Introduction
O-desmethylangolensin (O-DMA) is one
of the major metabolites of the isoflavone daidzein, and is
produced by intestinal bacteria. O-DMA was first identified
in 1986 (1), and 80–90% of the
population produce this metabolite (2–5).
Although several researchers have speculated that
the metabolites of isoflavones may be biologically important
(6–8), few studies have been conducted.
Epidemiological studies have led to the acceptance of the theory
that isoflavones provide protection against breast cancer (9,10).
Experimental studies have demonstrated that the daidzein precursor
of O-DMA shows activity against various cancers, including
breast cancer. Daidzein exhibits antiproliferative activity in
breast cancer through the induction of apoptosis and cell cycle
arrest (11–13). Moreover, as we reported previously
(14), daidzein exerts its
anticancer effects in human breast cancer cells via cell cycle
arrest at the G1 and G2/M phases.
The present study investigated the possible
anticarcinogenic effects of O-DMA through the inhibition of
cell proliferation and cell cycle arrest in human breast carcinoma
MCF-7 cells, which are regarded as an in vitro breast cancer
model.
Materials and methods
Cell culture and O-DMA treatment
Human breast MCF-7 cells were purchased from the
Korean Cell Line Bank (Seoul, Korea). MCF-7 were routinely
maintained in RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA)
supplemented with 10% fetal bovine serum and antibiotics (50 U/ml
penicillin and 50 μg/ml streptomycin; Sigma-Aldrich) at 37°C in a
humidified atmosphere containing 5% CO2. The synthesized
O-DMA was a gift from Dr Lee (Department of Chemistry,
Duksung Women’s University, Seoul, Republic of Korea) and dissolved
in dimethyl sulfoxide (DMSO; final concentration, 0.1% in
medium).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
MCF-7 cell proliferation was assessed using the MTT
assay. The cells were plated at a density of 1×105
cells/well in a 96-well tissue culture plate (Corning Inc.,
Corning, NY, USA) and incubated at 37°C for 24 h. The plated cells
were then treated with 5–200 μM O-DMA for 24, 48 and 72 h.
Next, the plated cells were incubated with MTT (Sigma-Aldrich;
final concentration, 0.5 mg/ml) for 4 h at 37°C. Subsequent to
discarding all the medium from the plates, 100 μl DMSO was added to
each well. The plates were placed at room temperature for 5 min
with agitation, so that complete dissolution of the formazan was
achieved. The absorbance of the MTT formazan was determined at 540
nm by a UV spectrophotometric plate reader (Emax; Molecular
Devices, Sunnyvale, CA, USA). The IC50 value (the
concentration of the extract required to inhibit cancer cell growth
by 50% of the control level, which was cells treated with 0.1%
DMSO) was estimated from the plot.
Cell cycle distribution and detection of
apoptosis
Cell cycle distribution and apoptosis were
determined by fluorescence-activated cell sorting (FACS) analysis
using propidium iodide (PI) staining to measure the DNA content.
The MCF-7 cells were plated at a density of 5×105
cells/well in a 6-well tissue culture plate (Corning Inc.) and
incubated at 37°C for 24 h. Next, the cells were treated with 50,
150 and 200 μM O-DMA for 72 h. The cells were then
harvested, washed with cold phosphate-buffered saline (PBS) and
processed for the cell cycle analysis. Briefly, the cells were
fixed in absolute ethanol and stored at −20°C for further analysis.
The fixed cells were centrifuged at 800 × g and washed with cold
PBS twice. RNase A (final concentration, 20 μg/ml; Sigma-Aldrich)
and PI staining solution (final concentration, 50 μg/ml) were added
to the cells and incubated for 30 min at 37°C in the dark. The
cells were analyzed using a FACSCalibur instrument (BD Biosciences,
San Jose, CA, USA) equipped with CellQuest 3.3 software.
For the detection of apoptosis, the cells were
treated and harvested as aforementioned. As an apoptosis biomarker,
phosphatidylserine, which is located on the cytoplasmic surface of
the cell membrane, was detected using the Annexin V-FITC Apoptosis
Detection kit (Calbiochem, EMD Chemicals Inc., Darmstadt, Germany).
