Introduction
Treatment options for metastatic renal cell
carcinoma (RCC) have been limited because tumors are inherently
chemotherapy-and radiotherapy-resistant (1). Based on the importance of immune
mechanisms in the regulation of tumor growth and progression in
metastatic RCC, the therapeutic potential of immunotherapeutic
agents such as interleukin-2 (IL2) and interferon-α (IFNα) has been
evaluated. However, only a limited subset of patients benefit from
cytokine therapy with objective response rates (ORR) of up to 20%
(2). New treatment strategies for
metastatic RCC have therefore been investigated. One of these is to
block the signals triggered by angiogenic growth factors, such as
vascular endothelial growth factor (VEGF) and platelet-derived
growth factor (PDGF) (3). Three
angiogenesis-targeting agents have been developed clinically for
the management of RCC, namely, the monoclonal antibody bevacizumab
(Avastin) and the small-molecule multi-kinase inhibitors (MKIs)
sorafenib tosylate (Nexavar) and sunitinib malate (Sutent).
Sorafenib is an oral MKI that inhibits VEGF
receptors (VEGFRs) 1–3, PDGF receptor (PDGFR)-β, and the serine
threonine kinase Raf-1, which acts through the mitogen-activated
protein kinase/extracellular signal-regulated kinase
kinase/extracellular signal-regulated kinase (Raf/MEK/ERK)
signaling pathway and plays a crucial role in cell proliferation
and tumorigenesis (4). Sunitinib is
an oral MKI against several tyrosine kinase receptors, including
VEGFRs 1–3, PDGFR-β, stem cell factor receptor (KIT), and FMS-like
tyrosine kinase-3 (5). Preclinical
studies have shown that sorafenib and sunitinib significantly
inhibited tumor growth in various carcinomas through the mediation
of suppressed angiogenesis and direct antitumor effects (1). Several phase II and III studies of
sorafenib and sunitinib revealed that each of these agents is
effective as a monotherapy in cytokine-refractory, metastatic RCC
(1,5–7). In a
pivotal phase III trial of sorafenib, 905 patients were randomly
assigned to receive sorafenib (400 mg, orally, twice daily) versus
the placebo, and the trial investigators demonstrated the efficacy
and safety of sorafenib treatment for advanced RCC (7). A phase III trial evaluating first-line
sunitinib (50 mg, orally, once daily for 4 weeks, followed by 2
weeks without treatment) versus IFNα (9 million units 3 times
weekly) in 750 patients with metastatic clear cell RCC demonstrated
statistically significant improvements in the clinical outcomes
with sunitinib compared to those with IFNα (8). However, for further improvement of
prognosis, agents with different mechanisms of action such as
chemotherapeutic agents are required. In addition to promising
preclinical and clinical data culminating in the approval of
sorafenib and sunitinib, their mechanism of action, good safety and
tolerability indicate that they may be a useful treatment option in
combination with conventional chemotherapy for advanced solid
cancers. Several studies have evaluated the effect of sorafenib or
sunitinib in combination with a variety of anticancer agents in
various types of tumor (9–14).
RCC is highly resistant to conventional
chemotherapy: vinblastine has been reported to achieve a 6–9% ORR,
and 5-fluorouracil (5FU) achieved a 5–8% ORR (15). Response rates of immunochemical
therapies combining 5FU with IL2 and IFNα ranged from 1.8 to 48%
(16,17). Recently, a phase II multicenter
trial involving 45 patients with metastatic RCC revealed the
efficacy and safety of S-1, an oral fluorinated pyrimidine that
includes tegafur, a prodrug of 5FU (18). These data suggest that 5FU is likely
a good candidate for combination therapy with molecular-targeting
agents. However, there are currently few reports addressing the
activity of 5FU in combination with sorafenib and sunitinib.
In this study, we evaluated the therapeutic activity
of two MKIs, sorafenib and sunitinib, in combination with 5FU in
vitro and in vivo. As 5FU acts via different mechanisms
from those of MKIs, we hypothesized that combination therapy
exploiting the antiangiogenic and antiproliferative properties of
MKIs along with the cytotoxic properties of 5FU is likely to
provide beneficial results.
Materials and methods
Cell culture
The three established human RCC cell lines, ACHN,
Caki-1 and Caki-2, were obtained from the ATCC (Manassas, VA, USA).
Cells were maintained in RPMI-1640 growth medium (Nissui, Tokyo,
Japan) supplemented with 10% fetal bovine serum (ICN Biomedicals,
Aurora, OH, USA), 100 U/ml penicillin, and 100 μg/ml
streptomycin (Gibco, Grand Island, NY, USA) in a standard
humidified incubator at 37°C in an atmosphere of 5% CO2.
The study was approved by the ethics committee of Nara Medical
Unversity.
Antitumor reagents
5FU (Sigma-Aldrich, St. Louis, MO, USA) and the 2
MKIs, sorafenib tosylate and sunitinib malate (LC Laboratories,
Woburn, MA, USA), were dissolved in dimethyl sulfoxide (DMSO) at a
concentration of 50, 200 and 40 mg/ml, respectively. The stock
solutions were stored at −20°C prior to use.
