Introduction
Gastric cancer is one of the most common forms of
cancer worldwide with ∼989,600 new cases and 738,000 mortalities
annually, which accounts for ∼8% of new cancers (1). Epidemiological studies of the
incidence and prevalence of gastric cancer indicate that the male
and female ratio is 2:1 (1–3). The gender difference may not be
accounted for by environmental risk factors for gastric cancer,
including Helicobacter pylori infection, smoking and diet
(4–8). The incidence of gastric cancer has
been reported to be higher in males than females prior to
menopause, however, following menopause, the incidence in females
is similar to that of males (9). In
patients with prostate cancer, the risk of developing gastric
cancer has been identified to be lower in individuals treated with
estrogen therapy compared with those who have not received
treatment (10–14). In breast cancer, patients treated
with anti-estrogen tamoxifen have been identified to exhibit a
significantly increased risk of subsequent gastric cancer (15,16).
In addition, ovariectomy also significantly increases the risk of
gastric cancer in females (17).
Taken together, it has been hypothesized that female sex hormones
play a protective role against the development of gastric
cancer.
It has been well established that the functions of
estrogen are mediated by estrogen receptor–α (ER-α) and ER-β
(18). ER-α mainly exists in three
isoforms, namely ER-α66, ER-α46 and ER-α36 (19). ER-α66 functions as a
ligand-dependent transcription factor, regulating gene expression
by binding estrogen response elements (EREs) in DNA (18). ER-α46 lacks an AF-1 domain, however,
it is able to bind to the ERE and form heterodimers with ER-α66
(20,21). ER-α46 is localized to the plasma
membrane, in the cytosol and to the nucleus and mediates rapid
estrogen signaling, including activation of the Src/PI3K/AKT
pathway (22–24), indicating a possible role of ER-α46
in rapid non-genomic estrogen signaling. ER-α36 differs from ER-α66
in that it lacks the two transcriptional activation domains (AF-1
and AF-2) but retains the DNA-binding, dimerization and the
majority of the ligand-binding domains. ER-α36 also mediates rapid
estrogen signaling (19). It is a
paradox that, on the one hand, estrogen is associated with gastric
cancer cells and, on the other hand, the expression of ER-α66 is
low and the presence of ER-β in gastric cancer may have a
protective effect against the invasiveness of gastric cancer
(25,26).
Previously, we reported that ER-α36 protein is
expressed in human gastric adenocarcinoma tissues and gastric
cancer cell lines, and that ER-α36 expression significantly
correlates with tumor invasion and lymph node metastasis in gastric
cancer (27). In the present study,
the underlying mechanisms by which ER-α36 functions in gastric
cancer SGC7901 cells were investigated and the role of the
c-src/cyclin D1 pathway was assessed.
Materials and methods
Reagents
17β-estradiol (E2β) and PP2 (a Src inhibitor) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Rabbit polyclonal
anti-ER-α36 antibody was generated and characterized as described
previously (28). Anti-c-Src
(sc-19), anti-p-c-Src (sc-81521 and sc-16846-R), anti-cyclin D1
(sc-718) and anti-β-actin (sc-47778) antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). RIPA buffer
and the Enhanced BCA Protein Assay kit were from the Beyotime
Institute of Biotechnology (Shanghai, China). PVDF membranes were
purchased from Millipore (Billerica, MA, USA). Lipofectamine 2000
reagent was from Invitrogen Life Technologies (Carlsbad, CA, USA)
and Protein A agarose was from Santa Cruz Biotechnology, Inc.
(sc-2001).
Cell lines
The human gastric cancer cell line, SGC7901, was
obtained from the Chinese Academy of Medical Sciences Cell Center
of Basic Medicine (Beijing, China). Recombinant cell lines (low and
high ER-α36 expression) of gastric cancer SGC7901 cells were
generated in the Pathology and Pathophysiology Key Laboratory of
Wuhan (China) as described previously (27).
Cell culture
All cells were maintained in RPMI-1640 medium
(Invitrogen Life Technologies) containing 10% fetal bovine serum
(FBS) at 37°C in a 5% CO2 atmosphere. Prior to treatment
with E2β, the cells were changed to phenol-red-free RPMI-1640
medium and 2% FBS for 2–3 days and then maintained in serum-free
medium for 6 h prior to experimentation.
Cell proliferation assay
To examine cell growth in the presence or absence of
estrogen, the cells maintained for 3 days in phenol red-free
RPMI-1640 medium plus 2% FBS were treated with E2β (0.1 nM) and/or
PP2 (10 μM) or ethanol vehicle as a control. Following
treatment for 5, 7, 9 and 11 days, the cells were trypsinized and
counted with the Scepter™ 2.0 handheld automated cell counter
(Merck KGaA, Darmstadt, Germany). Assays were performed in 3 dishes
for each time point and all experiments were repeated 3 times.
Western blot analysis
For the western blot analysis, the cells were washed
with cold PBS and lysed in lysis buffer [50 mM Tris-HCl (pH 8.0),
150 mM NaCl, 0.25 mM EDTA (pH 8.0), 0.1% SDS, 1% Triton X-100 and
50 mM NaF] supplemented with protease and phosphatase inhibitors
purchased from Sigma-Aldrich. Protein concentrations were
determined with the Enhanced BCA Protein Assay kit. The cell
lysates were mixed with loading buffer, separated by 12% SDS-PAGE
gels and transferred to a PVDF membrane. The membranes were probed
with various primary antibodies, appropriate secondary antibodies
and visualized with enhanced chemiluminescence detection reagents
(DNR Bio-Imaging Systems Ltd., Jerusalem, Israel). The densities of
the protein bands were assessed using the TotalLab analysis
software (Nonlinear Dynamics Ltd., Durham, NC, USA).