Annexin V and PI solution were added to the cell preparations and
incubated for 25 min in the dark. Binding buffer (400 μl) was then
added to each well and the samples were analyzed by flow
cytometry.
Immunoblotting and immunoprecipitation
assay
The MCF-7 cells were plated at a density of
7.5×105 cells/well in a 60-well culture plate and
incubated at 37°C for 24 h. Next, the cells were treated with 50,
150 and 200 μM O-DMA for 72 h, then lysed in
radioimmunoprecipitation assay buffer [1% NP-40, 150 mM NaCl, 0.05%
sodium deoxycholate, 1% sodium dodecyl sulfate (SDS) and 50 mM
Tris; pH 7.5) containing protease inhibitor mixture (Bio-Rad
Laboratories, Hercules, CA, USA) for 1 h at 4°C. The supernatant
was centrifuged at 13,000 × g and the protein concentration was
determined using Bradford Protein Assay kit II (Bio-Rad
Laboratories). Proteins (25 μg/well) denatured with sample buffer
were separated by 10–12% SDS-polyacrylamide gel. The proteins were
then transferred onto nitrocellulose membranes (0.45 μm). The
membranes were blocked with a 1% bovine serum albumin solution for
3 h and washed twice with PBS containing 0.2% Tween-20, then
incubated with primary antibodies overnight at 4°C. Rabbit
polyclonal CDK1, goat polyclonal CDK2 and CDK4, rabbit polyclonal
CDK6, mouse monoclonal cyclin A and D, rabbit polyclonal cyclin B
and E, mouse monoclonal p15INK4b, p16INK4a,
p18INK4c and p19INK4d, rabbit polyclonal
p21Cip1, p27Kip1 and p57Kip2 and
goat polyclonal β-actin were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA) and used to probe the
separate membranes. The following day, the immunoreaction was
continued with the the respective secondary antibodies (goat
anti-mouse IgG-HRP, goat anti-rabbit IgG-HRP, donkey anti-goat
IgG-HRP, Santa Cruz Biotechnology Inc. after washing for 2 h at
room temperature. The specific protein bands were detected by an
Opti-4CN Substrate kit (Bio-Rad Laboratories). The relative
intensity of the bands was analyzed by Quantity One Software
(Bio-Rad Laboratories).
For the CDK-cyclin binding assay, the cells was
treated and lysed as described in the immunoblotting section. The
cell lysates (250 μg) were then incubated with the primary
antibodies overnight at 4°C. The immune complexes were collected by
incubation with protein A/G-plus agarose beads (Santa Cruz
Biotechnology, Inc.) for 90 min at 4°C. The precipitates were
washed with lysis buffer, denatured in sample buffer and analyzed
by immunoblotting as described previously.
Statistical analyses
All the experiments were repeated four times. Data
are presented as the mean ± SD (n=4–7), with the exception of the
reactive oxygen species scavenging and antiproliferative activity
data, which are expressed as percentages compared with the
vehicle-treated control cells, which were arbitrarily assigned
100%. Data were analyzed by one-way analysis of variance followed
by Dunnett’s multiple comparison test using SigmaStat (Jandel
Scientific, San Rafael, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Inhibition of cell proliferation by
O-DMA
The effect of O-DMA on the proliferation of
the human breast cancer MCF-7 cells was measured using an MTT
assay. Treatment of the MCF-7 cells with O-DMA for 24 h
increased cell proliferation by up to 10%, however, this difference
was not significant (Fig. 1).
O-DMA significantly decreased cell proliferation after 48
and 72 h in a dose- and time-dependent manner (306.34 and 178.52 μM
at IC50 for 48 and 72 h, respectively; P<0.05). Cell
proliferation was decreased by 55.15% compared with the controls
after treatment with 200 μM O-DMA for 72 h.
