenized
in lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM othovanadate, 10 mM
NaF, 1 mM PMSF, 0.5 μg/ml leupeptin, 1 μg/ml
pepstatin, 10 KIU/ml aprotinin and 1% triton X-100). Lysates were
rocked at 4°C for 30 min and then centrifuged at 12,000 rpm for 15
min. Supernatant protein concentrations were measured using a BAC
Protein Assay kit (Merck KGaA; Darmstadt, Germany), and 50
μg of protein were separated by 8 or 12% SDS-polyacrylamide
gel electrophoresis and transferred to a polyvinylidene difluoride
membrane. The membrane was blocked for 60 min at room temperature
with 5% skimmed milk or bovine serum albumin (BSA) and then reacted
with anti-KIT antibody (DakoCytomation), anti-SCF antibody (Abcam),
or anti-c-kit (phospho Y703) (Abcam), as the primary antibody.
Peroxidase-labeled anti-rabbit IgG was used as the secondary
antibody. The Western Lightning chemiluminescence reagent (Santa
Cruz Biotechnology, Inc.; Santa Cruz, CA, USA) was used for the
detection of proteins.
Results
SCF expression in GISTs and its
correlation with tumor proliferation
The expression of KIT and its ligand SCF were
detected by immunohistochemical staining in 68 GIST samples. All
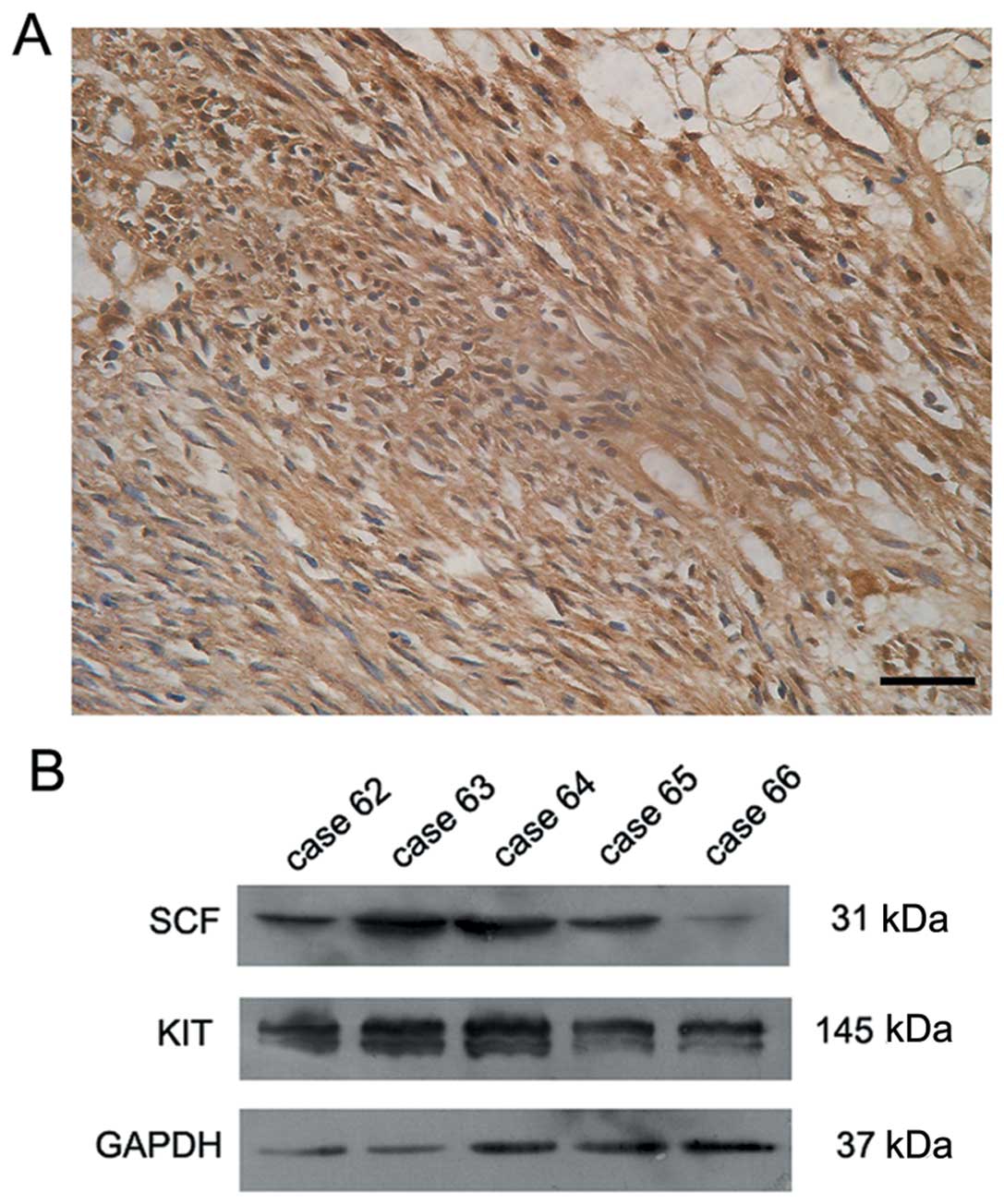
GIST samples demonstrated KIT positivity. Expression of SCF was
observed in 52 cases (Fig. 2A). The
SCF-positive cases presented moderate or strong staining in the
membrane and cytoplasm of GIST cells. Expression of SCF was further
demonstrated by western blot analysis in fresh GIST tissues that
stained positive for SCF by immunohistochemistry. As shown in
Fig. 2B, a positive signal was
detected at 31 kDa that corresponded to the membrane-bound form of