Nude mouse xenograft assay
Male nude mice (BALB/c nu/nu nude mice, 20–25 g)
were purchased from the Hubei Experimental Animal Center (Wuhan,
China). All experimental procedures were approved by the Animal
Care and Use Committee at the School of Medicine (Wuhan University,
Wuhan, China). All experimental procedures were performed in
compliance with the National Institutes of Health guidelines on the
ethical use of animals. The following cell lines were used:
SGC7901, ER-α36 upregulated SGC7901 (High36) and ER-α36-knockdown
SGC7901 (Low36) cells. Cells (∼5×105) suspended in PBS
were dorsally implanted into nude mice subcutaneously. The tumor
volume (V) was measured with a caliper every 4 days and was
calculated as V = length × width (cm2). After 24 days,
all the animals were sacrificed. The tumors were removed and
weighed. All tumor tissues were retained for western blot analysis
and immunohistochemistry (IHC).
IHC
Paraffin-embedded tissue sections (5 μm) were
dewaxed in xylene and rehydrated in graduated concentrations of
ethanol (100, 95, 90, 80 and 70% in PBS, 5 min each solution).
Antigen retrieval was performed by incubating the slides with 100
mM sodium citrate solution (pH 6.0) for 20 min. The tumor tissues
were stained with antibodies against ER-α36, c-Src and cyclin D1,
followed by avidin-biotin-immunoperoxidase visualization. The cell
nuclei were stained with hematoxylin. Positively-stained cells were
observed using an Olympus microscope (Olympus Corporation, Tokyo,
Japan). Immunostained slides were evaluated by two pathologists
independently in a blind manner. In the majority of cases, the
evaluations of the two pathologists were identical. Any
discrepancies were resolved by re-examination and consensus.
Immunoprecipitation (IP)
E2β (0.1 nM) and/or PP2 (10 μM) were applied
for 10 min to stimulate the SGC7901 cells. The cells were washed
with cold PBS and lysed with lysis buffer supplemented with
protease and phosphatase inhibitors. The protein concentrations
were determined using the Enhanced BCA Protein Assay kit. The cell
lysates were used for IP. Briefly, the lysates were mixed with
antibodies against ER-α36, c-src, p416-c-Src and p527-c-Src in IP
buffer [10 g/l HEPES (pH 7.4), 150 g/l NaCl and 0.1% BSA]
supplemented with protease inhibitors and were incubated for 1 h at
4°C with gentle agitation. Protein A sepharose beads were added and
the samples were incubated for 2 h at 4°C using gentle agitation.
Unbound proteins were removed by washing the beads three times in
IP buffer. The bound proteins were eluted from the beads with
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) sample buffer and the IP samples were analyzed with
SDS-PAGE.
Statistical analysis
The statistical analysis was performed using SPSS
12.0 software (SPSS, Inc., Chicago, IL, USA). Data are presented as
the mean ± SD in three replicate samples and compared using the
Student’s t-test and analysis of variance. P<0.05 was considered
to indicate a statistically significant difference. All experiments
were performed at least 3 times to ensure the reproducibility of
the results.
Results
Estrogen stimulates proliferation of
gastric cancer cells via ER-α36
We previously identified that ER-α36 was expressed
in a number of gastric cancer cell lines and tissue specimens from
gastric cancer patients (27). In
connection with these observations, in the present study, the role
of ER-α36-mediated estrogen signaling in the proliferation of
gastric cancer cells was studied. For this purpose, cell lines were
established from the gastric cancer SGC7901 cells that highly
expressed recombinant ER-α36 (High36) or exhibited knocked down
levels of ER-α36 expression (Low36). Next, E2β (0.1 nM) was used to
treat the SCG7901, High36 and Low36 cell lines for various time
periods. E2β was demonstrated to promote the proliferation of these
cells. High36 had the highest growth rate in response to estrogen
treatment and Low36 had the lowest. These results indicate that
ER-α36 mediates the estrogen-stimulated proliferation of gastric
cancer cells.