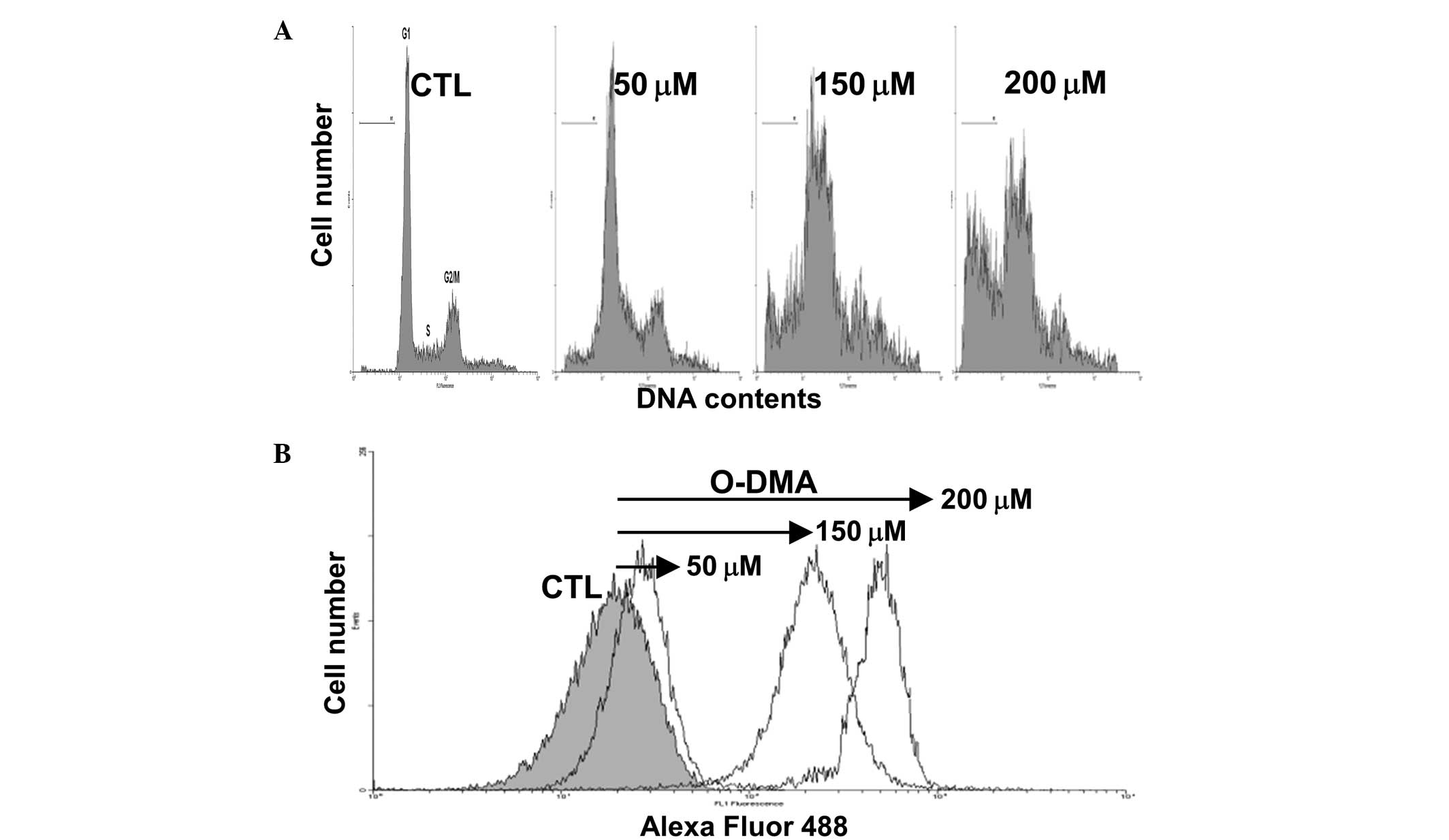
O-DMA induces cell cycle arrest and
apoptosis
The MCF-7 cells were treated with 50, 150 and 200 μM
O-DMA for 72 h, and DNA synthesis arrest was determined by
FACS analysis (Fig. 2).