SCF in 17 out of 21 cases. In those 17 tumors, the signal for KIT,
which has a molecular weight of 145 kDa, was also confirmed in the
tumor tissues (Fig. 2B).
We then examined the association between SCF
expression and the proliferative activity of tumors. The Ki-67
index and the number of mitotic cells in 50 HPF were used to
evaluate the proliferation potential of the GIST cells. The
expression rate of Ki-67 and the number of mitotic cells in
SCF-positive cases were significantly higher than those of
SCF-negative cases (P<0.001 and P<0.05, respectively;
Mann-Whitney U test; Table II).
| Table II.Number of mitotic cells and Ki-67
index in SCF-positive cases compared with SCF-negative cases. |
Table II.
Number of mitotic cells and Ki-67
index in SCF-positive cases compared with SCF-negative cases.
| No. of SCF-positive
cases | No. of SCF-negative
cases | P-value |
|---|
| No. of mitotic cells
(per 50 HPF) | | | 0.049 |
| ≤10 | 36 | 15 | |
| >10 | 16 | 1 | |
| Ki-67 index (%) | | | 0.001 |
| <5 | 15 | 12 | |
| 5–10 | 23 | 4 | |
| >10 | 14 | 0 | |
Correlation between c-kit mutations and
SCF expression
A total of 54 GIST samples were investigated for
mutations in the KIT proto-oncogene (exons 9 and 11). Exons
9 and 11 have been demonstrated to be frequently mutated in GISTs.
DNA sequencing results obtained for all 54 samples are listed in
Table III. The correlation of
KIT mutations with SCF expression was analyzed by a
McNemar’s test. No correlation was observed between the presence of
KIT mutations and the expression of SCF (P>0.05).
| Table III.KIT mutation details of the 54 cases
of GIST. |
Table III.
KIT mutation details of the 54 cases
of GIST.