c-Src is involved in ER-α36-mediated
mitogenic estrogen signaling in gastric cancer cells
To observe the mechanisms by which ER-α36 mediates
the estrogen-stimulated growth of gastric cancer cells, the c-Src
inhibitor, PP2 (10 μM), was used to analyze gastric cancer
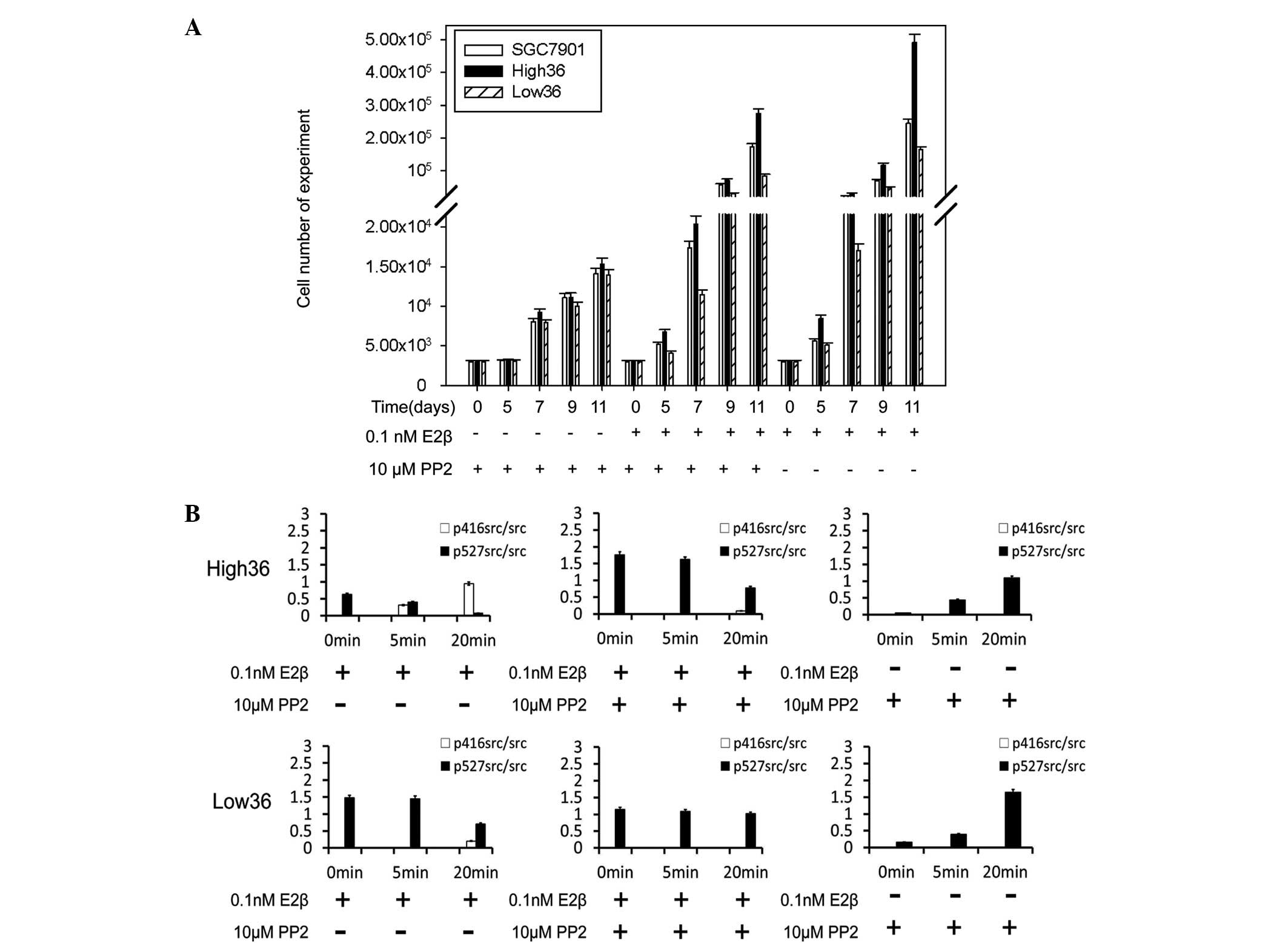
cell (SGC7901) proliferation. PP2 inhibited the cell proliferation
stimulated by E2β in all cell lines (Fig. 1A). PP2 blocked 68.91 and 91.56% of
proliferation in the High36 and Low36 cell lines, respectively
(Fig. 1A). Western blot analysis
using phospho-specific c-Src antibodies revealed that E2β induced
phosphorylation of Tyr416 in c-Src and reduced phosphorylation of
Tyr527 (Fig. 1B). PP2 inhibited the
phosphorylation of Tyr416 induced by E2β and increased the
phosphorylation of Tyr527 (Fig.
1B). The phosphorylation of Tyr416 was shown to correlate with
the expression levels of ER-α36, indicating that c-Src is involved
in the non-genomic estrogen signaling mediated by ER-α36 in gastric
cancer cells.
c-Src is involved in induction of cyclin
D1 expression by estrogen in gastric cancer cells
It is well known that cyclin D1 is an estrogen
responsive gene that contributes to the estrogen-stimulated
proliferation of breast cancer cells. To examine whether
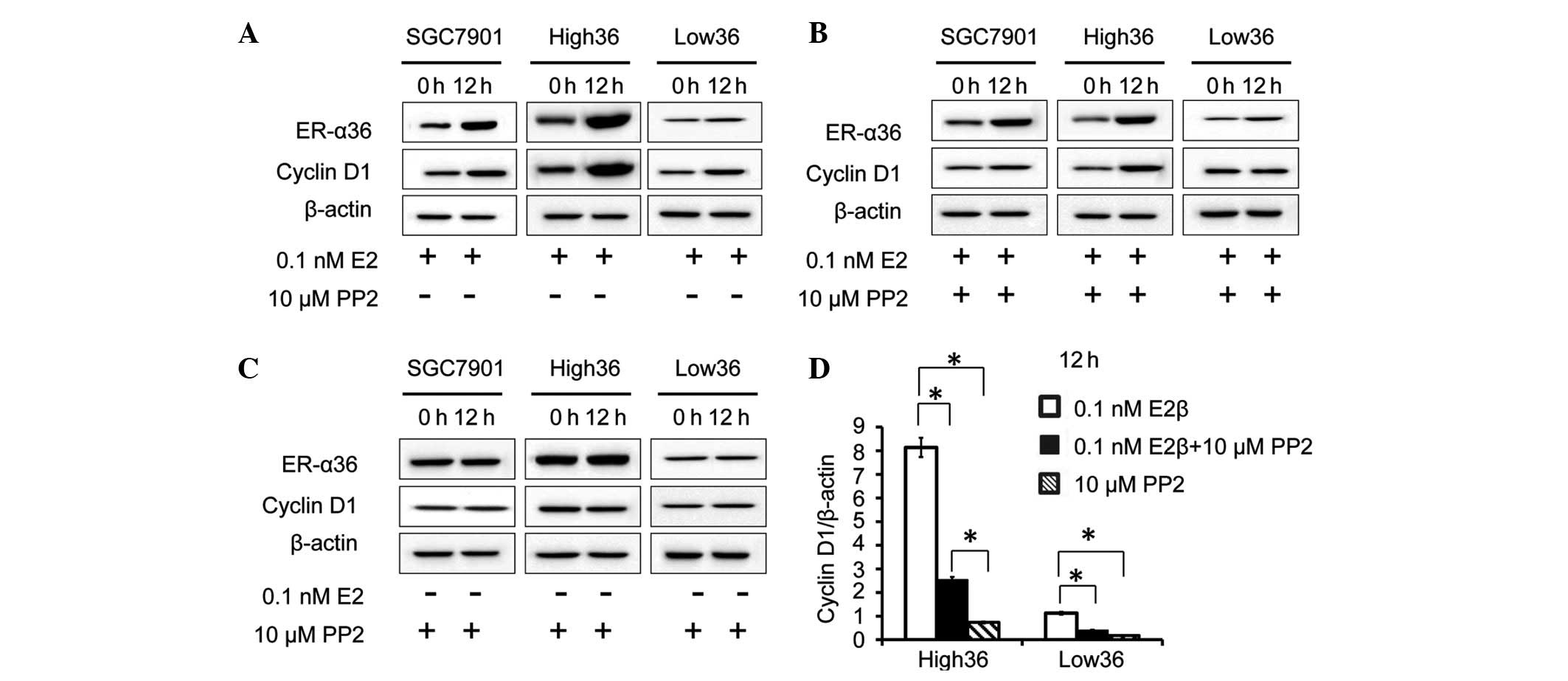
