1. Introduction
Peutz-Jeghers syndrome (PJS) is an autosomal
dominant disease that is characterized by hamartomatous polyposis
in the gastrointestine and mucocutaneous melanin spots in the oral
mucosa, lips, wings of the nose, fingers and toes. The first case
was reported in a Dutch family by Peutz in 1921 (1). Jeghers et al described the
details of another family in 1948 (2) and the syndrome was defined as an
independent disease. The clinical features of PJS include
gastrointestinal polyps that increase in number and size and the
appearance of abdominal symptoms, including ileus and
gastrointestinal hemorrhage. Among the types of hereditary
gastrointestinal polyposis, the incidence of PJS is second highest
following that of familial adenomatous polyposis. PJS develops at
an incidence rate of 1 in 5–25×104 individuals.
The tumor suppressor gene, STK11/LKB1, which
is located on chromosome 19p13.3, is responsible for PJS.
STK11 is believed to be involved in cellular energy
metabolism, cell proliferation, cell polarity, p53-dependent
apoptosis, the regulation of vascular endothelial growth factor
(VEGF) and Wnt signal transduction. Loss of heterozygosity (LOH)
due to a mutation, including a deletion in the normal allele, in
addition to a germline STK11 mutation, may cause the
clinical features of PJS, including gastrointestinal polyposis and
cancerization of other organs. PJS is complicated by benign and
malignant tumors of various organs and by rare diseases, including
sex cord tumor with annular tubules (SCTAT) and minimal deviation
adenocarcinoma (MDA). The development of endometrial carcinoma in
patients with PJS has also attracted attention in the field of
gynecology. Lobular endocervical glandular hyperplasia (LEGH) in a
PJS patient with a germline STK11 mutation has recently been
reported, which has led to the hypothesis that LEGH is a prodromal
lesion of MDA (3).
2. Clinical characteristics of PJS
A disease resembling PJS was described by Peutz in
1921 and by Jeghers et al in 1949 (1,2). The
pathology of PJS is considered to be hamartomatous, since PJS
polyps are not cancerous and their histology is similar to the
normal mucosal structure. A more accurate description of the
lesions is hyperplasia of the lacunar epithelium. However, the
morphology differs from that of a general hyperplastic polyp. The
gland opening is dilated toward the outer side due to the
hyperplastic mucoepithelium and muscular fibers overgrow in a
dendritic pattern along the epithelium. These changes are preceded
by epithelial hyperplasia. Adjacent lamina muscularis mucosae are
believed to bend and fuse to give the characteristic appearance and
small lesions undergo hyperplastic changes (4,5).
Clinically, PJS is characterized by the development
of abdominal symptoms, including pain, ileus and gastrointestinal
hemorrhage, which occur with an increase in the number and size of
the gastrointestinal polyps. The incidence of PJS is secondary to
that of familial adenomatous polyposis among the types of
hereditary gastrointestinal polyposis. Malignant and benign tumor
complications may occur in PJS, with studies indicating that
malignant tumors develop in approximately half of patients by the
age of 57 years. The incidence of digestive organ malignant tumors
is highest in the colorectum, followed by the stomach, small
intestine, duodenum and pancreas. In regions other than the
digestive organs, the incidence is reported to be highest in the
uterine cervix, followed by the lung and ovary in female PJS
patients (Table I) (3). Associations with ovarian SCTAT and
uterine cervical LEGH and MDA have been highlighted in the field of
gynecology, and an association with endometrial cancer has recently
gained interest based on case reports and genetic studies (6–11).
 | Table ICancer cases reported in patients
with PJS. |
Table I
Cancer cases reported in patients
with PJS.