| Case no. | Gender | Age (years) | SCF | Origin | Tumor size
(cm) | KIT
mutation |
|---|
| 1 | F | 40 | − | Small
intestine | 4.0 | Wt |
| 2 | M | 36 | + | Stomach | 5.0 | Wt |
| 3 | M | 61 | + | Stomach | 5.5 | Exon 11 V560D |
| 4 | M | 43 | + | Stomach | 5.0 | Exon 11
DEL555–556 |
| 8 | F | 50 | + | Small
intestine | 5.0 | Wt |
| 9 | F | 56 | + | Stomach | 4.5 | Exon 11
DEL556–582 |
| 10 | F | 72 | + | Stomach | 2.0 | Wt |
| 13 | M | 59 | − | Stomach | 4.0 | Wt |
| 15 | M | 34 | + | Small
intestine | 3.5 | Exon 9
INS502–503 |
| 16 | F | 49 | + | Stomach | 4.0 | Exon 11
DEL555–558 |
| 17 | F | 52 | + | Unknown | 9.0 | Exon 9
INS502–503 |
| 18 | M | 43 | + | Small
intestine | 3.5 | Exon 11
DEL553–554 |
| 19 | F | 78 | − | Stomach | 5.0 | Exon 11
DEL555–558 |
| 20 | F | 61 | + | Small
intestine | 5.0 | Exon 11
DEL555–559 |
| 21 | F | 59 | + | Stomach | 3.0 | Wt |
| 22 | M | 49 | + | Small
intestine | 2.5 | Wt |
| 23 | F | 53 | + | Stomach | 2.5 | Wt |
| 27 | M | 63 | − | Stomach | 5.0 | Exon 11 V559D |
| 28 | M | 48 | + | Stomach | 3.0 | Exon 11
DEL565–572 |
| 29 | F | 39 | + | Stomach | 18.0 | Exon 11
INS577–579 |
| 30 | F | 48 | + | Small
intestine | 12.0 | Wt |
| 31 | M | 71 | + | Stomach | 5.5 | Wt |
| 32 | M | 53 | + | Rectum | 6.0 | Exon 11 W557R |
| 33 | M | 45 | + | Small
intestine | 2.0 | Wt |
| 34 | F | 73 | + | Stomach | 6.0 | Exon 11 V559D |
| 37 | F | 57 | + | Stomach | 6.5 | Wt |
| 38 | M | 54 | − | Small
intestine | 6.0 | Exon 11 DEL579 |
| 40 | M | 68 | − | Small
intestine | 7.0 | Exon 11
DEL557–558 |
| 41 | F | 48 | + | Stomach | 3.0 | Exon 11
DEL557–558 |
| 42 | F | 73 | − | Stomach | 2.5 | Exon 11
DEL555–557 |
| 43 | M | 50 | + | Stomach | 4.0 | Wt |
| 45 | F | 37 | + | Small
intestine | 3.0 | Exon 11
INS557–582 |
| 47 | F | 51 | + | Small
intestine | 2.5 | Exon 9
INS502–503 |
| 48 | F | 49 | + | Stomach | 5.0 | Exon 11
INS577–582 |
| 49 | M | 78 | + | Stomach | 14.0 | Exon 11
DEL555–558 |
| 50 | F | 41 | + | Unknown | 8.0 | Exon 11 V560D |
| 53 | M | 77 | + | Stomach | 5.5 | Exon 11 V559D |
| 54 | F | 55 | + | Stomach | 22.0 | Exon 11
DEL555–559 |
