Introduction
Neuroendocrine tumors (NETs) frequently metastasize
to the liver, but it is rare to find them there as primary tumors.
NETs represent 2% of all tumors in the gastrointestinal tract and
they have an annual incidence of 2–3 individuals out of 100,000 per
year, with an overall slight preponderance in females (1). The most common primary sites are the
lung, stomach, small intestine, pancreas, meckel, appendix, colon,
rectum, breast, prostate, ovary, ileum, duodenum and the jejunum
(1,2).
Isolated polycystic liver disease (PCLD) is a rare
autosomal dominant disease with an incidence of <0.01%. The
majority (>80%) of PCLD patients are clinically asymptomatic.
Certain patients develop symptoms of a cystic enlarging liver,
which causes morbidity and mortality (3). In PLCD, the cysts arise from
malformation of the embryonic ductal plate, with formation of von
Meyenburg complexes (hamartomas) that are lined with functional
biliary epithelium (4). There is no
known association between PCLD and neuroendocrine or other
tumors.
The study was approved by the ethics committee of
General Hospital of Naoussa, Naoussa, Greece. Written informed
consent was obtained from the patient’s family.
Case report
A 64-year-old female presented with increasing
abdominal pain over a two-week period. The patient was a
non-smoker, did not consume alcohol and denied any systemic
symptoms of fever, weight loss or anorexia. No cardiac,
neurological or other symptoms were reported. The patient had a
past medical history of isolated PCLD, which was discovered two
years previously, and an ectopic left kidney. No family history of
polycystic kidney or liver disease was reported. A clinical
examination revealed massive hepatomegaly, and this was confirmed
by ultrasonography. No other abnormalities were detected.
Laboratory investigations revealed that the patient
had normal renal function with an estimated glomerular filtration
rate of >60 ml/min. The hemoglobin, white cell count
differentials and coagulation screen were all within the normal
range. Liver function tests revealed raised alkaline phosphatase
and γ-glutamyltransferase levels of level of 475 and 531 IU/l,
respectively, and a slightly low albumin level of 2.2 g/dl with a
total protein level of 6.4 g/dl. The levels of total bilirubin and
alanine transaminase were 2.5 mg/dl and 50 IU/l, respectively.
Tumor marker levels, including those of AFP, CEA, CA 19-9 and CA
125, were not observed to be elevated.
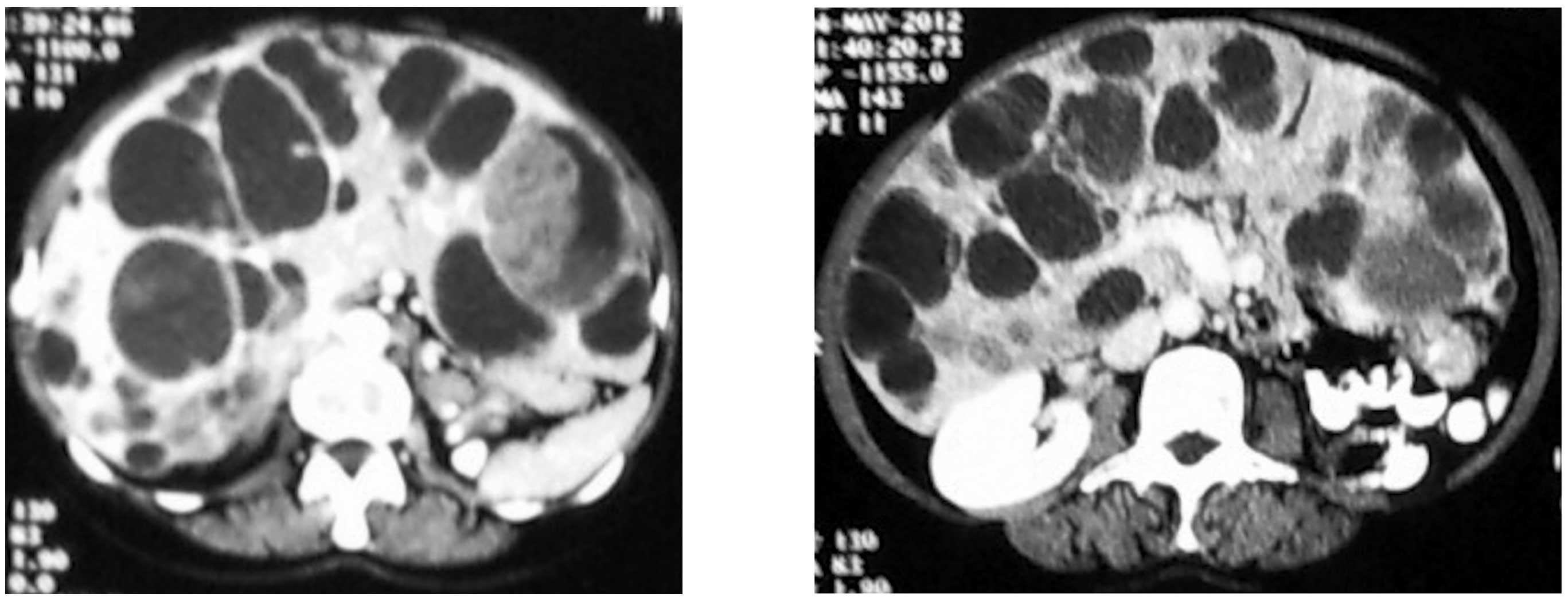
Contrast computed tomography (CT) examination of the
abdomen confirmed massive hepatomegaly and a polycystic liver with
multiple scattered cystic formations. A number of these included
septals, hemorrhagic material and several nodular-compact internal
lesions, which was corroborated following intravenous contrast
injection (Fig. 1).
The patient underwent open surgical biopsy one month