| Location | Number of
patients |
|---|
|
Gastrointestinal |
| Esophagus | 1 |
| Stomach | 16 |
| Small
intestine | 22 |
| Large
intestine | 26 |
| Pancreas | 8 |
|
Extraintestinal |
| Breast | 17 |
| Uterine
cervix | 10 |
| Ovary | 7 |
| Uterus | 2 |
| Fallopian
tube | 1 |
| Testis | 1 |
| Prostate | 1 |
| Lung | 9 |
| Thyroid | 2 |
|
Leiomyosarcoma | 2 |
| Gall bladder | 1 |
| Liver | 1 |
| Basal cell | 1 |
| Osteosarcoma | 1 |
| Multiple
myeloma | 1 |
3. STK11/LKB1 and PJS
The gene that is responsible for causing PJS is the
tumor suppressor gene, STK11/LKB1, which is located on the
short arm of chromosome 19 (19p13.3) (12). The gene is 23 kb in size and is
comprised of nine coding exons and one non-coding exon. The gene
encodes a serine threonine kinase containing 433 amino acids
(12). The mRNA is 3.0–3.3 kb in
size and is expressed in almost all human tissues. Germline
mutations of STK11/LKB1 are observed in more than half of
PJS patients. However, the somatic development of PJS in patients
with no familial medical history and cases without a mutation in
STK11/LKB1 have also been described.
In 1997, Hemminki et al identified the
responsible gene region in PJS to be near the chromosome 19 short
arm marker using a linkage analysis of families with PJS (13). Amos et al confirmed this
finding (14). The STK11
gene was known to be present on chromosome 19 (15,16)
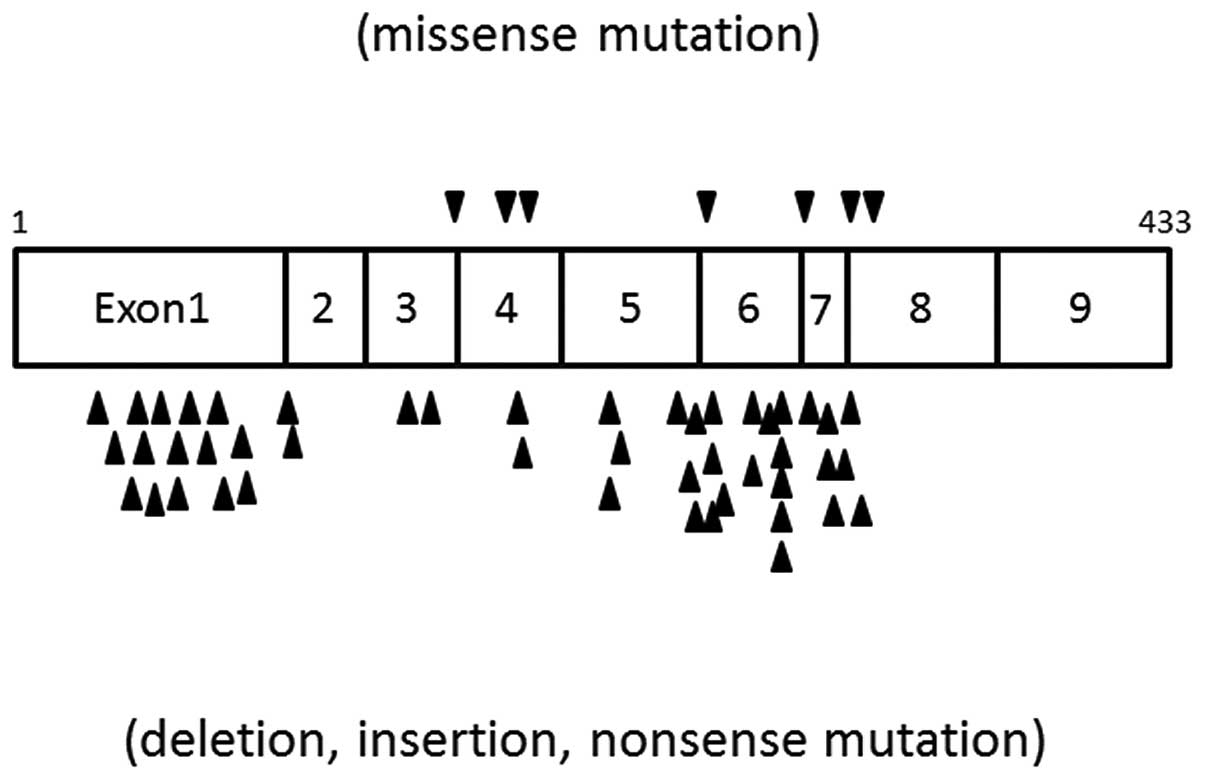
and Yoon et al subsequently identified a number of mutation
types in this gene, including missense, frame shift, nonsense and
splicing site mutations, in 10 PJS patients using polymerase chain
reaction-single strand conformation polymorphism (PCR-SSCP;
Fig. 1) (17). Germline mutations were present in
five of these patients. Tseng et al analyzed STK11
mRNA expression in twin sisters with PJS with the same allele using
PCR (18) and revealed that
STK11 gene expression was absent in the two subjects. These
results suggest that STK11 on chromosome 19 is responsible
for PJS and that a mutation or decreased expression of STK11
is the cause of the disease.