ER-α36-mediated estrogen signaling induces cyclin D1 expression in
gastric cancer cells, the cells were treated with E2β (0.1 nM) for
12 h and a western blot analysis was performed to examine cyclin D1
expression. As a result, in the SCG7901 and High36 cell lines, E2β
upregulated the expression levels of cyclin D1, whereas in the
Low36 cells, E2β failed to induce cyclin D1 expression, indicating
that estrogen induces cyclin D1 expression via ER-α36 in gastric
cancer cells. Next, the role of c-Src in the induction of cyclin D1
by estrogen was investigated in the gastric cancer cells. The
effect of the c-Src inhibitor, PP2, on cyclin D1 induction by E2β
was investigated. The cells were treated with E2β and PP2, and a
western blot analysis was performed to examine ER-α36 and cyclin D1
expression. PP2 did not alter the ER-α36 expression induced by E2β.
The increased levels of cyclin D1 expression induced by E2β were
inhibited by PP2, indicating that c-Src iss involved in the
induction of cyclin D1 expression induced by E2β in gastric cancer
cells (Fig. 2A–D).
ER-α36 and cyclin D1 are expressed in
tumor xenografts
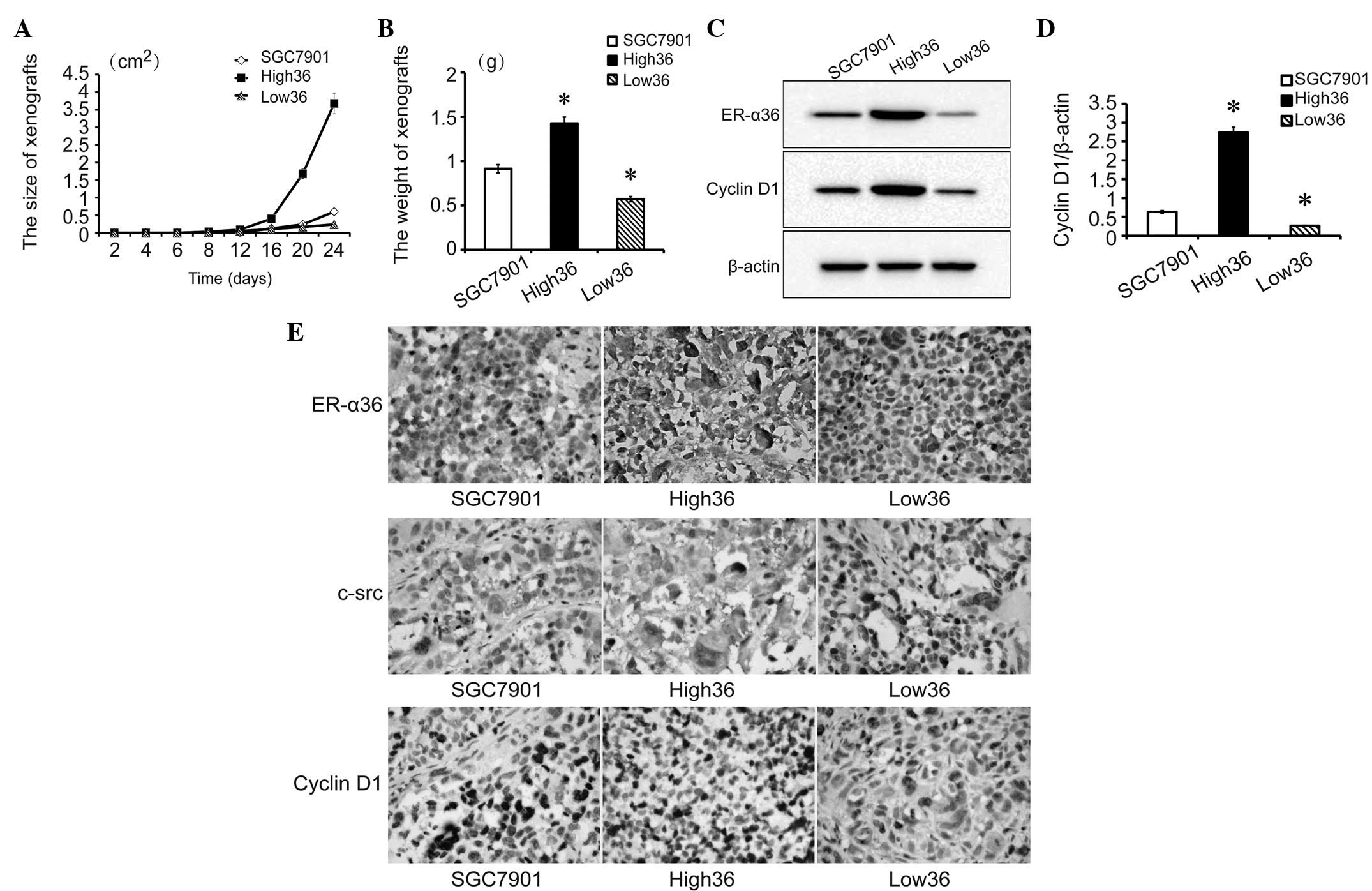
To determine the tumor growth of the cell lines, all
cell lines (1×106 cells/nude mice) were transplanted
subcutaneously into the skin of the dorsal body of 2 nude mice/cell
line. The growth of the transplanted tumors was monitored every 4