following presentation. The histological examination and
immunohistochemical findings suggested an intermediate grade
neuroendocrine tumor (mitotic index ki-67, 10–15%; positive for
keratin 8/18, CD 56 and synaptophysin).
A 24-h delayed whole-body scintigraphy technique for
the identification and localization of neuroendocrine tumors via
the administration of In-111-labeled OctreoScan was used; however,
no extrahepatic accumulation was observed.
Two weeks after the biopsy, the patient presented
with dyspnea. A large right pleural effusion was confirmed by X-ray
of the chest and triplex, while ultrasound examination of the legs
revealed deep venous thrombosis (DVT). The patient succumbed a few
days later.
Discussion
Despite significant physical examination and
radiological findings, the majority of patients remain asymptomatic
with preserved liver function, and only mild elevations in the
γ-glutamyltransferase and alkaline phosphatase levels are observed
(4). Certain patients with PLCD
will develop symptoms including abdominal pain, distension or early
satiety due to massive hepatomegaly. More rarely, complications of
cysts may include cyst infection, hemorrhage or rupture with
hemoperitoneum, cyst torsion, portal hypertension, hepatic vein
compression or jaundice due to bile duct compression (5,6).
Surgery remains the mainstay of treatment when
patients become symptomatic. Surgical intervention comprises cyst
aspiration and sclerosis, fenestration with and without hepatic
resection and orthotopic liver transplantation (7).
Primary neuroendocrine tumors of the liver are rare.
The frequency of primary NETs reported to occur in the liver or
biliary tract is <1%. Unknown primary sites or uncommon sites
account for 11–14% of cases (8).
Neuroendocrine tumors originate from neuroendocrine cells from the
embryological neural crest. In the gastrointestinal system,
neuroendocrine cells are located from the mouth to the anus,
including in the pancreas (9).
Proposed theories on the origin of neuroendocrine cells that give
rise to primary hepatic neuroendocrine tumors include ectopic
neuroendocrine cells of pancreatic or adrenal origin, and
neuroendocrine cells from within the intrahepatic biliary tree or
from neuroendocrine-programmed ectoblasts (10).
There is no known association between PCLD and
primary or metastatic neuroendocrine tumors. A rare correlation
between PCLD and intracranial meningiomas was recently described,
which was likely to have occurred by chance rather than to
represent a previously unrecognized association between PCLD and
cranial meningioma (11).
Additionally, a case of liver failure in a patient
affected by PCLD and liver metastases from breast carcinoma was
described (12). Extrahepatic
abnormalities in PCLD have been described, such as mitral valve
prolapse and intracranial aneurysms, but there is no known
connection between PCLD and cancer (13).