The protein product of STK11 is involved in
cellular energy metabolism, cell proliferation, cell polarity,
p53-dependent apoptosis, the regulation of VEGF and Wnt signal
transduction (3). LOH due to a
mutation, including a deletion in the normal allele, in addition to
the germline STK11 mutation, results in gastrointestinal
polyposis and cancerization of other organs, which are common
clinical features of PSJ.
As noted previously, Yoon et al identified
germline mutations in five of 10 PJS patients, which suggests other
developmental mechanisms in the remaining five patients, based on
STK11 gene mutation-induced development (somatic case) and
the association with other genes. Several studies have suggested
the presence of a gene that is associated with PJS other than
STK11. In an investigation of 21 PJS patients from 13
families, Papp et al identified that 8 (62%) of the 13 cases
of PJS had familial medical histories and that the remaining five
cases (38%) were due to de novo mutations (12). Germline mutations were screened in
the 21 patients and 13 pathogenic mutations of STK11 were
identified. Three of these were frameshift mutations, three were
nonsense mutations, two were mutations of the splicing sequence and
five were deletions of exons 1–7. This deletion was noted in five
of the 13 families, showing a high frequency, and was also shown to
affect two genes that were located upstream of the STK11
gene, SBNO2 and GPX4, which are considered to modify
STK11. This finding suggests that an abnormality in the
genes that modify STK11 function may promote the development
of PJS, even in the absence of a mutation in STK11
itself.
Souza et al initially reported a contiguous
genetic syndrome in which a developmental disorder, heart
malformation and facial dysplasia appear as phenotypes, in addition
to the symptoms of PSJ (19). This
syndrome is caused by a 19p13.3 chromosomal deletion of a region of
~1.1 Mb that includes STK11(19). Scollon et al also reported a
syndrome with a similar gene deletion at a similar site, but the
development of a cleft lip and gastrointestinal polyposis differed
between the syndromes, suggesting that the phenotypes may vary,
despite a common STK11 deletion between the two syndromes
(20).
4. SCTAT in PJS
Histologically, SCTAT is characterized by the
annular growth of sex cord cells while hyaline bodies form around
the nucleus. SCTAT includes ovarian Sertoli cell tumors, ovarian
mucous/serous epithelial tumors and ovarian mature teratoma. Scully
et al suggested that SCTAT is derived from ovarian granulosa
cells and grows in a pattern that is characteristic of Sertoli
cells (21). In another hypothesis,
SCTAT has been considered to be comprised of sex cord-derived
immature cells, which have the possibility to differentiate into
granulosa and Sertoli cells. These tumors are positive for
immunohistochemical staining of testosterone and estradiol and are
diagnosed based on endocrinological symptoms in the majority of
cases, including precocious puberty and irregular menstruation,
which may involve amenorrhea, hypermenorrhea and postmenopausal
hemorrhage (3).
SCTAT is an ovarian tumor that is most likely to
complicate PJS, with ~36% of SCTAT cases reported to be associated
with the syndrome (22). The
clinical features of SCTAT differ between patients with and without
PJS (22). In a comparison between
SCTAT in 21 patients with PJS and 47 patients without PJS, Young
et al identified that SCTAT with PJS was multifocal,
bilateral, small and required microscopy to confirm the diagnosis
in the majority of the cases. More than half of the cases were
accompanied by calcification and the prognosis was favorable. In
contrast, sporadic SCTAT was unilateral, large enough for palpation
and calcified only in 12% of cases. In total, 20% of cases
progressed to malignancy (22).