O-DMA-induced cell cycle arrest in the G1/S and
G2/M phases was observed in a dose-dependent manner.
Additionally, a sub-G1 peak was observed, indicating
O-DMA-induced apoptosis. A significant increase in the
number of sub-G1 phase cells was observed following
exposure to 50, 150 and 200 μM O-DMA (8.4, 17.1 and 37.8% of
the cell population, respectively, compared with the controls,
1.3%; P<0.05).
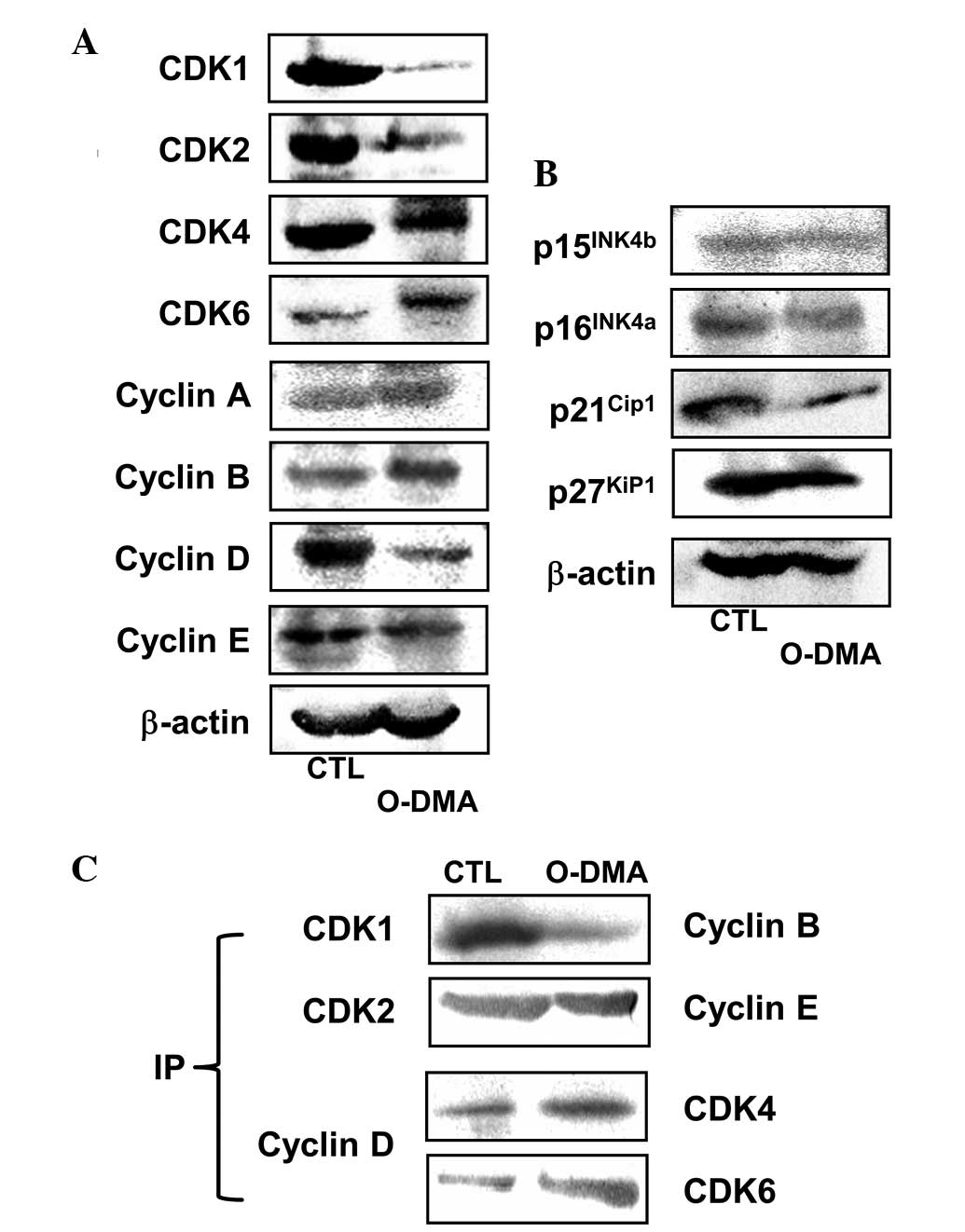
Modulation of G1/S and
G2/M checkpoint regulators by O-DMA
To investigate the effect of O-DMA on the
expression of the cell cycle regulatory proteins, the MCF-7 cells
were treated with 150 μM O-DMA for 72 h (Fig. 3A). The O-DMA treatment
resulted in marked reductions in the expression of CDK1 and CDK2,
given as expression densities; i.e., the ratio of each protein to
β-actin was <5% compared with the control level. CDK4 and CDK6
expression changed in opposite fashions, with CDK4 decreased by
76.9% and CDK6 increased by 152.3% compared with the controls
(P<0.05). O-DMA increased the expression of cyclins A and
B, with only cyclin B increased significantly by 118.7% compared
with the control (P<0.05). By contrast, the expression of
cyclins D and E decreased following exposure to O-DMA (by
14.2 and 46.7%, respectively, compared with the control level;
P<0.05).