| 55 | F | 82 | + | Small
intestine | 3.5 | Exon 11
DEL553–554 |
| 56 | F | 55 | + | Small
intestine | 5.0 | Exon 9
INS502–503 |
| 57 | F | 84 | − | Stomach | 5.0 | Wt |
| 58 | F | 55 | + | Stomach | 4.0 | Exon 11
DEL555–558 |
| 59 | M | 60 | + | Stomach | 2.8 | Exon 11
DEL565–572 |
| 60 | F | 75 | + | Stomach | 6.0 | Exon 11
INS577–579 |
| 61 | F | 63 | + | Small
intestine | 3.0 | Exon 9
INS502–503 |
| 62 | M | 47 | + | Stomach | 3.0 | Exon 11 DEL576 |
| 63 | M | 35 | + | Stomach | 6.5 | Exon 11
INS575–582 |
| 64 | M | 60 | + | Small
intestine | 3.2 | Exon 9 S451C |
| 65 | F | 59 | + | Stomach | 14.0 | Exon 11
DEL557–558 |
| 66 | M | 75 | − | Stomach | 15.0 | Exon 11
DEL550–558 |
| 67 | M | 53 | + | Stomach | 6.0 | Exon 11 DEL579 |
| 68 | M | 29 | − | Rectum | 5.0 | Wt |
Proliferation of GIST cells stimulated by
SCF
To investigate the function of SCF/KIT signaling in
GIST cells, primary GIST cells were incubated for 72 h with SCF at
concentrations of 0, 1, 10 and 100 ng/ml, and their viability and
rate of multiplication were determined by a cell count assay. The
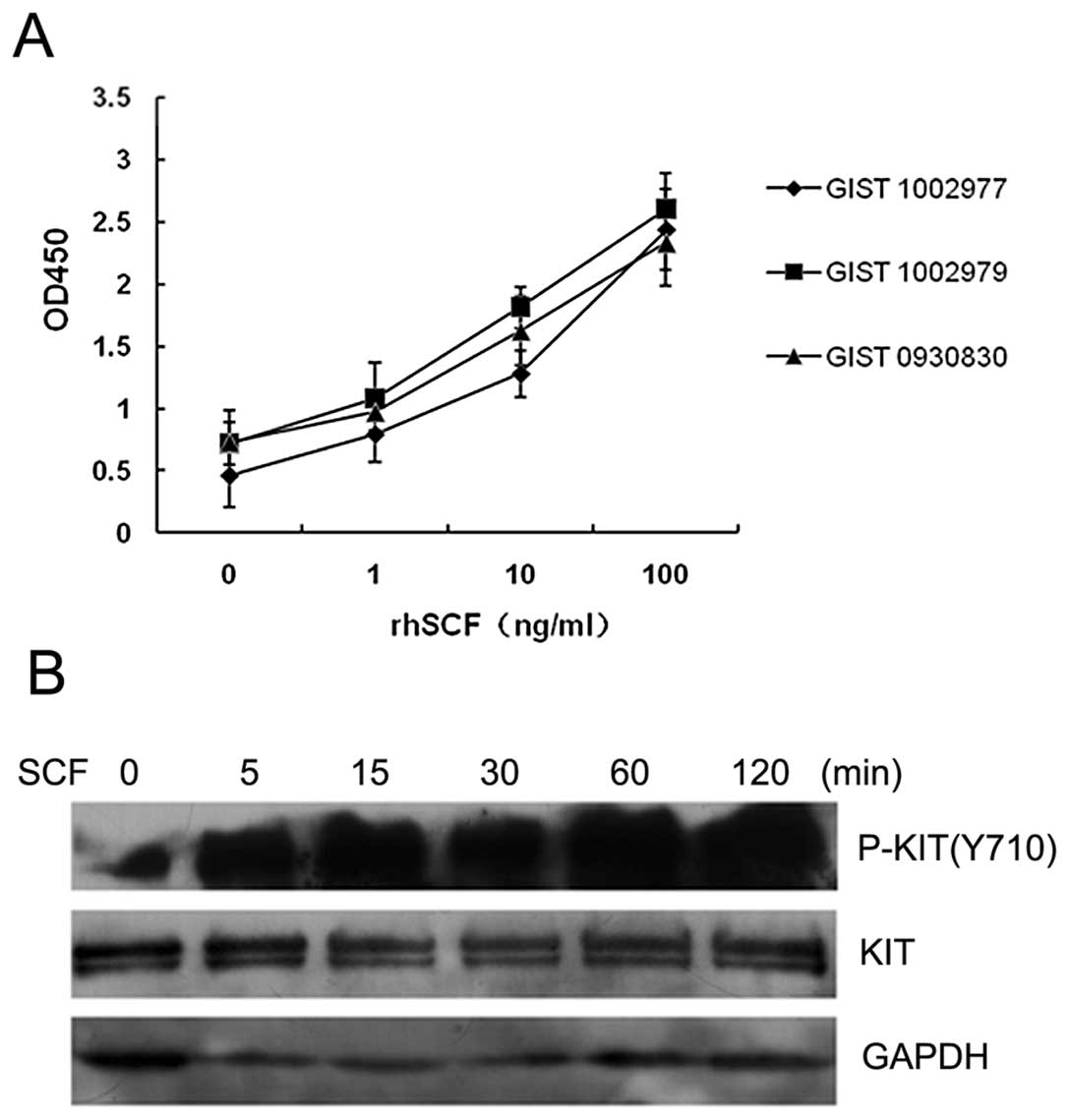
primary GIST cells from all three patients examined proliferated in
response to SCF in a dose-dependent manner (Fig. 3A). We subsequently examined KIT
activation in GIST cells following cultivation with or without SCF.
The levels of KIT phosphorylation in SCF-stimulated cells were
higher than those of unstimulated cells (Fig. 3B). These results suggest that SCF,
as the ligand of KIT, is capable of activating its receptor in GIST
cells. In addition, the results suggest that the SCF/KIT signaling
pathway plays a key role in the proliferation of GIST cells.
Effect of imatinib treatment on SCF
expression and SCF-stimulated KIT activation in GIST cells
Treatment with imatinib has been shown to result in
an increased SCF concentration in the serum of GIST patients
(20). To test the effect of
imatinib treatment on SCF expression of primary GIST cells in
vitro, FCS-starved GIST cells were treated with imatinib for 72
h and then analyzed by western blot analysis for SCF expression.