days and tumors were detected from day 8. High36 cells formed the
largest tumors while Low36 cells formed the smallest tumors
(Fig. 3A–B). After 24 days, the
nude mice were sacrificed and the tumors were removed. The levels
of ER-α36 and cyclin D1 expression in the xenografted tumors were
examined with western blot analysis (Fig. 3C and D). Next, the expression of
ER-α36, c-Src and cyclin D1 in the xenografted tumors was tested by
IHC (Fig. 3E). The expression of
ER-α36, c-Src and cyclin D1 was higher in the High36, moderate in
the SGC7901 and lower in the Low36 cell lines. In addition, c-Src
and cyclin D1 expression was shown to be associated with the
expression of ER-α36. These results further indicated that
ER-α36-mediated signaling is important for the development of
gastric cancer, presumably through c-Src and cyclin D1.
ER-α36-c-Src interaction is induced by
E2β
ER-α36 is known to physically interact with the
EGFR/Src/Shc complex and mediate estrogen-induced phosphorylation
of epidermal growth factor receptor (EGFR) and c-Src in breast
cancer cells (28). Therefore, in
the present study, the direct interaction of ER-α36 with c-Src in
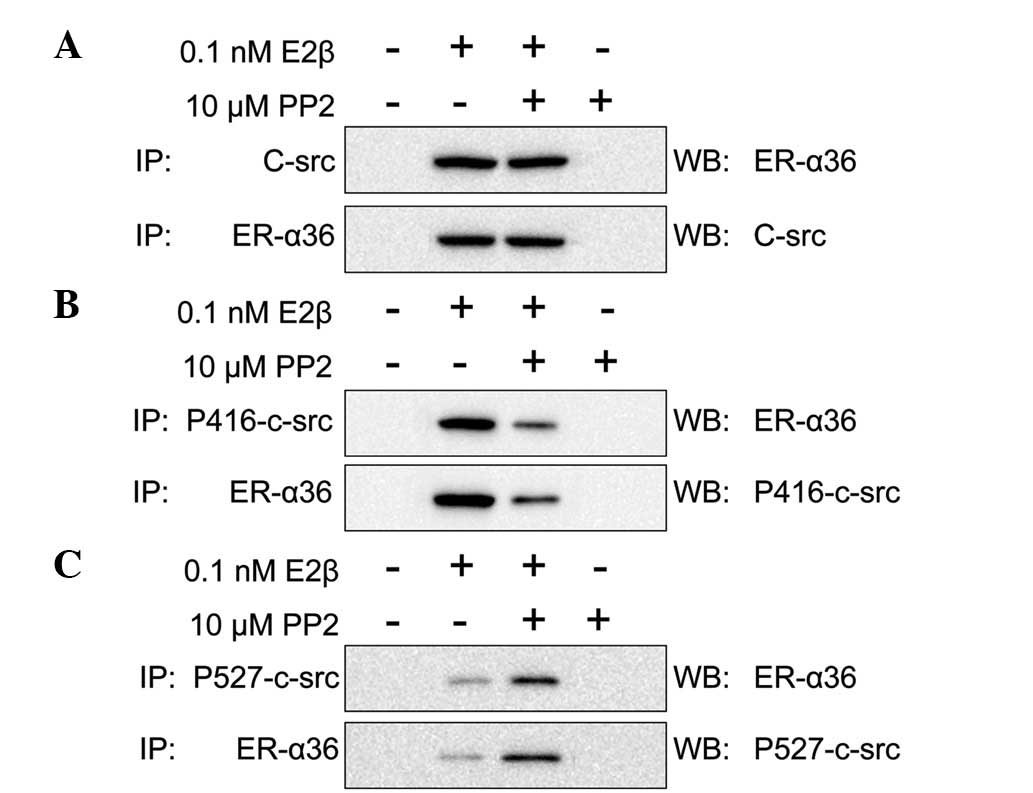
the SGC7901 cells was analyzed. E2β (0.1 nM) and/or PP2 (10
μM) were used to stimulate the SGC7901 cells for 10 min.