Cell viability assay
Cells were seeded in a 96-well plate at a density of
2000 cells/well in growth medium, incubated for 24 h and treated
with the indicated concentrations of 5FU, sorafenib, and sunitinib,
alone or in combination. Following incubation of the plates for 72
h, a cell viability assay was performed using the Cell Counting
Kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s
instructions. Sensitivity against antitumor reagents was expressed
as the 50% inhibitory concentration (IC50) as described previously
(19). The data were expressed as
relative values to untreated cells, which were set to 100.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Cells were seeded in 6-well plates at a density of
1×105 cells/well in growth medium and incubated for 24
h. Total RNA was extracted using the RNeasy mini kit (Qiagen,
Hilden, Germany). Total RNA (1 μg) was reverse transcribed
in a final volume of 20 μl with the High Capacity cDNA
Reverse Transcription Kit (Applied Biosystems, Foster City, CA,
USA). Primer sequences used in this study and the annealing
temperatures are shown in Table I.
PCR was performed with cDNA, 0.2 μM of each primer, and 10
μl of AmpliTaq Gold® PCR Master Mix (Applied
Biosystems) under the following conditions: denaturation at 95°C
for 10 min; 35 cycles of 96°C for 3 sec, annealing temperature (as
listed in Table I) for 3 sec, and
68°C for 15 sec; and a final extension at 72°C for 10 sec. PCR
products were then electrophoresed in 1.5% agarose gel and
visualized by a transilluminator. Subsequent to verifying the mRNA
expression of PDGFR-β, semi-quantitative RT-PCR for this gene was
performed in the three cell lines. The pixel intensity for each
band was determined using the ImageJ program (NIH Image, Bethesda,
MD, USA) and normalized to the amount of hypoxanthine
phosphoribosyltransferase (HPRT).
 | Table ISequence of primers used for reverse
transcription-polymerase chain reaction. |
Table I
Sequence of primers used for reverse
transcription-polymerase chain reaction.
| Primer | Sequence | Annealing temperature
(°C) | Fragment size
(bp) |
|---|
| VEGFR-1 |
5′-TCATGAATGTTTCCCTGCAA
5′-GGAGGTATGGTGCTTCCTGA | 55 | 123 |
| VEGFR-2 |
5′-GGTGTTTTGCTGTGGGAAAT
5′-AAACGTGGGTCTCTGACTGG | 60 | 186 |
| VEGFR-3 |
5′-CCCACGCAGACATCAA
5′-TGCACAACTCCACGA | 50 | 380 |
| PDGFR-α |
5′-CTCCTGAGAGCATCTTTGAC
5′-AAGTGGAAGGAACCCCTCGA | 50 | 712 |
| PDGFR-β |
5′-AATGTCTCCAGCACCTTCGT
5′-AGCGGATGTGGTAAGGCATA | 58 | 688 |
| HPRT |
5′-GTTGGATATAAGCCAGACTTTGTTG
5′-ACTCAACTTGAACTCTCATCTTAGGC | 55 | 164 |
Immunocytochemical (ICC) staining
Cells were seeded at a density of 50,000 cells/well
in a Lab-Tek II 4-well Chamber Slide (Nalge Nunc International,
Rochester, NY, USA) and incubated in growth medium for 24 h. To fix
the cells, the slides were immersed in 4% paraformaldehyde solution
(Wako, Osaka, Japan) for 20 min at 4°C. ICC staining was performed
with a streptavidin-biotin complex method using the Histofine
SAB-PO kit (Nichirei Co., Tokyo, Japan), according to the
manufacturer’s instructions. To detect the cell expression of
PDGFR-β, mouse monoclonal anti-PDGFR-β (clone 28; BD Transduction
Laboratories, San Diego, CA, USA) was used as the primary antibody.
The specificity of the antibody was assessed by performing a
secondary antibody-only control experiment. Slides were
counterstained with Meyer’s hematoxylin (Muto Chemical, Tokyo,
Japan) and mounted with malinol (Muto Chemical).
Treatment in mouse xenograft models
Animal experiments for this study were approved by
the institutional animal care and use committee at Nara Medical
University. Female athymic BALB/c nu/nu mice (8 weeks old) were
maintained under pathogen-free conditions and provided with sterile
food and water. Caki-1 (2×106) in 100 μl
RPMI-1640 and 100 μl of Matrigel (Becton Dickinson, Bedford,
MA, USA) were injected subcutaneously into each mouse. Fourteen
days following inoculation of the cells, the animals were divided
randomly into 6 groups (placebo, 5FU, sorafenib, sunitinib, 5FU
plus sorafenib, and 5FU plus sunitinib), and treatment was
initiated. MKIs were administered orally once daily using a
disposable soft catheter tube (Fuchigami Co., Kyoto, Japan), and
5FU was injected intraperitoneally for 5 consecutive days each week
for 4 weeks. Doses of sorafenib, sunitinib and 5FU were 15, 20 and
8 mg/kg, respectively. Control mice received vehicle alone on the
same schedule. Tumor diameters were measured twice per week with
electronic calipers, and tumor volumes were calculated using the
formula [(width)2 × length] / 2 (mm3). The
weight of mice was measured once each week. The mice were
sacrificed on day 42, and the tumors were resected and subjected to
immunohistochemical (IHC) analysis.
IHC analysis of xenograft tumors
Tumors were fixed in 10% formaldehyde solution and
embedded in paraffin. IHC staining was performed as previously
described (18). The primary
antibodies and incubation conditions were mouse monoclonal
anti-Ki-67 (clone MIB-1, Dako Japan, Kyoto, Japan) in ready-to-use
form at room temperature for 30 min and rabbit polyclonal
anti-PECAM1 (CD31) (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
1:500 dilution, 4°C overnight. For verification purposes, some
tumor cells were counterstained with hematoxylin and eosin.