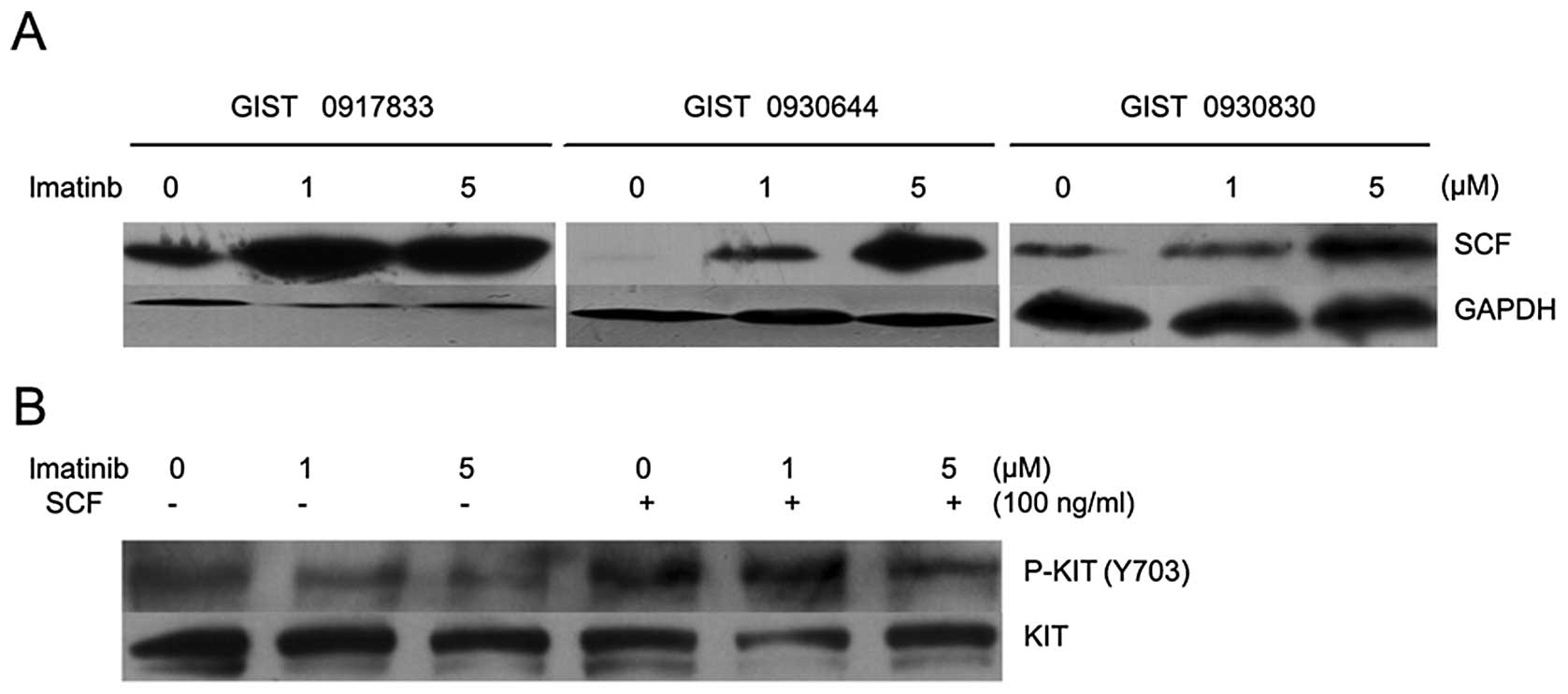
The SCF signal was stronger in imatinib-treated compared with
untreated GIST cells (Fig. 4A).
These results suggest that imatinib is capable of increasing
endogenously produced SCF in cultured GIST cells. We then examined
the inhibitory effects of imatinib on KIT activation that has been
induced by stimulation with exogenous SCF. GIST cells were
pre-treated with or without imatinib and then stimulated with 100
ng/ml SCF. Following the 100 ng/ml SCF stimulation, higher levels
of KIT phosphorylation remained present in GIST cells. An increased
dose of 5 μM imatinib was not able to inhibit KIT
phosphorylation in SCF-stimulated cells in vitro (Fig. 4B). These results suggest that
imatinib may not effectively inhibit KIT phosphorylation that has
been induced by its ligand (SCF).
Discussion
In this study, we observed abundant SCF expression
in GIST tissues, which suggested an autocrine mechanism of SCF.