Granulosa cell, Seltori-Lydig cell, borderline malignant and mucous
tumors have been reported as other histological types of
PJS-associated ovarian tumors, in addition to SCTAT.
5. MDA in PJS
MDA is a novel disease type that has been proposed
for a condition that was previously termed adenoma malignum by
Silverberg and Hurt in 1975 (23).
MDA may be a subtype of adenocarcinoma, but it has a
well-differentiated histology that is indistinguishable from that
of the normal cervical gland. Certain patients that are diagnosed
with MDA have a favorable prognosis, and in 1999, Nucci et
al proposed the disease type lobular endocervical glandular
hyperplasia (LEGH) for these cases (24). LEGH has been assumed to be a
precancerous lesion of MDA in one study; however, a conclusion has
not been established until recently (6).
MDA is a well-known gynecological tumor that is
likely to complicate PJS. The incidence of MDA in PJS patients is
estimated to be 15–30%, while ~10% of MDA cases are complicated by
PJS. The mean age of MDA patients with PJS has been reported to be
33 years old, whereas that of MDA patients without PJS is 55 years
old, indicating a young onset age for the dual disease (25). MDA complicating PJS is considered to
be a malignant tumor with the poorest prognosis among all the
gynecological tumors complicating PJS.
A mutation in the gene that is responsible for PJS,
STK11, and LOH on chromosome 19p13.3, the location of
STK11, have been analyzed in studies of MDA without PJS
(26,27). LOH at 19p13.3 was identified in ~50%
of these cases. In another study, an allelic deletion of >3.5 Mb
was noted on chromosome 19p13.3 in nine MDA patients without PJS
characteristics (28). Furthermore,
in an investigation of the association between STK11
mutation and LOH on chromosome 19p13.3, Connolly et al
identified no somatic mutation in the STK11 coding region in
eight MDA patients without PJS, but LOH was present on chromosome
19p13.3 in three of those patients. Therefore, somatic mutations in
STK11 are absent in MDA without PJS, but LOH may occur at
chromosome 19p13.3 (25). These
results demonstrate that LOH may occur at chromosome 19p13.3, the
location of the STK11 gene that is responsible for PJS, in
certain MDA patients without PJS, suggesting the involvement of an
unknown tumor suppressor gene on chromosome 19p13.3. Lee et
al also identified LOH on the chromosome on which STK11
is present in six of nine MDA patients without PJS and at a site
190 kb away from STK11 in two patients. These findings also
suggest the presence of a tumor suppressor gene that is associated
with the development of MDA, other than STK11, on chromosome
19p13.3. Linkage with 19q13.4 has also been reported (29).
Kuragaki et al(27) identified STK11 mutations in
six out of 11 MDA patients without PJS and also confirmed LOH in
all six of the patients with STK11 mutations, suggesting
that STK11 on 19p13.3 is associated with MDA, in addition to
PJS. With regard to the presence of the STK11 mutations in
only six of the 11 patients with mucous MDA, Kuragaki et al
suggested that the development of mucous MDA involves the loss of
MDA expression, the post-translational modification of the STK11
protein and a gene other than STK11(27). Further analysis of STK11 at
the exon level in the six MDA patients with STK11 mutations
revealed that a mutation was present in the exons of one out of
three of the patients (27). In
other studies, Nakagawa et al identified a germline mutation
in exon six of STK11 in five out of ten PJS patients
(30), Connolly et al
observed mutations in exons 4 and 6 in two SCTAT patients with PJS
(25) and Hemminki et al
identified a mutation in exon 1 in seven out of 12 PJS patients
(29). These results suggest that
the significant exons of STK11 for PJS are exons 1, 4 and
6.
Hirasawa et al recently described the first
case of LEGH in a PJS patient with a germline STK11
mutation. The mutation was a deletion of four bases at codon 263 of
exon 6 (6). This is significant in
terms of the development of LEGH as a uterine cervical tumor in a
PJS patient and also with regard to LEGH as a potential
precancerous lesion of MDA.