Microvessel density (MVD) analysis
MVD of xenograft tumors was determined as described
previously (20). Briefly, slides
that were stained with anti-CD31 antibody were scanned at a low
magnification (x40) to identify the highest MVD. The number of
stained blood vessels in each of these areas was estimated using a
high magnification field (x200). Subsequently, the MVD score was
calculated as the mean.
Statistical analysis
PRISM software version 5.00 (GraphPad Software, San
Diego, CA, USA) was used for drawing graphs and statistical
analyses. P<0.05 was considered to be statistically
significant.
Results
Sensitivity against antitumor reagents in
RCC cell lines
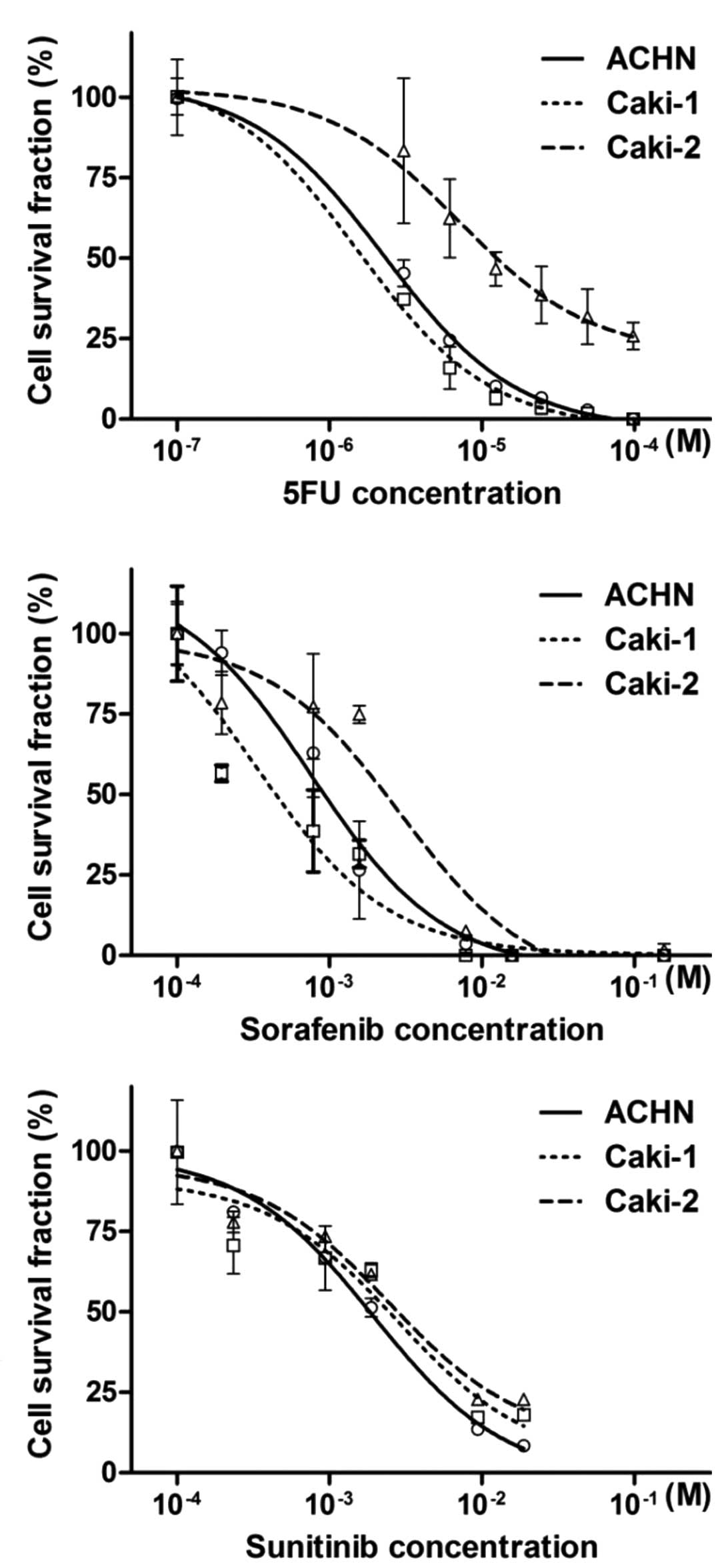
The sensitivity of RCC cell lines to the three
antitumor reagents, 5FU, sorafenib and sunitinib, was determined by
the cell viability assays (Fig.
1A-C), and the sensitivity of the three cell lines was compared
by calculating the IC50 and 95% confidence interval (95% CI) of the
antitumor reagents (Table II). The
IC50 for the treatment with 5FU and sorafenib was significantly
higher in Caki-2 cells than that in ACHN and Caki-1. The IC50s of
sunitinib in ACHN, Caki-1, and Caki-2 were 1.95, 2.80, and 2.48,
respectively. Taken together, these results show that Caki-2 cells
are more resistant to the antitumor reagents than ACHN and Caki-1
cells.
| Table IIAntitumor reagent concentration for
50% cell survival of renal cell carcinoma cell lines. |
Table II
Antitumor reagent concentration for
50% cell survival of renal cell carcinoma cell lines.