Patients with neuroendocrine tumors may be evaluated
by computed tomography (CT) or magnetic resonance imaging (MRI),
and the functional status of these tumors is assessed by
physiological imaging via scintigraphy, with agents such as
radiolabeled meta-iodobenzylguanidine (MIBG) or radiolabeled
octreotide (Octreoscan) (14).
An-111-labeled OctreoScan is highly sensitive for the detection of
gastroenteropancreatic neuroendocrine tumors and their metastases,
and provides important aspects in the evaluation of these tumors
for patient management (1).
No studies in the literature describe a patient with
autosomal dominant isolated PCLD and a primary or metastatic
neuroendocrine tumor of the liver. The death of our patient did not
allow for further diagnostic examination, such as a positron
emission tomography (PET) scan, to exclude any primary site other
than the liver. However, the scintigraphy test and the MRI of the
chest and abdomen did not reveal any suspicious regions in organs
other than the liver.
References
|
1
|
Ramage JK, Ahmed A, Ardill J, et al:
Guidelines for the management of gastroenteropancreatic
neuroendocrine (including carcinoid) tumours (NETs). Gut. 61:6–32.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Modlin IM, Kidd M, Latich I, Zikusoka MN
and Shapiro MD: Current status of gastrointestinal carcinoids.
Gastroenterology. 128:1717–1751. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qian Q: Isolated polycystic liver disease.
Adv Chronic Kidney Dis. 17:181–189. 2010. View Article : Google Scholar
|
|
4
|
Bistritz L, Tamboli C, Bigam D and Bain
VG: Polycystic liver disease: experience at a teaching hospital. Am
J Gastroenterol. 100:2212–2217. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arnold HL and Harrison SA: New advances in
evaluation and management of patients with polycystic liver
disease. Am J Gastroenterol. 100:2569–2582. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Everson GT, Taylor MR and Doctor RB:
Polycystic disease of the liver. Hepatology. 40:774–782. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schnelldorfer T, Torres VE, Zakaria S,
Rosen CB and Nagorney DM: Polycystic liver disease: a critical
appraisal of hepatic resection, cyst fenestration, and liver
transplantation. Ann Surg. 250:112–118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hauso O, Gustafsson BI, Kidd M, et al:
Neuroendocrine tumor epidemiology: contrasting Norway and North
America. Cancer. 113:2655–2664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gurung A, Yoshida EM, Scudamore CH, Hashim
A, Erb SR and Webber DL: Primary hepatic neuroendocrine tumour
requiring live donor liver transplantation: case report and concise
review. Ann Hepatol. 11:715–720. 2012.PubMed/NCBI
|
|
10
|
Gravante G, De Liguori Carino N, Overton
J, Manzia TM and Orlando G: Primary carcinoids of the liver: a
review of symptoms, diagnosis and treatments. Dig Surg. 25:364–368.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Neto AB, Zanini MA, DA Silva AP, Winckler
C, Dos Santos RM and Furtado ML: Meningeal tumor: A rare
extrahepatic association in patients with polycystic liver disease
enrolled for liver transplantation. Oncol Lett. 3:1007–1010.
2012.PubMed/NCBI
|
|
12
|
Fadda GM, Santeufemia DA, Cossu-Rocca P,
et al: Fulminant liver failure in a patient affected by polycystic
liver disease and liver metastases from breast carcinoma. Tumori.
95:557–561. 2009.PubMed/NCBI
|
|
13
|
Hoevenaren IA, Wester R, Schrier RW, et
al: Polycystic liver: clinical characteristics of patients with
isolated polycystic liver disease compared with patients with
polycystic liver and autosomal dominant polycystic kidney disease.
Liver Int. 28:264–270. 2008. View Article : Google Scholar
|
|
14
|
Le Duc-Pennec A, Thol C, Cavarec M, et al:
Octreotide imaging plus bone scintigrams to optimally localize
gastroenteropancreatic neuroendocrine tumors. Clin Nucl Med.
28:5–8. 2003.PubMed/NCBI
|