Formed complexes were then pulled down and probed using antibodies
against ER-α36, c-Src, p416-c-Src and p527-c-Src. When the SGC7901
cells were stimulated by E2β or E2β and PP2 together, ER-α36 was
observed to interact with c-Src. However, when the SGC7901 cells
were stimulated by PP2 alone, ER-α36 did not interact with c-Src
(Fig. 4A). In addition, PP2
decreased the activation of c-Src, which showed a high expression
of p527-c-Src and a low expression of p416-c-Src. These
observations indicate that p527-c-Src expression is higher than
p416-c-Src when SCGC7901 cells are stimulated by E2β and PP2
together (Fig. 4B–C). Therefore, we
hypothesized that ER-α36 and c-Src interact in the presence of E2β
and that this interaction is not inhibited by PP2. However, PP2
does inhibit the activation of c-Src.
Discussion
Previous epidemiological studies have reported that
the gender difference in the incidence and prevalence of gastric
cancer cannot be explained by other factors except estrogen levels.
However, a previous study demonstrated that gastric tumor tissues
were negative for or showed extremely low levels of ER-α66 (the
traditional estrogen receptor) expression (29), generating the query of how the
estrogen concentration correlates with the incidence and prevalence
of gastric cancer. Our previous study showed that a variant of
ER-α, ER-α36, was highly expressed in human gastric tissues and
mainly expressed on the plasma membrane and in the cytoplasm of
gastric cancer cells. ER-α36 expression was associated with lymph
node metastasis, indicating that ER-α36 may be a marker of gastric
cancer metastasis (27). Consistent
with these observations, in the present study, ER-α36-mediated
estrogen signaling was shown to promote the growth of SGC7901 cells
in vitro and in vivo. In addition, ER-α36-mediated
estrogen signaling stimulated proliferation of the gastric cancer
cells through the activation of the c-Src signaling pathway and the
upregulation of cyclin D1 expression.
However, the function of estrogen that stimulated
the growth of gastric cancer cells was associated with the
concentration of estrogen. Physiologically low concentrations of
estrogen (0.1 nM) were identified to promote the growth of gastric
cancer cells and the expression of ER-α36. In addition,
physiologically high concentrations of estrogen (5 μM)
inhibited the growth of gastric cancer cells and the expression of
ER-α36 (30). This may explain the
male predominance of gastric cancer (9). The pathogenesis of gastric cancer is a
multi-step process affected by a number of risk factors. The
dysregulation of multiple signaling pathways involved in cell
proliferation, invasion and metastasis had been described in
gastric cancer (31,32). Results of the current study indicate
that ER-α36-mediated estrogen-signaling is important for the
development of human gastric cancer.
When we identified that ER-α36 was associated with
the incidence and prevalence of gastric cancer, we continued to
study the possible downstream signaling mechanisms involved. In a
previous study in ER-negative breast cancer cells, c-Src was
identified to function as a switch in ER-α36-mediated biphasic
estrogen signaling through the EGFR/STAT5 pathway (33). In addition, ER-α36 has been reported
to physically interact with the EGFR/Src/Shc complex (28). Consistent with these observations,
we hypothesize that c-Src also functions in this manner in
ER-α36-positive gastric cancer cells.
c-Src is a non-receptor protein tyrosine kinase that
transduces signals involved in a variety of cellular processes,
including cell adhesion, invasion, growth and differentiation
(34). An important regulatory
mechanism of c-Src tyrosine kinase activity involves the control of
its phosphorylation status. There are two major phosphorylation
sites in the c-Src protein, Tyr416 and Tyr527. When Tyr416 is
phosphorylated, it positively regulates c-Src activity and when
Tyr416 is dephosphorylated, it negatively regulates c-Src activity
(35–37).
By probing the underlying mechanisms of E2β
signaling in gastric cancer cells in the present study, 0.1 nM E2β
was demonstrated to induce the phosphorylation of c-Src at Tyr416
and the dephosphorylation of c-Src at Tyr527 in all cell lines.
These results were more profound in cells with upregulated ER-α36
expression, consistent with the observation that the PP2 c-Src
inhibitor inhibited proliferation in these cells. Therefore, the
results indicated that the phosphorylation state of c-src-Tyr416
and c-src-Tyr527 functions as a switch to turn on and off
non-genomic estrogen signaling depending on the concentration of
estrogen.
Cell growth is regulated by proliferation and
apoptosis. Cyclin D1 is an important regulatory factor for cell
cycle progression and is required to mediate the G1 to S
transition, in turn leading to DNA synthesis and cell cycle
progression (38). The
overexpression of cyclin D1 has been documented in a number of
carcinomas, including gastric cancer (39–41). A
previous study identified a gender difference in MNNG-induced rat
gastric carcinogenesis that was hypothesized to be associated with
gender differences in cyclin D1/cdk4 expression (42). However, the mechanisms linked to
this observation remain unknown. In the present study, E2β induced
c-src-Tyr416 phosphorylation in cells with upregulated ER-α36
expression and failed to induce c-src-Tyr527 phosphorylation in
cells with knocked down ER-α36 expression. c-Src-Tyr416
phosphorylation increased the levels of cyclin D1 expression and
promoted cell proliferation in the upregulated ER-α36 SGC7901
cells, while the opposite occurred in SGC7901 cells with knocked
down ER-α36. To further confirm these observations, the expression
of ER-α36 and cyclin D1 was analyzed in xenografts of nude mice,
which included upregulated ER-α36, knocked down ER-α36 and control
SGC7901 cell lines. Cyclin D1 expression was shown to be positively
correlated with ER-α36 expression in these xenografts. The results
demonstrated that E2β-ER-α36 regulates the phosphorylation of
c-src-Tyr-416 and -Tyr-527 to promote the growth of gastric cancer
and further indicates that E2β-ER-α36-c-Src is important for
proliferation in gastric cancer.