Western blot analysis verified that the SCF protein expressed in
GISTs is predominantly present as the membrane-bound form, which
has the ability to stably and continuously activate its receptor,
KIT. Our data also demonstrated co-expression of SCF and KIT in
GISTs, and SCF expression was significantly correlated with an
increased proliferative potential of GISTs. A previous study
(21) also SCF expression in GISTs,
as well as a larger tumor size and higher MIB-1 index in
SCF-positive cases. The present data, together with these findings,
suggest that SCF may be a potential marker for GIST
proliferation.
There are two mechanisms of activation of KIT in
malignant tumors. One is autophosphorylation of KIT due to
gain-of-function mutations of the c-kit gene, and the other is
ligand-dependent activation. The present study did not detect a
correlation between SCF expression and the status of KIT
mutation in GISTs. These data suggest that SCF-induced KIT
activation is an independent mechanism in GISTs, regardless of KIT
autophosphorylation. A previous study (22) demonstrated that the fraction of
activated KIT was not correlated with the fraction of mutant KIT in
GISTs. The absence of a correlation may be explained by
ligand-dependent SCF/KIT signaling in GISTs.
SCF stimulation of GIST544 cells, which express a
heterozygous c-kit exon 9 mutation, induces stronger KIT
phosphorylation (23). SCF
treatment of GIST882 cells, which contain a homozygous c-kit exon
13 mutation, does not induce high levels of KIT phosphorylation
(24). One isoform of KIT that
contains a 4 amino acid sequence, GNNK, has been demonstrated to be
the most abundant isoform in GISTs (25). Following stimulation with SCF,
wild-type GNNK-negative KIT induced an improvement in cell survival
and stronger proliferation (26).
In the present study, we also demonstrated that SCF was capable of
inducing GIST cell proliferation in vitro. Additionally, a
high level of KIT phosphorylation in SCF-stimulated cells was
observed. All GIST cells were verified to contain heterozygous,
functional mutations of c-kit. Thus, this study has demonstrated
that SCF may activate wild-type KIT in GISTs and participate in the
activation of heterozygous KIT mutants.
Notably, the results of the present study
demonstrated increased SCF expression levels in imatinib-treated
GIST cells. Tumor growth is complicated and multiple signaling
pathways are involved in the survival and proliferation of tumor
cells. Therefore, it is possible that in GISTs, after activation of
the mutant KIT is inhibited by imatinib, other compensatory signals
may become involved in maintaining tumor cell survival. As a
result, increased SCF expression levels may be a response that
facilitates preservation of the wild-type KIT signaling in GISTs. A
previous study (27) demonstrated
that mutant KIT was mainly retained within the endoplasmic
reticulum and Golgi compartments in an immature constitutively
phosphorylated form, whereas wild-type KIT was expressed at the
plasma membrane in a mature non-phosphorylated form.
Imatinib-induced inhibition of the phosphorylation of mutant KIT
proteins resulted in the restoration of KIT expression at the cell
surface. Our data, together with those findings, suggest that
increased expression of SCF and KIT at the cell surface due to
imatinib treatment results in abundant SCF/KIT signaling
activation. Although the inhibitor imatinib blocked the
ligand-independent signaling of KIT, it simultaneously enhanced the
ligand-dependent signaling. Negri et al(28) also demonstrated that the surgical
samples of imatinib-treated GISTs were characterized by high
expression levels and activation of the wild-type KIT receptor,
together with high expression levels of its ligand. These findings
provide a favorable compliment to our study in vivo.
In this study, we verified the inefficiency of
imatinib for the inhibition of KIT phosphorylation stimulated by
SCF in vitro. In clinical studies, it has been confirmed
that GISTs with no KIT mutations demonstrate resistance to
imatinib. A clinical study (29)
also found that there was no clinical efficacy of imatinib in uveal
melanomas expressing SCF/KIT without mutations. These results
suggest that the ligand-dependent activation of KIT is likely to
have primary resistance to imatinib, while the stronger
ligand-independent activation of wild-type KIT due to imatinib
treatment may contribute to the development of acquired
resistance.