The prognosis of MDA has been reported to vary
between being extremely poor (31)
to being mostly equal to that of well-differentiated uterine
cervical adenocarcinoma of the same stage (32). Kuragaki et al investigated
the association between STK11 mutations and prognosis in 11
MDA patients without PJS (27). The
clinical stages of the patients were IB and IIB in five and one out
of six patients with STK11 mutations, respectively, and IB
and IIB in four and one out of five patients with no STK11
mutations, respectively, showing no difference in the clinical
stage between the two groups. However, four of the six patients
with STK11 mutations succumbed within 24 months following
surgery and the tumor recurred in one of the two surviving
patients. In contrast, all five of the patients without
STK11 mutations survived for >50 months following
surgery, suggesting that the prognosis of MDA with STK11
mutations is poorer than that of MDA without STK11 mutations
(P=0.039). The prognosis of PJS patients whose condition is
complicated by MDA has also been recorded to be poor in numerous
other studies (3,33). This may have been due to a higher
level of STK11 mutations in the MDA patients with PJS than
in those without PJS.
6. PJS and endometrial carcinoma
PJS is characterized by the development of numerous
gastrointestinal hamartomatous polyps and mucocutaneous
pigmentation and a higher risk of malignant tumors in the digestive
tract and other organs compared with the general population. The
risk of gynecological cancer is also high, with the lifetime risk
of endometrial carcinoma in females with PJS reported to be 9%
(34). However, this risk in PJS
patients is lower than that of patients with Lynch syndrome and
surveillance for endometrial carcinoma in PJS patients is
considered unnecessary, unlike for ovarian, cervical and breast
cancers (35).
7. Conclusion
Benign or malignant tumors readily develop in organs
such as the digestive tract, mammary glands, ovaries and uterus in
PJS patients, with a risk that is 10–18 times higher than that of
the general population (36). In
females with PJS, the incidence of two rare gynecological tumors,
SCTAT and MDA, also tends to increase. In a few patients, several
gynecological tumors may complicate PJS simultaneously. One example
of this may be observed in the female PJS patient described by
Mangili et al, in whom microscopic ovarian SCTAT and uterine
cervical MDA were identified (37).
With regard to the association between MDA and PJS,
STK11 on chromosome 19p13.3 is likely to be the gene
responsible for the two diseases. Gene analyses have been performed
separately for PJS and MDA, but only case reports are available for
MDA patients with PJS and no comparative gene analysis has been
described. Until recently, there have also been no studies of
concomitant PJS and LEGH, which is considered to be a precancerous
lesion of MDA (6).
The clarification of the association between PJS and
LEGH, MDA, SCTAT and endometrial carcinoma requires the elucidation
of the developmental mechanism at the genetic level. However, PJS
is a rare disease and patients with SCTAT and MDA complications are
rarer, making the number of patients very small. A complete gene
analysis requires a large number of patients and will require a
multicenter study. However, probing of the genetic mechanisms of
SCTAT and MDA complicated by PJS may lead to greater understanding
of the developmental mechanisms of uterine cervical adenocarcinoma
and simultaneous multiple gynecological tumors that complicate PJS.
Further studies are anticipated in this field.
References
|
1
|
Peutz JLA: Very remarkable case of
familial polyposis of mucous membrane of intestinal tract and
nasopharynx accompanied by peculiar pigmentations of skin and
mucous membrane. Nederl Maandschr Geneesk. 10:134–146. 1921.(In
Dutch).
|
|
2
|
Jeghers H, McKusick VA and Katz KH:
Generalized intestinal polyposis and melanin spots of the oral
mucosa, lips and digits: A syndrome of diagnostic significance. N
Engl J Med. 241:993–1005. 1949. View Article : Google Scholar
|
|
3
|
Ueki A, Kisu I, Banno K, et al:
Gynecological tumors in patients with Peutz-Jeghers syndrome (PJS).
OJGen. 1:65–69. 2011. View Article : Google Scholar
|
|
4
|
Buck JL, Harned RK, Lichtenstein JE and
Sobin LH: Peutz-Jeghers syndrome. Radiographics. 12:365–378. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gatalica Z and Torlakovic E: Pathology of
hereditary colorectal carcinoma. Fam Cancer. 7:15–26. 2008.