In a previous study, ER-α36 and c-Src were reported
to be associated in MDA-MB-231 breast cancer cells (43). In the current study, the interaction
between ER-α36 and c-Src was demonstrated in SGC7901 gastric cancer
cells. ER-α36 and c-Src were identified to interact in the presence
of E2β, and PP2 did not affect this interaction. However, PP2 was
observed to inhibit the activation of c-Src. In addition, the
association between ER-α36 and cyclin D1 in the SGC7901 gastric
cancer cells was induced by E2β.
Since 1983, a number of studies have examined the
expression of the ER in gastric cancer (44,45).
However, considerable controversy remains with regard to the
expression levels of ER and their prognostic value in gastric
cancer. Studies have shown that the traditional ER, ER-α66, is
absent in gastric cancer (25). The
ER has been hypothesized to be associated with gastric cancer,
however, to date, no studies have explained the inconsistent
negative expression of ER-α66 (25). The identification of the E2-ER
α36-c-Src pathway revealed that E2 promotes proliferation in
gastric cancer cells by activating ER-α36.
In summary, the results of the present study have
demonstrated that ER-α36-mediated estrogen signaling promotes the
proliferation of gastric cancer cells, indicating that ER-α36 is
important for the development of human gastric cancer. In addition,
the study also provides further evidence that c-Src is involved in
ER-α36-mediated mitogenic estrogen signaling in gastric cancer
cells.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (no.
30870981) and the Science Foundation of Health Office of Hubei
Province (no. NX200727). The authors would like to thank Dr
Hong-yan Zhen for her advice.
References
|
1.
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
3.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics for Hispanics/Latinos, 2012. CA Cancer J Clin.
62:283–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Chandanos E and Lagergren J: Oestrogen and
the enigmatic male predominance of gastric cancer. Eur J Cancer.
44:2397–2403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.
|
|
6.
|
Kelley JR and Duggan JM: Gastric cancer
epidemiology and risk factors. J Clin Epidemiol. 56:1–9. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lindblad M, Rodriguez LA and Lagergren J:
Body mass, tobacco and alcohol and risk of esophageal, gastric
cardia, and gastric non-cardia adenocarcinoma among men and women
in a nested case-control study. Cancer Causes Control. 16:285–294.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Parkin DM, Whelan SL, Ferlay J, et al:
Cancer incidence in five continents. VIII. IARC Press; Lyon: pp.
1–781. 2002
|
|
9.
|
Sipponen P and Correa P: Delayed rise in
incidence of gastric cancer in females results in unique sex ratio
(M/F) pattern: etiologic hypothesis. Gastric Cancer. 5:213–219.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Fernandez E, Gallus S, Bosetti C, et al:
Hormone replacement therapy and cancer risk: a systematic analysis
from a network of case-control studies. Int J Cancer. 105:408–412.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Frise S, Kreiger N, Gallinger S, et al:
Menstrual and reproductive risk factors and risk for gastric
adenocarcinoma in women: findings from the canadian national
enhanced cancer surveillance system. Ann Epidemiol. 16:908–916.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kaneko S, Tamakoshi A, Ohno Y, et al:
Menstrual and reproductive factors and the mortality risk of
gastric cancer in Japanese menopausal females. Cancer Causes
Control. 14:53–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lindblad M, García Rodríguez LA, Chandanos
E and Lagergren J: Hormone replacement therapy and risks of
oesophageal and gastric adenocarcinomas. Br J Cancer. 94:136–141.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lindblad M, Ye W, Rubio C and Lagergren J:
Estrogen and risk of gastric cancer: a protective effect in a
nationwide cohort study of patients with prostate cancer in Sweden.
Cancer Epidemiol Biomarkers Prev. 13:2203–2207. 2004.PubMed/NCBI
|
|
15.
|
Chandanos E, Lindblad M, Jia C, et al:
Tamoxifen exposure and risk of oesophageal and gastric
adenocarcinoma: a population-based cohort study of breast cancer
patients in Sweden. Br J Cancer. 95:118–122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Matsuyama Y, Tominaga T, Nomura Y, et al:
Second cancers after adjuvant tamoxifen therapy for breast cancer
in Japan. Ann Oncol. 11:1537–1543. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Duell EJ, Travier N, Lujan-Barroso L, et
al: Menstrual and reproductive factors, exogenous hormone use, and
gastric cancer risk in a cohort of women from the European
Prospective Investigation Into Cancer and Nutrition. Am J
Epidemiol. 172:1384–1393. 2010. View Article : Google Scholar
|
|
18.
|
Kong EH, Pike AC and Hubbard RE: Structure
and mechanism of the oestrogen receptor. Biochem Soc Trans.