| Antitumor
reagent | ACHN | Caki-1 | Caki-2 |
|---|
|
|
|
|---|
| IC50 | 95% CI | IC50 | 95% CI | IC50 | 95% CI |
|---|
| 5FU
(μM) | 2.28 | 1.99–2.62 | 1.55 | 1.23–1.96 | 7.09 | 3.42–14.7 |
| Sorafenib
(μM) | 0.76 | 0.46–1.24 | 0.33 | 0.14–0.81 | 2.91 | 1.43–5.92 |
| Sunitinib
(μM) | 1.95 | 1.39–2.72 | 2.80 | 1.36–4.52 | 2.48 | 0.98–8.02 |
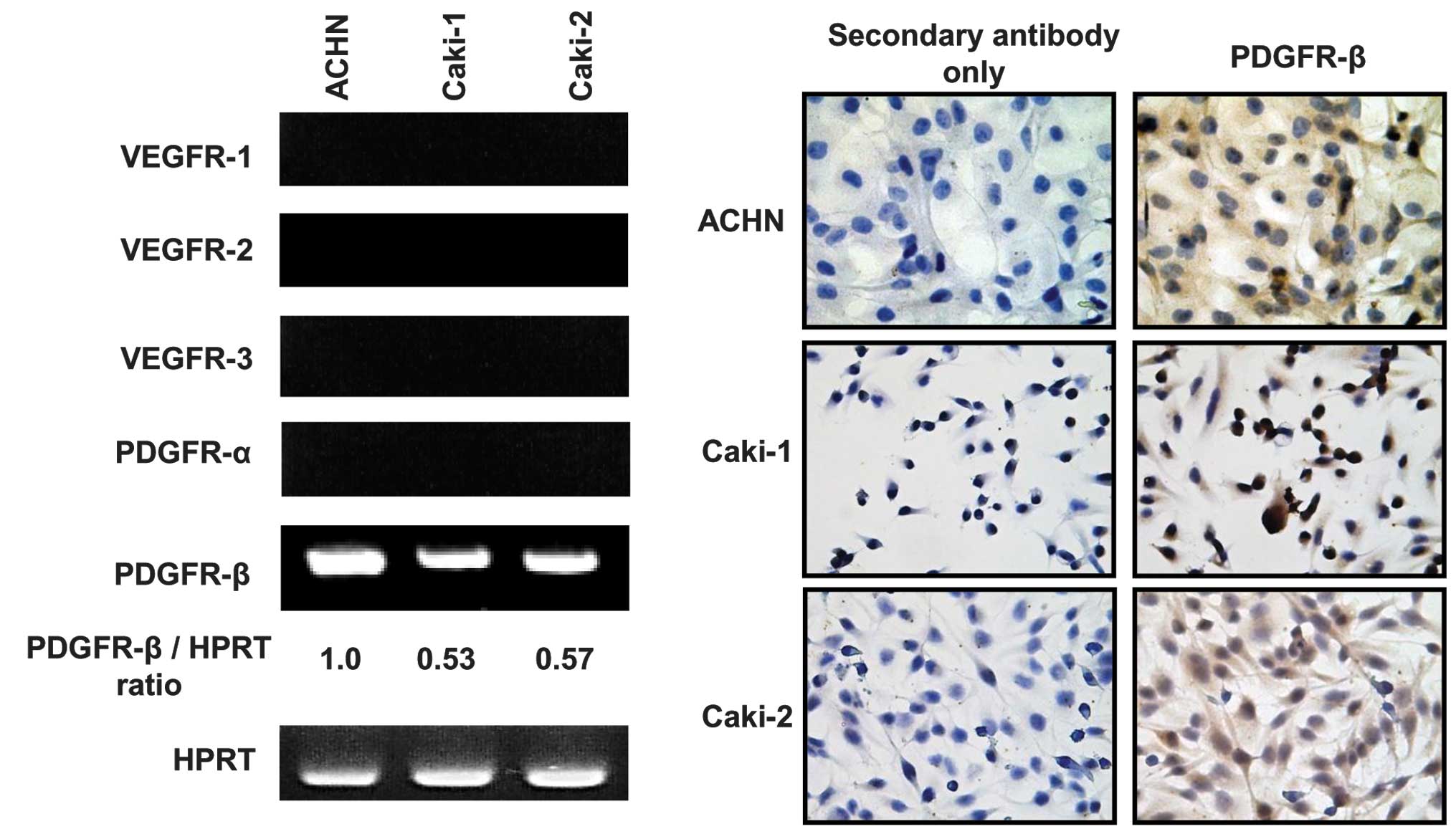
Expression of receptor tyrosine kinases
in RCC cell lines
Sorafenib and sunitinib are capable of inhibiting
multiple receptor tyrosine kinases (RTKs) (4,5).
RT-PCR, performed to investigate the expression of RTKs in RCC cell
lines (Fig. 2A), demonstrated that
PDGFR-β was expressed in the three cell lines, whereas the
expression of VEGFR 1–3 and PDGFR-α was not observed in the cell
lines used in our study. Densitometric analysis revealed that the
PDGFR-β expression levels in ACHN, Caki-1 and Caki-2 cells were
1.0, 0.53 and 0.57, respectively (Fig.
2A). There was a tendency of a positive correlation between the
sensitivity to sunitinib and PDGFR-β expression levels (p=0.09,
Pearson’s correlation). IHC staining was performed in
logarithmically growing cells to verify the protein expression of
PDGFR-β. A strong expression of PDGFR-β was detected in the cell
membrane of the three RCC cell lines (Fig. 2B). These results suggest that
PDGFR-β is a key target molecule for predicting the effect of
sunitinib in RCC.