O-DMA treatment significantly altered the
expression of members of the INK4 and CIP/KIP families compared
with vehicle-treated MCF-7 cells (P<0.05; Fig. 3B). O-DMA did not change the
p16INK4a and p15INK4b expression levels,
while p21Cip1 and p27Kip1 expression was
decreased slightly. In addition, formation of the CDK4/6-cyclin D
complex was increased in the MCF-7 cells in response to
O-DMA, while the CDK1-cyclin B complex levels were decreased
(Fig. 3C).
Discussion
Although there are only minor differences in the
structures of the isoflavones, genistein and daidzein, and their
metabolites, O-DMA and equol, the anticancer effects and
mechanisms are diverse. The present study first examined the
antiproliferative effect of O-DMA on human breast cancer
MCF-7 cells. O-DMA significantly decreased cell
proliferation in a dose-dependent manner following exposure for 48
and 72 h. This was consistent with previous studies that
demonstrated that daidzein inhibits the growth of human breast
carcinoma cells (11–14). Similar to phytoestrogen, daidzein,
the precursor of O-DMA, may possess biphasic activity
(inhibitory at high concentrations and stimulatory at low
concentrations) (15,16). Although O-DMA has only a weak
affinity for the estrogen receptor (17–19),
the exposure to O-DMA in the present study resulted in
slightly increased MCF-7 cell growth after only 24 h. Thus,
O-DMA may be a more promising anticancer candidate than its
precursor.
To further understand the mechanisms underlying the
anticancer activity of O-DMA, apoptosis induction and the
cell cycle distribution of the MCF-7 cells exposed to 50, 150 and
200 μM O-DMA for 72 h were analyzed in the present study.
Similar to the cell proliferation data, O-DMA induced
significant apoptosis and cell cycle arrest at the G1/S
and G2/M phases in a dose-dependent manner. Numerous
studies have demonstrated that the majority of flavonoids induce
G1 phase arrest in human cancer cells and that certain
flavonoids inhibit the cell cycle at either the G1/S or
G2/M phases in various human cancer cells (20–22).
In our previous study, daidzein caused cell cycle arrest in the
G1/S or G2/M phases, depending on the cell
line (14). Thus, cell cycle arrest
at the G1/S and G2/M phases and apoptosis
induction by O-DMA support the hypothesis that O-DMA
has useful anticancer properties.
The cell cycle is divided into four distinct phases,
G1, S, G2 and M, and is tightly controlled by
catalytic complexes of CDKs/cyclins that coordinate internal and
external signals at several key checkpoints. Activated CDK-cyclin
complexes may be changed to an inactive state by binding to CDK
inhibitory subunits (CKIs), which are divided into two classes,
namely the CIP/KIP (p21Cip1, p27Kip1 and
p57Kip2) and INK4 (p16INK4a,
p15INK4b, p18INK4c and p19INK4d)
families (23,24). Cell cycle arrest leads to either the
inhibition of proliferation or the activation of the apoptosis
pathway (25,26). Therefore, the discovery of
anticancer candidate compounds that may function to regulate CDKs,
is a therapeutic strategy that may result in improved cancer
therapies.