View Article : Google Scholar
|
|
6
|
Hirasawa A, Akahane T, Tsuruta T, et al:
Lobular endocervical glandulara hyperplasia and peritoneal
pigmentation associated with Peutz-Jeghers syndrome due to a
germline mutation of STK11. Annal Oncol. 23:2990–2992. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tanwar PS, Kaneko-Tarui T, Zhang L, Tanaka
Y, Crum CP and Teixeira JM: Stromal liver kinase B1 [STK11]
signaling loss induces oviductal adenomas and endometrial cancer by
activating mammalian target of rapamycin complex 1. PLoS Genet.
8:e10029062012.PubMed/NCBI
|
|
8
|
Ito M, Minaguchi M, Mikami Y, Ueda Y,
Sekiyama K, Yamamoto T and Takakura K: Peutz-Jeghers
syndrome-associated atypical mucinous proliferation of the uterine
cervix: a case of minimal deviation adenocarcinoma (‘adenoma
malignum’) in situ. Pathol Res Pract. 208:623–627. 2012.PubMed/NCBI
|
|
9
|
Tsuji T, Togami S, Nomoto M, Higashi M,
Fukukura Y, Kamio M, Yonezawa S and Douchi T: Uterine cervical
carcinomas associated with loblar endocervical glandular
hyperplasia. Histopathology. 59:55–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koo YJ, Lee JE, Hong SR and Kwon YS:
Co-occurrence of an adenoma malignum and an endocervical-type
adenocarcinoma of the uterine cervix in a woman with Peutz-Jeghers
syndrome. J Gynecol Oncol. 21:203–206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clements A, Robison K, Granai C, Steinhoff
NM, Scalia-Wibur J and Moore RG: A case of Peutz-Jeghers syndrome
with breast, bilateral sex cord tumor with annular tubules, and
adenoma malgnum caused by STK11 gene mutation. Int J Gynecol
Cancer. 19:1591–1594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Papp J, Kovacs ME, Solyom S, et al: High
prevalence of germline STK11 mutations in Hungarian Peutz-Jeghers
Syndrome patients. BMC Med Genet. 11:1692010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hemminki A, Tomlinson I, Markie D, et al:
Localization of a susceptibility locus for Peutz-Jeghers syndrome
to 19p using comparative genomic hybridization and targeted linkage
analysis. Nat Genet. 15:87–90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amos CI, Bali D, Thiel TJ, et al: Fine
mapping of a genetic locus for Peutz-Jeghers syndrome on chromosome
19p. Cancer Res. 57:3653–3656. 1997.PubMed/NCBI
|
|
15
|
Jenne DE, Reimann H, Nezu J, et al:
Peutz-Jeghers syndrome is caused by mutations in a novel serine
threonine kinase. Nat Genet. 18:38–43. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buchet-Poyau K, Mehenni H, Radhakrishna U
and Antonarakis SE: Search for the second Peutz-Jeghers syndrome
locus: exclusion of STK13, PRKCG, KLK10, and PSCD2 genes on
chromosome 19 and the STK11IP gene on chromosome 2. Cytogenet
Genome Res. 97:171–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoon KA, Ku JL, Choi HS, et al: Germline
mutations of the STK11 gene in Korean Peutz-Jeghers syndrome
patients. Br J Cancer. 82:1403–1406. 2000.PubMed/NCBI
|
|
18
|
Tseng CJ, Chen SF, Liou SI, et al: Lack of
STK11 gene expression in homozygous twins with Peutz-Jeghers
syndrome. Ann Clin Lab Sci. 34:154–158. 2004.PubMed/NCBI
|
|
19
|
Souza J, Faucz F, Sotomaior V, Filho AB,
Rosenfeld J and Raskin S: Chromosome 19p13.3 deletion in a child
with Peutz-Jeghers syndrome, congenital heart defect, high myopia,
learning difficulties and dysmorphic features: Clinical and
molecular characterization of a new contiguous gene syndrome. Genet
Mol Biol. 34:557–561. 2011. View Article : Google Scholar
|
|
20
|
Scollon S, McWalter K, Abe K, King J,
Kimata K and Slavin TP: Haploinsufficiency of STK11 and neighboring
genes cause a contiguous gene syndrome including Peutz-Jeghers
phenotype. Am J Med Genet A. 158A:2959–2962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scully RE: Sex cord tumor with annular
tubules: a distinctive ovarian tumor of the Peutz-Jeghers syndrome.