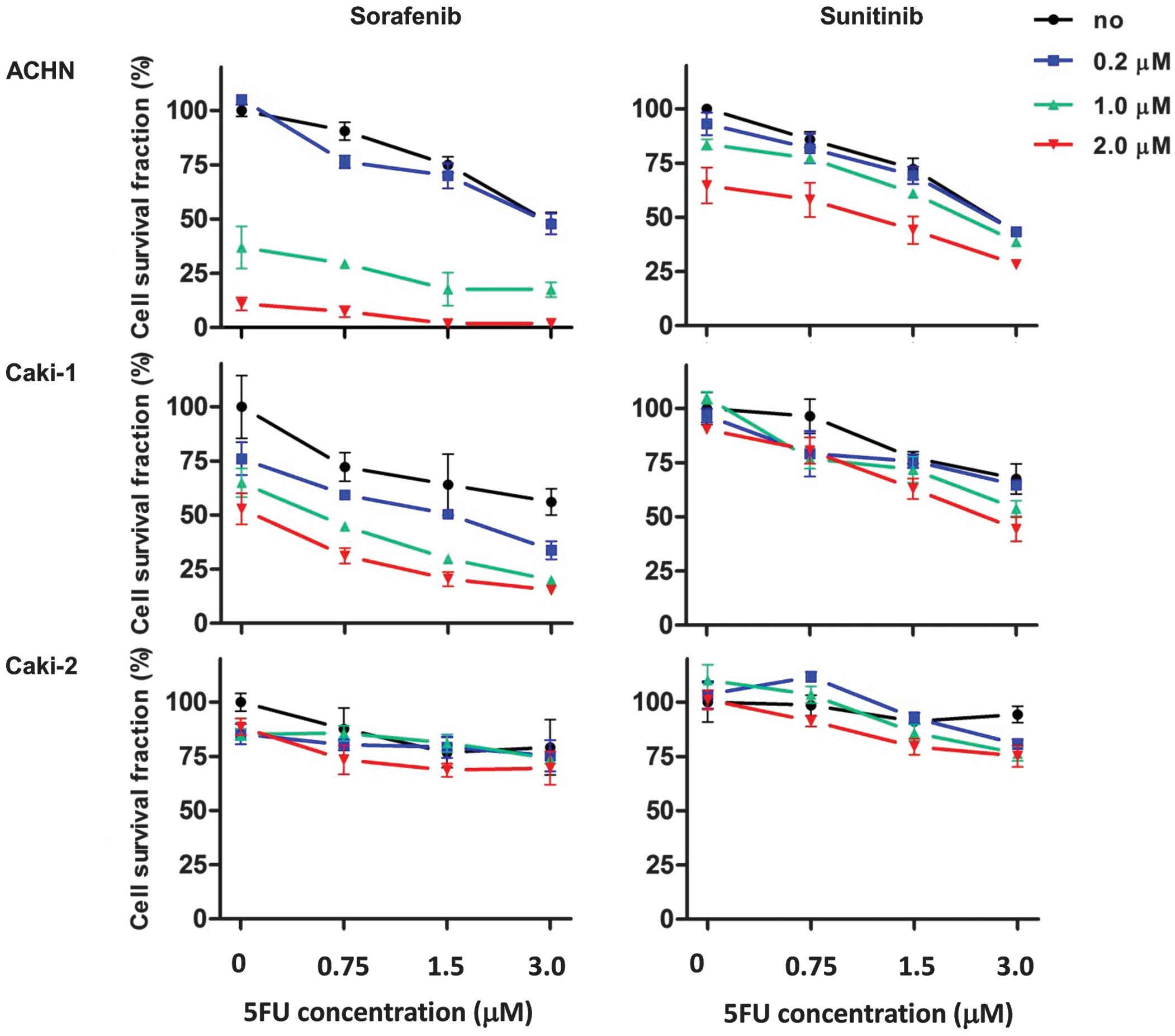
Combination of 5FU with MKIs in
vitro
To examine whether 5FU enhanced the effect of MKIs
when used in combination, cell viability assays were carried out
using various concentrations of antitumor agents (Fig. 3). The treatment concentrations of
5FU were 0.75, 1.5 and 3.0 μM. The treatment concentrations
of sorafenib and sunitinib were 0.2, 1.0 and 2.0 μM. In ACHN
and Caki-1 cells, MKI treatment combined with 5FU led to additive
growth inhibitory effects (Fig. 3,
upper and middle panels). The effect of the combination treatment
with MKIs and 5FU was lower in Caki-2 cells than in ACHN and Caki-1
cells.