In conclusion, the expression of SCF suggests an
autocrine mechanism in GISTs. It is likely that ligand-independent
KIT activation due to gain-of-function mutations in the KIT
gene is the main mechanism of GIST oncogenesis, whereas the
ligand-dependent activating mechanism is the crucial reason for
tumor proliferation. The activation of SCF/KIT signaling may be a
considerable contributing factor in imatinib resistance. Our study
suggests that the simultaneous inhibition of ligand-dependent and
ligand-independent activation of KIT may be a more effective
strategy for GIST therapy.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (Grant No.s
30700809 and 30972876), awarded to Professor Da-Lie Ma.
References
|
1.
|
Rousset D, Agnès F, Lachaume P, André C
and Galibert F: Molecular evolution of the genes encoding receptor
tyrosine kinase with immunoglobulin like domains. J Mol Evol.
41:421–429. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Anderson DM, Lyman SD, Baird A, Wignall
JM, Eisenman J, Rauch C, March CJ, Boswell HS, Gimpel SD and Cosman
D: Molecular cloning of mast cell growth factor, a hematopoietin
that is active in both membrane bound and soluble forms. Cell.
63:235–243. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Miyazawa K, Williams DA, Gotoh A,
Nishimaki J, Broxmeyer HE and Toyama K: Membrane-bound steel factor
induces more persistent tyrosine kinase activation and longer life
span of c-kit gene-encoded protein than its soluble form. Blood.
85:641–649. 1995.PubMed/NCBI
|
|
4.
|
Cohen PS, Chan JP, Lipkunskaya M, Biedler
JL and Seeger RC: Expression of stem cell factor and c-kit in human
neuroblastoma. The children’s cancer group. Blood. 84:3465–3472.
1994.
|
|
5.
|
Hassan S, Kinoshita Y, Kawanami C, Kishi
K, Matsushima Y, Ohashi A, Funasaka Y, Okada A, Maekawa T, He-Yao W
and Chiba T: Expression of proto-oncogene c-kit and its ligand stem
cell factor (SCF) in gastric carcinoma cell lines. Dig Dis Sci.
43:8–14. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hines SJ, Organ C, Kornstein MJ and
Krystal GW: Coexpression of the c-kit and stem cell factor genes in
breast carcinomas. Cell Growth Differ. 6:769–779. 1995.PubMed/NCBI
|
|
7.
|
Inoue M, Kyo S, Fujita M, Enomoto T and
Kondoh G: Coexpression of the c-kit receptor and the stem cell
factor in gynecological tumors. Cancer Res. 54:3049–3053.
1994.PubMed/NCBI
|
|
8.
|
Krystal GW, Hines SJ and Organ CP: Organ.
Autocrine growth of small cell lung cancer mediated by coexpression
of c-kit and stem cell factor. Cancer Res. 56:370–376.
1996.PubMed/NCBI
|
|
9.
|
Pietsch T, Kyas U, Steffens U, Yakisan E,
Hadam MR, Ludwig WD, Zsebo K and Welte K: Effects of human stem
cell factor (c-kit ligand) on proliferation of myeloid leukemia
cells: Heterogeneity in response and synergy with other
hematopoietic growth factors. Blood. 80:1199–206. 1992.PubMed/NCBI
|
|
10.
|
Toyota M, Hinoda Y, Takaoka A, Makiguchi
Y, Takahashi T, Itoh F, Imai K and Yachi A: Expression of c-kit and
kit ligand in human colon carcinoma cells. Tumour Biol. 14:295–302.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kindblom LG, Remotti HE, Aldenborg F and
Meis-Kindblom JM: Gastrointestinal pacemaker cell tumor (GIPACT):
gastrointestinal stromal tumors show phenotypic characteristics of
the interstitial cells of Cajal. Am J Pathol. 152:1259–1269.
1998.
|
|
12.
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, Kiese B, et al: Kinase mutations and
imatinib response in patients with metastatic gastrointestinal
stromal tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
Muhammad Tunio G, et al: Gain-of-function mutations of c-kit in
human gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Rubin BP, Singer S, Tsao C, Duensing A,
Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri
GD, et al: KIT activation is a ubiquitous feature of
gastrointestinal stromal tumors. Cancer Res. 61:8118–8121.
2001.PubMed/NCBI
|
|
15.
|
Wardelmann E, Losen I, Hans V, Neidt I,
Speidel N, Bierhoff E, Heinicke T, Pietsch T, Büttner R and
Merkelbach-Bruse S: Deletion of Trp-557 and Lys-558 in the
juxtamembrane domain of the c-kit protooncogene is associated with
metastatic behavior of gastrointestinal stromal tumors. Int J
Cancer. 106:887–895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Emile JF, Théou N, Tabone S, Cortez A,
Terrier P, Chaumette MT, Julié C, Bertheau P, Lavergne-Slove A,
Donadieu J, Barrier A, et al: A clinicopathologic, phenotypic, and
genotypic characteristics of gastrointestinal mesenchymal tumors.