31:56–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wang Z, Zhang X, Shen P, et al:
Identification, cloning, and expression of human estrogen
receptor-alpha36, a novel variant of human estrogen
receptor-alpha66. Biochem Biophys Res Commun. 336:1023–1027. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Flouriot G, Brand H, Denger S, et al:
Identification of a new isoform of the human estrogen
receptor-alpha (hER-alpha) that is encoded by distinct transcripts
and that is able to repress hER-alpha activation function 1. EMBO
J. 19:4688–4700. 2000. View Article : Google Scholar
|
|
21.
|
Penot G, Le Péron C, Mérot Y, et al: The
human estrogen receptor-alpha isoform hERalpha46 antagonizes the
proliferative influence of hERalpha66 in MCF7 breast cancer cells.
Endocrinology. 146:5474–5484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kim KH and Bender JR: Rapid, estrogen
receptor-mediated signaling: why is the endothelium so special? Sci
STKE. 2005:pe282005.PubMed/NCBI
|
|
23.
|
Li L, Haynes MP and Bender JR: Plasma
membrane localization and function of the estrogen receptor alpha
variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA.
100:4807–4812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Moriarty K, Kim KH and Bender JR:
Minireview: estrogen receptor-mediated rapid signaling.
Endocrinology. 147:5557–5563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Gan L, He J, Zhang X, et al: Expression
profile and prognostic role of sex hormone receptors in gastric
cancer. BMC Cancer. 12:5662012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ryu WS, Kim JH, Jang YJ, et al: Expression
of estrogen receptors in gastric cancer and their clinical
significance. J Surg Oncol. 106:456–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Deng H, Huang X, Fan J, et al: A variant
of estrogen receptor-alpha, ER-alpha36 is expressed in human
gastric cancer and is highly correlated with lymph node metastasis.
Oncol Rep. 24:171–176. 2010.PubMed/NCBI
|
|
28.
|
Zhang XT, Kang LG, Ding L, et al: A
positive feedback loop of ER-alpha36/EGFR promotes malignant growth
of ER-negative breast cancer cells. Oncogene. 30:770–780. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Wang M, Pan JY, Song GR, et al: Altered
expression of estrogen receptor alpha and beta in advanced gastric
adenocarcinoma: correlation with prothymosin alpha and
clinicopathological parameters. Eur J Surg Oncol. 33:195–201. 2007.
View Article : Google Scholar
|
|
30.
|
Teoh H and Man RY: Progesterone modulates
estradiol actions: acute effects at physiological concentrations.
Eur J Pharmacol. 378:57–62. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Yang L, Xie G, Fan Q and Xie J: Activation
of the hedgehog-signaling pathway in human cancer and the clinical
implications. Oncogene. 29:469–481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Yeh TS, Wu CW, Hsu KW, et al: The
activated Notch1 signal pathway is associated with gastric cancer
progression through cyclooxygenase-2. Cancer Res. 69:5039–5048.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Zhang XT, Ding L, Kang LG and Wang ZY:
Involvement of ER-alpha36, Src, EGFR and STAT5 in the biphasic
estrogen signaling of ER-negative breast cancer cells. Oncol Rep.
27:2057–2065. 2012.PubMed/NCBI
|
|
34.
|
Frame MC: Src in cancer: deregulation and
consequences for cell behaviour. Biochim Biophys Acta.
1602:114–130. 2002.PubMed/NCBI
|
|
35.
|
Chiang GG and Sefton BM: Phosphorylation
of a Src kinase at the autophosphorylation site in the absence of
Src kinase activity. J Biol Chem. 275:6055–6058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Xu W, Harrison SC and Eck MJ:
Three-dimensional structure of the tyrosine kinase c-Src. Nature.
385:595–602. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Xu W, Doshi A, Lei M, et al: Crystal
structures of c-Src reveal features of its autoinhibitory
mechanism. Mol Cell. 3:629–638. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Sherr CJ: D-type cyclins. Trends Biochem
Sci. 20:187–190. 1995. View Article : Google Scholar
|
|
39.
|
Sauter ER, Yeo UC, von Stemm A, et al:
Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer
Res. 62:3200–3206. 2002.PubMed/NCBI
|
|
40.
|
Udhayakumar G, Jayanthi V, Devaraj N and
Devaraj H: Interaction of MUC1 with beta-catenin modulates the Wnt
target gene cyclinD1 in H. pylori-induced gastric cancer. Mol
Carcinog. 46:807–817. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Arber N, Gammon MD, Hibshoosh H, et al:
Overexpression of cyclin D1 occurs in both squamous carcinomas and
adenocarcinomas of the esophagus and in adenocarcinomas of the
stomach. Hum Pathol. 30:1087–1092. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Motohashi M, Wakui S, Muto T, et al:
Cyclin D1/cdk4, estrogen receptors α and β, in
N-methyl-N’-nitro-N-nitrosoguanidine-induced rat gastric
carcinogenesis: immunohistochemical study. J Toxicol Sci.
36:373–378. 2011.
|
|
43.
|
Zhang X, Ding L, Kang L and Wang ZY:
Estrogen receptor-alpha 36 mediates mitogenic antiestrogen
signaling in ER-negative breast cancer cells. PloS One.
7:e301742012. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Kitaoka H: Sex hormone dependency and
endocrine therapy in diffuse carcinoma of the stomach. Gan To
Kagaku Ryoho. 10:2453–2460. 1983.(In Japanese).
|
|
45.
|
Furukawa H, Iwanaga T, Koyama H and
Taniguchi H: Effect of sex hormones on the experimental induction
of cancer in rat stomach - a preliminary study. Digestion.
23:151–155. 1982. View Article : Google Scholar : PubMed/NCBI
|