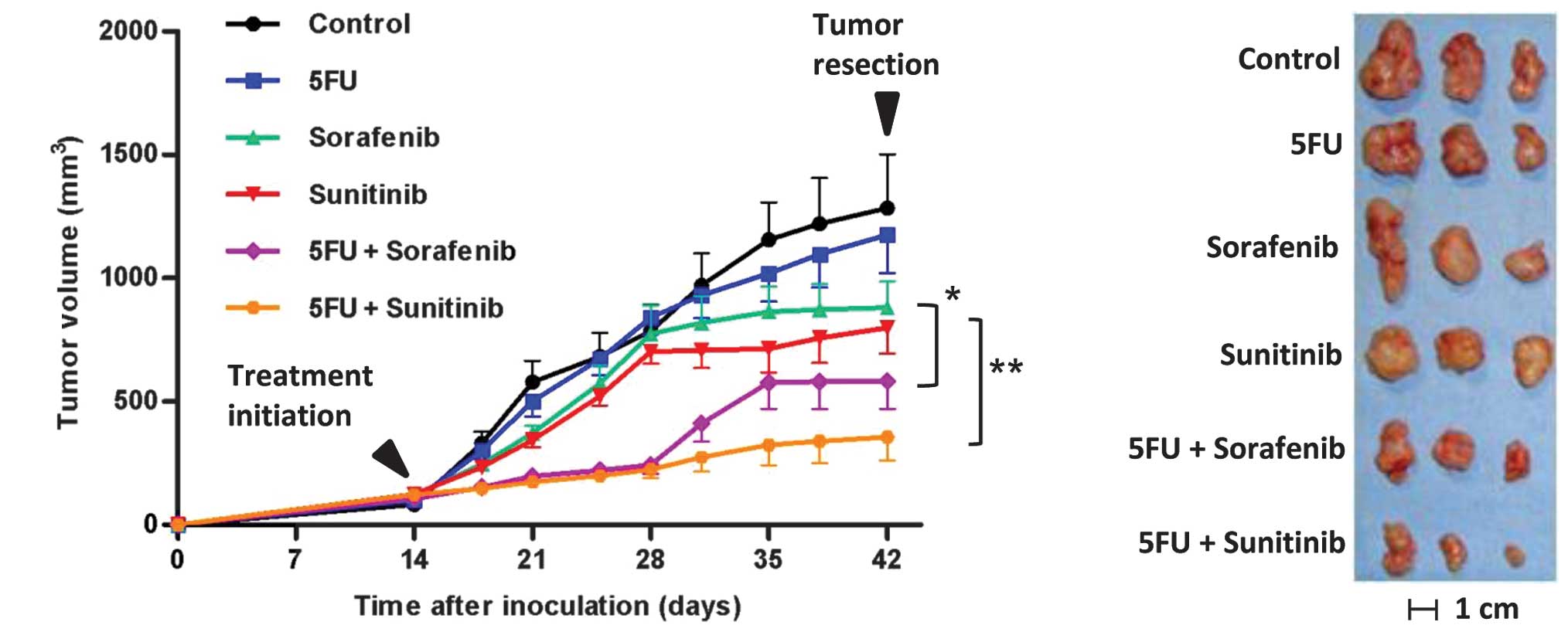
Combination effect of 5FU with MKIs in
Caki-1 xenograft tumors
Since our aim was to establish a new therapeutic
strategy for advanced metastatic clear cell RCC, we selected the
Caki-1 cell line, which is derived from skin metastases of clear
cell RCC (21), for the in
vivo study. To evaluate the enhanced antitumor effect of MKIs
by 5FU in vivo, xenograft mice bearing Caki-1 cells were
prepared and treated as described in Materials and methods. The
mean tumor volumes (mm3) on day 42 following the initial
treatment of the 6 groups i.e., placebo, 5FU, sorafenib, sunitinib,
5FU plus sorafenib, and 5FU plus sunitinib, were 1284±218,
1175±155, 880±105, 799±105, 583±113 and 355±95, respectively
(Fig. 4A, p=0.03, sorafenib
monotherapy versus sorafenib plus 5FU; p=0.008, sunitinib
monotherapy versus sunitinib plus 5FU). Tumors from mice treated
with MKIs were not only smaller, but also paler than the control
mice tumors (Fig. 4B). There were
no significant differences in the inhibitory effects of sorafenib
versus sunitinib monotherapies or sorafenib plus 5FU versus
sunitinib plus 5FU. Yellow skin discoloration was observed in all
the mice treated with sunitinib. From treatment initiation until
tumor resection, no significant loss of body weight was observed in
any of the groups. These findings suggested that the combination of
MKIs and 5FU is effective and tolerable as a novel therapeutic
modality for metastatic RCC.
Analyses of resected tumors using the
Ki-67 labeling index and MVD
To investigate the cell proliferation and
angiogenesis status of the treated and control tumors, IHC staining
for Ki-67 and CD31 was carried out (Fig. 4C). The Ki-67 labeling index data
revealed that tumors from mice treated with MKIs with and without
5FU had decreased levels of proliferation as compared to the
control mice. Combination treatment with sunitinib and 5FU led to a
significant decrease in the Ki-67 labeling index compared to
sunitinib monotherapy (Fig. 4D).
Assessment of MVD revealed that tumors from mice treated with MKIs
with and without 5FU demonstrated decreased MVD compared to the
control mice (Fig. 4E). The
angiogenesis status was not affected by 5FU alone or in combination
in this in vivo study. The effect of MKI treatment on
PDGFR-β expression in cancer tissues in vivo was assessed by
IHC staining for this receptor, and the results revealed no
differences between tissues resected from the 6 treatment
groups.
Discussion
Approaches that may maximize the clinical benefit of
antitumor therapies should include an evaluation of the appropriate
combination regimens, implementation of novel trial designs and the
identification of predictive markers of response. The different
mechanisms of the antitumor activity of sorafenib/sunitinib and 5FU
and their manageable single-agent safety profiles provide a strong
rationale for combining these agents. The findings of the present
in vivo study demonstrated that 5FU may enhance the
antitumor effect of the two MKIs available in clinical settings
(Fig. 4A).
Several studies have addressed the combination
therapy of sorafenib/sunitinib and chemotherapeutic reagents, such
as platinum, taxane, and gemcitabine (9–14),
whereas only a small number of studies have evaluated the
combination with fluorinated pyrimidine (11,22).