Clin Gastroenterol Hepatol. 2:597–605. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Debiec-Rychter M, Sciot R, Le Cesne A,
Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S,
Stul M, Casali PG, Zalcberg J, et al EORTC Soft Tissue and Bone
Sarcoma Group; Italian Sarcoma Group; Australasian GastroIntestinal
Trials Group: KIT mutations and dose selection for imatinib in
patients with advanced gastrointestinal stromal tumours. Eur J
Cancer. 42:1093–1103. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Heinrich MC, Corless CL, Blanke CD,
Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M,
Fletcher CD, Sandau K, McDougall K, et al: Molecular correlates of
imatinib resistance in gastrointestinal stromal tumors. J Clin
Oncol. 24:4764–4774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Demetri GD, von Mehren M, Blanke CD, Van
den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA,
Singer S, Janicek M, Fletcher JA, et al: Efficacy and safety of
imatinib mesylate in advanced gastrointestinal stromal tumors. N
Engl J Med. 347:472–480. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bono P, Krause A, von Mehren M, Heinrich
MC, Blanke CD, Dimitrijevic S, Demetri GD and Joensuu H: Serum KIT
and KIT ligand levels in patients with gastrointestinal stromal
tumors treated with Imatinib. Blood. 103:2929–2935. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Hirano K, Shishido-Hara Y, Kitazawa A,
Kojima K, Sumiishi A, Umino M, Kikuchi F, Sakamoto A, Fujioka Y and
Kamma H: Expression of stem cell factor (SCF), a KIT ligand, in
gastrointestinal stromal tumors (GISTs): a potential marker for
tumor proliferation. Pathol Res Pract. 204:799–807. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Théou-Anton N, Tabone S, Brouty-Boyé D,
Saffroy R, Ronnstrand L, Lemoine A and Emile JF: Co expression of
SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an
autocrine/paracrine mechanism. Br J Cancer. 94:1180–1185.
2006.PubMed/NCBI
|
|
23.
|
Duensing A, Medeiros F, McConarty B,
Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD and
Fletcher JA: Mechanisms of oncogenic KIT signal transduction in
primary gastrointestinal stromal tumors (GISTs). Oncogene.
23:3999–4006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lux ML, Rubin BP, Biase TL, Chen CJ,
Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD and Fletcher
JA: KIT extracellular and kinase domain mutations in
gastrointestinal stromal tumors. Am J Pathol. 156:791–795. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Théou N, Tabone S, Saffroy R, Le Cesne A,
Julié C, Cortez A, Lavergne-Slove A, Debuire B, Lemoine A and Emile
JF: High expression of both mutant and wild-type alleles of c-KIT
in gastrointestinal stromal tumors. Biochim Biophys Acta.
1688:250–256. 2004.PubMed/NCBI
|
|
26.
|
Pedersen M, Rönnstrand L and Sun J: The
c-Kit/D816V mutation eliminates the differences in signal
transduction and biological responses between two isoforms of
c-Kit. Cell Signal. 21:413–418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Tabone-Eglinger S, Subra F, El Sayadi H,
Alberti L, Tabone E, Michot JP, Théou-Anton N, Lemoine A, Blay JY
and Emile JF: KIT mutations induce intracellular retention and
activation of an immature form of the KIT protein in
gastrointestinal stromal tumors. Clin Cancer Res. 14:2285–2294.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Negri T, Bozzi F, Conca E, Brich S,
Gronchi A, Bertulli R, Fumagalli E, Pierotti MA, Tamborini E and
Pilotti S: Oncogenic and ligand-dependent activation of KIT/PDGFRA
in surgical samples of imatinib-treated gastrointestinal stromal
tumours (GISTs). J Pathol. 217:103–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hofmann UB, Kauczok-Vetter CS, Houben R
and Becker JC: Overexpression of the KIT/SCF in uveal melanoma does
not translate into clinical efficacy of imatinib mesylate. Clin
Cancer Res. 15:324–329. 2009. View Article : Google Scholar : PubMed/NCBI
|