O-DMA decreased CDK4 and cyclin D expression
in the present study, however, it also increased CDK6 expression.
In addition, the expression levels of CDK2 and cyclin E were
decreased by O-DMA. These data are consistent with the
ability of O-DMA to arrest the cell cycle at the
G1/S phase. G1 phase progression and
G1/S phase transition are regulated by CDK2 and CDK4,
which assemble with cyclin E and D. CDK4-cyclin D and CDK2-cyclin E
act predominantly during the G1/S transition (27).
Activated CDK-cyclin complexes are inactivated by
binding to CKIs. The CIP/KIP family has a preference for CDK2- and
CDK4-cyclin complexes, and the INK4 family is specific for CDK4-
and CDK6-cyclin complexes (28,29).
O-DMA had effects on p21Cip1 and
p27Kip1 of the CIP/KIP family in the present study. It
has been previously reported that p21Cip1 promotes cell
cycle arrest in response to a number of antiproliferative signals
(30). Moreover, in the present
study, O-DMA reduced the levels of CDK1 and significantly
increased those of cyclin B, but not cyclin A. CDK1 is a catalytic
subunit of the M phase promoting factor, which is activated at the
G2/M transition and controls the onset of mitosis
(31). A previous study
demonstrated that CDK1, in combination with cyclin A and B, is
critical for the G2/M phase transition (32). Moreover, the present study detected
significantly increased levels of CDK4/6-cyclin D and decreased
levels of CDK1-cyclin B. Based on these data, O-DMA acted on
multiple cell cycle regulators, including affecting the interaction
between the CDK4/6-cyclin D and CDK1-cyclin B complexes.
In conclusion, O-DMA may exert anticancer
activity through the inhibition of cell proliferation and the
induction of apoptosis in human breast cancer MCF-7 cells.
Moreover, the present study indicates for the first time that the
regulation of the CDK4/6-cyclin D and CDK1-cyclin B complexes may
participate in the anticancer activity of O-DMA.
Acknowledgements
This study was supported by the Basic Research
Program through the National Research Foundation of Korea, funded
by the Ministry of Education, Science and Technology
(NRF-2010-0023766 and NRF-2009-0094017).
References
|
1
|
Adlercreutz H, Musey PI, Fotsis T, et al:
Identification of lignans and phytoestrogens in urine of
chimpanzees. Clin Chim Acta. 158:147–154. 1896. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
L’homme R, Brouwers E, Al-Maharik N, et
al: Time-resolved fluoroimmunoassay of plasma and urine
O-desmethylangolensin. J Steroid Biochem Mol Biol. 81:353–361.
2002.PubMed/NCBI
|
|
3
|
Arai Y, Uehara M, Sato Y, et al:
Comparison of isoflavones among dietary intake, plasma
concentration and urinary excretion for accurate estimation of
phytoestrogen intake. J Epidemiol. 10:127–135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Frankenfeld CL, McTiernan A, Aiello EJ, et
al: Mammographic density in relation to daidzein-metabolizing
phenotypes in overweight, postmenopausal women. Cancer Epidemiol
Biomarkers Prev. 13:1156–1162. 2004.PubMed/NCBI
|
|
5
|
Kelly GE, Joannou GE, Reeder AY, et al:
The variable metabolic response to dietary isoflavones in humans.
Proc Soc Exp Biol Med. 208:40–43. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Setchell KD, Brown NM and Lydeking-Olsen
E: The clinical importance of the metabolite equol - a clue to the
effectiveness of soy and its isoflavones. J Nutr. 132:3577–3584.
2002.PubMed/NCBI
|
|
7
|
Bushinsky DA, Sessler NE and Krieger NS:
Greater unidirectional calcium efflux from bone during metabolic,
compared with respiratory, acidosis. Am J Physiol. 262:F425–F431.