The present findings are consistent with the result reported by
Takeuchi et al (22), who
examined the combination effect of S-1 (an oral fluorinated
pyrimidine) and sorafenib for RCC treatment in vitro and in
tumor-bearing murine models. Synergistic anti-proliferative effects
were observed in vitro and in vivo, and the authors
revealed that suppression of TS and E2F-1 expression contributed to
the synergism. A phase I study, including 73 patients with advanced
solid tumors, assessed the tolerability and effect of sunitinib in
combination with capecitabine, which is an oral fluorinated
pyrimidine (11). There were no
clinically significant pharmacokinetic drug-drug interactions, and
most adverse events, which were mild to moderate in severity, were
manageable with dose reduction and rarely led to study withdrawal.
Partial responses were confirmed in 9 (12.3%) patients. The authors
concluded that the combination of sunitinib and capecitabine
resulted in an acceptable safety profile in patients with advanced
solid tumors. However, further evaluation of sorafenib/sunitinib in
combination with fluorinated pyrimidine antitumor reagents may be
required.
Sorafenib and sunitinib bind reversibly to the ATP
binding site of their target kinases, thereby inhibiting their
catalytic activity. The two inhibitors show significant activity
against neovascularization-related RTKs, such as VEGFR2, VEGFR3,
and PDGFR-β (23,24). Although one of the most noteworthy
target kinases of sorafenib is Raf-1, the contribution of the Raf
kinase inhibitory properties of sorafenib to the antitumor effect
in RCC is not completely understood (1). In the present study, to identify
markers predicting the sensitivity to MKIs, we investigated the
mRNA expression levels of major RTKs in RCC cell lines. Our results
revealed that PDGFR-β was expressed in all the cell lines (Fig. 2). A previous study demonstrated that
PDGFR-β is found in pericytes that surround capillary endothelial
cells, and is involved in stabilizing the vascular endothelium
(23). MKIs exert tumor growth
inhibitory effects through the suppression of neovascularization,
and through direct cytotoxic or cytostatic effects (1). Our findings suggested that the direct
cytotoxic or cytostatic effects of sorafenib/sunitinib in RCC cells
are mediated by a block in the signaling pathway triggered by
PDGFR-β phosphorylation. Additionally, our analysis revealed that
the higher sensitivity to sunitinib tended to be correlated to a
higher PDGFR-β expression. These results suggested that PDGFR-β
expression levels serve as a predictive marker for the efficacy of
sunitinib treatment in RCC. More studies, including analyses of the
phosphorylation levels of PDGFR-β and other protein kinases, are
required to clarify the mechanism underlying the direct cytotoxic
effect of MKIs against RCC cells.
Examination of the sensitivity of the three RCC cell
lines against 5FU and the two MKIs revealed that Caki-2 showed a
higher resistance to 5FU and sorafenib compared to the other two
cell lines, which was not the case with sunitinib (Table II). The multidrug-resistant (MDR)
phenotype has been widely recognized in cancer chemotherapy. One of
the main causes of the MDR phenotype is the overexpression of
P-glycoprotein, encoded by the multidrug resistance gene
(MDR1) and multidrug resistance-associated proteins (MRP)
(23). Findings of a recent study
have shown that of the two MKIs, only sorafenib is a substrate for
MRP2, a clinically significant member of the MRP transporter family
(24). In addition, MRP2 is known
to be linked to resistance against 5FU (25). Considering the evidence from
previous reports, it is suggested that the overexpression of MRP2
in Caki-2 cells was responsible for the high resistance to both 5FU
and sorafenib.
The major antitumor activity of MKIs is mediated by
inhibition of vasculogenesis and angiogenesis, which cannot be
reflected in in vitro experiments. Our in vitro
experiments revealed the additive effect of combination treatment
with 5FU and MKIs in Caki-1 cells (Fig.
2), whereas in vivo experiments showed that the
combination acted synergistically to inhibit the growth of
subcutaneous xenograft tumors of Caki-1 cells (Fig. 4A). Assessment of the Ki-67 labeling
index and MVD confirmed that the combination therapy decreased cell
proliferation as compared to monotherapy (Fig. 4D and E). There is a notable
limitation to the present study. The in vivo data were
generated only in Caki-1 xenograft tumors. The use of other RCC
cell lines is required to accurately assess the clinical potential
of the combination of 5FU and MKIs.
In conclusion, we showed that the combination
therapy of 5FU and sorafenib/sunitinib exerts a synergistic
anti-tumor effect in human RCC cells. Our findings support the
feasibility of the clinical evaluation of combination therapy with
fluorinated pyrimidines and MKIs. The regimen should be modified to
suit clinical needs, and this protocol may provide an effective and
tolerable therapeutic modality for advanced RCC patients.
References
|
1
|
Rini BI: Vascular endothelial growth
factor-targeted therapy in renal cell carcinoma: current status and
future directions. Clin Cancer Res. 13:1098–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagstaff J: Renal cell cancer: is
immunotherapy dead? Ann Oncol. 18(Suppl 9): 94–97. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara N: Vascular endothelial growth
factor as a target for anticancer therapy. Oncologist. 9(Suppl 1):
2–10. 2004. View Article : Google Scholar
|
|
4
|
Wilhelm SM, Carter C, Tang L, et al: BAY
43-9006 exhibits broad spectrum oral antitumor activity and targets
the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in
tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roskoski R Jr: Sunitinib: a VEGF and PDGF
receptor protein kinase and angiogenesis inhibitor. Biochem Biophys
Res Commun. 356:323–328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hutson TE: Targeted therapies for the
treatment of metastatic renal cell carcinoma: clinical evidence.