Cancer. 25:1107–1121. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Young RH, Welch WR, Dickersin GR and
Scully RE: Ovarian sex cord tumor with annular tubules: Review of
74 cases including 27 with Peutz-Jeghers syndrome and four with
adenoma malignum of the cervix. Cancer. 50:1384–1402. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Silverberg SG and Hurt WG: Minimal
deviation adenocarcinoma (‘adenoma malignum’) of the cervix : a
reappraisal. Am J Obstet Gynecol. 121:971–975. 1975.
|
|
24
|
Nucci MR, Clement PB and Young RH: Lobular
endocervical glandular hyperplasia, not otherwise specified: a
clinicopathologic analysis of thirteen cases of a distinctive
pseudoneoplastic lesion and comparison with fourteen cases of
adenoma malignum. Am J Surg Pathol. 23:886–891. 1999. View Article : Google Scholar
|
|
25
|
Connolly DC, Katabuchi H, Cliby WA and Cho
KR: Somatic mutations in the STK11/LKB1 gene are uncommon in rare
gynecological tumor types associated with Peutz-Jegher’s syndrome.
Am J Pathol. 156:339–345. 2000.PubMed/NCBI
|
|
26
|
Nishioka Y, Kobayashi K, Sagae S, et al:
Mutational analysis of STK11 gene in ovarian carcinomas. Jpn J
Cancer Res. 90:629–632. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuragaki C, Enomoto T, Ueno Y, et al:
Mutations in the STK11 gene characterize minimal deviation
adenocarcinoma of the uterine cervix. Lab Invest. 83:35–45. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JY, Dong SM, Kim HS, et al: A distinct
region of chromosome 19p13.3 associated with the sporadic form of
adenoma malignum of the uterine cervix. Cancer Res. 58:1140–1143.
1998.PubMed/NCBI
|
|
29
|
Hemminki A, Markie D, Tomlinson I, et al:
A serine/threonine kinase gene defective in Peutz-Jeghers syndrome.
Nature. 391:184–187. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakagawa H, Koyama K, Miyoshi Y, et al:
Nine novel germline mutations of STK11 in ten families with
Peutz-Jeghers syndrome. Hum Genetics. 103:168–172. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gilks CB, Young RH, Aguirre P, DeLellis RA
and Scully RE: Adenoma malignum (minimal deviation adenocarcinoma)
of the uterine cervix: A clinicopathological and
immunohistochemical analysis of 26 cases. Am J Surg Pathol.
13:717–729. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kaminski PF and Norris HJ: Minimal
deviation carcinoma (adenoma malignum) of the cervix. Int J Gynecol
Pathol. 2:141–152. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
von Hochstetter AR, Ess D, Bannwart F and
Bühler H: Adenocarcinoma of the cervix in Peutz-Jeghers syndrome.
Case report and review of the literature. Schweiz Med Wochenschr.
117:1910–1914. 1987.(In German).
|
|
34
|
Giardiello FM, Brensinger DJ, Tersmette
AC, et al: Very high risk of cancer in familial Peutz-Jeghers
syndrome. Gastroenterology. 119:1147–1153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Banno K, Kisu I, Yanokura M, et al:
Epigenetics and genetics in endometrial cancer: new carcinogenic
mechanisms and relationship with clinical practice. Epigenomics.
4:147–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Boardman LA, Thibodeau SN, Schaid DJ, et
al: Increased risk for cancer in patients with the Peutz-Jeghers
syndrome. Ann Intern Med. 128:896–899. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mangili G, Taccagni G, Garavaglia E,
Carnelli M and Montoli S: An unusual admixture of neoplastic and
metaplastic lesions of the female genital tract in the
Peutz-Jeghers syndrome. Gynecol Oncol. 92:337–342. 2004. View Article : Google Scholar : PubMed/NCBI
|