1992.PubMed/NCBI
|
|
8
|
Jackman KA, Woodman OL and Sobey CG:
Isoflavones, equol and cardiovascular disease: pharmacological and
therapeutic insights. Curr Med Chem. 14:2824–2830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nagata C: Factors to consider in the
association between soy isoflavone intake and breast cancer risk. J
Epidemiol. 20:83–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Messina M, Nagata C and Wu AH: Estimated
Asian adult soy protein and isoflavone intakes. Nutr Cancer.
55:1–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin S, Zhang QY, Kang XM, et al: Daidzein
induces MCF-7 breast cancer cell apoptosis via the mitochondrial
pathway. Ann Oncol. 21:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang WH, Liu JJ, Chen CH, et al: Growth
inhibition and induction of apoptosis in MCF-7 breast cancer cells
by fermented soy milk. Nutr Cancer. 43:214–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Constantinou AI, Kamath N and Murley JS:
Genistein inactivates bcl-2, delays the G2/M phase of the cell
cycle, and induces apoptosis of human breast adenocarcinoma MCF-7
cells. Eur J Cancer. 34:1927–1934. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi EJ and Kim GH: Daidzein causes cell
cycle arrest at the G1 and G2/M phases in human breast cancer MCF-7
and MDA-MB-453 cells. Phytomedicine. 15:683–690. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balabhadrapathruni S, Thomas TJ, Yurkow
EJ, et al: Effects of genistein and structurally related
phytoestrogens on cell cycle kinetics and apoptosis in MDA-MB-468
human breast cancer cells. Oncol Rep. 7:3–12. 2000.PubMed/NCBI
|
|
16
|
Guo JM, Xiao BX, Liu DH, et al: Biphasic
effect of daidzein on cell growth of human colon cancer cells. Food
Chem Toxicol. 42:1641–1646. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang BY and Adams NR: Effect of equol on
oestrogen receptors and on synthesis of DNA and protein in the
immature rat uterus. J Endocrinol. 85:291–297. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shutt DA and Cox RI: Steroid and
phyto-oestrogen binding to sheep uterine receptors in vitro. J
Endocrinol. 52:299–310. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pfitscher A, Reiter E and Jungbauer A:
Receptor binding and transactivation activities of red clover
isoflavones and their metabolites. J Steroid Biochem Mol Biol.
112:87–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Casagrande F and Darbon JM: Effects of
structurally related flavonoids on cell cycle progression of human
melanoma cells: regulation of cyclin-dependent kinases CDK2 and
CDK1. Biochem Pharmacol. 61:1205–1215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Woo HH, Jeong BR and Hawes MC: Flavonoids:
from cell cycle regulation to biotechnology. Biotechnol Lett.
27:365–374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh RP and Agarwal R: Natural flavonoids
targeting deregulated cell cycle progression in cancer cells. Curr
Drug Targets. 7:345–354. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
24
|
Collins I and Garrett MD: Targeting the
cell division cycle in cancer: CDK and cell cycle checkpoint kinase
inhibitors. Curr Opin Pharmacol. 5:366–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Z, Li M, Rayburn ER, et al:
Oncogenes as novel targets for cancer therapy (part IV): regulators
of the cell cycle and apoptosis. Am J Pharmacogenomics. 5:397–407.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee B, Kim CH and Moon SK: Honokiol causes
the p21WAF1-mediated G(1)-phase arrest of the cell cycle through
inducing p38 mitogen activated protein kinase in vascular smooth
muscle cells. FEBS Lett. 580:5177–5184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng C, Zhang P, Harper JW, et al: Mice
lacking p21CIP1/WAF1 undergo normal development, but are defective
in G1 checkpoint control. Cell. 82:675–684. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takizawa CG and Morgan DO: Control of
mitosis by changes in the subcellular location of cyclin-B1-Cdk1
and Cdc25C. Curr Opin Cell Biol. 12:658–665. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Porter LA and Donoghue DJ: Cyclin B1 and
CDK1: nuclear localization and upstream regulators. Prog Cell Cycle
Res. 5:335–347. 2003.PubMed/NCBI
|