Oncologist. 16(Suppl 2): 14–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Escudier B, Eisen T, Stadler WM, et al:
Sorafenib for treatment of renal cell carcinoma: final efficacy and
safety results of the phase III treatment approaches in renal
cancer global evaluation trial. J Clin Oncol. 27:3312–3318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Motzer RJ, Hutson TE, Tomczak P, et al:
Overall survival and updated results for sunitinib compared with
interferon alfa in patients with metastatic renal cell carcinoma. J
Clin Oncol. 27:3584–3590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dal Lago L, D’Hondt V and Awada A:
Selected combination therapy with sorafenib: a review of clinical
data and perspectives in advanced solid tumors. Oncologist.
13:845–858. 2008.PubMed/NCBI
|
|
10
|
Ulahannan SV and Brahmer JR:
Antiangiogenic agents in combination with chemotherapy in patients
with advanced non-small cell lung cancer. Cancer Invest.
29:325–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sweeney CJ, Chiorean EG, Verschraegen CF,
Lee FC, Jones S, Royce M, Tye L, Liau KF, Bello A, Chao R and
Burris HA: A phase I study of sunitinib plus capecitabine in
patients with advanced solid tumors. J Clin Oncol. 28:4513–4520.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castillo-Avila W, Piulats JM, Garcia Del
Muro X, Vidal A, Condom E, Casanovas O, Mora J, Germà JR, Capellà
G, Villanueva A and Viñals F: Sunitinib inhibits tumor growth and
synergizes with cisplatin in orthotopic models of
cisplatin-sensitive and cisplatin-resistant human testicular germ
cell tumors. Clin Cancer Res. 15:3384–3395. 2009. View Article : Google Scholar
|
|
13
|
Zhang D, Hedlund EM, Lim S, Chen F, Zhang
Y, Sun B and Cao Y: Antiangiogenic agents significantly improve
survival in tumor-bearing mice by increasing tolerance to
chemotherapy-induced toxicity. Proc Natl Acad Sci USA.
108:4117–4122. 2011. View Article : Google Scholar
|
|
14
|
Yoon CY, Lee JS, Kim BS, Jeong SJ, Hong
SK, Byun SS and Lee SE: Sunitinib malate synergistically
potentiates anti-tumor effect of gemcitabine in human bladder
cancer cells. Korean J Urol. 52:55–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hartmann JT and Bokemeyer C: Chemotherapy
for renal cell carcinoma. Anticancer Res. 19:1541–1543.
1999.PubMed/NCBI
|
|
16
|
Ravaud A, Trufflandier N, Ferrière JM,
Debled M, Palussière J, Cany L, Gaston R, Mathoulin-Pélissier S and
Bui BN: Subcutaneous interleukin-2, interferon alpha-2b and
5-fluorouracil in metastatic renal cell carcinoma as second-line
treatment after failure of previous immunotherapy: a phase II
trial. Br J Cancer. 89:2213–2218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miyake M, Fujimoto K, Tanaka M, Hirao Y,
Uemura H, Otani T and Yoshii M: Immunochemotherapy with
interferon-α, interleukin-2, 5-fluorouracil, and cimetidine for
patients with advanced renal cell carcinoma. J Nara Med Assoc.
60:37–47. 2009.
|
|
18
|
Naito S, Eto M, Shinohara N, Tomita Y,
Fujisawa M, Namiki M, Nishikido M, Usami M, Tsukamoto T and Akaza
H: Multicenter phase II trial of S-1 in patients with
cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol.
28:5022–5029. 2010. View Article : Google Scholar
|
|
19
|
Miyake M, Fujimoto K, Anai S, Ohnishi S,
Nakai Y, Inoue T, Matsumura Y, Tomioka A, Ikeda T, Okajima E,
Tanaka N and Hirao Y: Inhibition of heme oxygenase-1 enhances the
cytotoxic effect of gemcitabine in urothelial cancer cells.
Anticancer Res. 30:2145–2152. 2010.PubMed/NCBI
|
|
20
|
Miyake M, Fujimoto K, Anai S, Ohnishi S,
Kuwada M, Nakai Y, Inoue T, Matsumura Y, Tomioka A, Ikeda T, Tanaka
N and Hirao Y: Heme oxygenase-1 promotes angiogenesis in urothelial
carcinoma of the urinary bladder. Oncol Rep. 25:653–660. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koch I, Depenbrock H, Danhauser-Riedl S,
Rastetter JW and Hanauske AR: Interleukin 1 modulates growth of
human renal carcinoma cells in vitro. Br J Cancer. 71:794–800.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takeuchi A, Shiota M, Tatsugami K,
Yokomizo A, Eto M, Inokuchi J, Kuroiwa K, Kiyoshima K and Naito S:
Sorafenib augments cytotoxic effect of S-1 in vitro and in vivo
through TS suppression. Cancer Chemother Pharmacol. 68:1557–1564.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Naito S, Yokomizo A and Koga H: Mechanisms
of drug resistance in chemotherapy for urogenital carcinoma. Int J
Urol. 6:427–439. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shibayama Y, Nakano K, Maeda H, Taguchi M,
Ikeda R, Sugawara M, Iseki K, Takeda Y and Yamada K: Multidrug
resistance protein 2 implicates anticancer drug-resistance to
sorafenib. Biol Pharm Bull. 34:433–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang EJ and Johnson WW: The farnesyl
protein transferase inhibitor lonafarnib (SCH66336) is an inhibitor
of multidrug resistance proteins 1 and 2. Chemotherapy. 49:303–308.
2003. View Article : Google Scholar : PubMed/NCBI
|