Introduction
Tumor-induced osteomalacia (TIO) is an acquired type
of hypophosphatemia that is frequently associated with mesenchymal
tumors (1). In 1947, McCance et
al described the first case of TIO, and ~300 cases have been
reported in the literature (1,2). TIO
is characterized clinically by fractures, bone pain, phosphaturia,
hypophosphatemia, low serum 1.25(OH)2D concentrations
and high serum alkaline phosphatase (ALP) concentrations (1,3,4).
Fibroblast growth factor 23 (FGF-23) has been identified as a major
pathophysiological factor responsible for phosphaturia (1,5–7).
Surgical resection of the tumor results in the dramatic improvement
of symptoms in the majority of cases (1,4). In
1987, Weidner et al (8)
described the pathological features of a series of 17 mesenchymal
tumors classified as a single entity that caused TIO, and labeled
them as phosphaturic mesenchymal tumors (PMTs). However, the
majority of clinicians and pathologists are not aware of the
existence of this type of tumor, and it is often misdiagnosed as
another type of tumor. The majority of PMTs are benign, and
malignant PMTs resulting in mortality are extremely rare. The
current study presents the cases of two patients who succumbed to
malignant PMTs of the pelvis. Patients provided written informed
consent.
Case report
Case one
In March 2008, a 35-year-old female was referred to
the Osaka University Hospital (Osaka, Japan) with lower back pain.
Laboratory analysis revealed high ALP levels (1,080 mg/ml), with
severe hypophosphatemia (1.4 mg/ml), high thyroglobulin (Tg) levels
(65.2 mg/ml), normal calcium levels (8.7 mg/dl) and normal
parathyroid hormone levels (58.7 mg/ml). Upon physical examination,
a mass was identified in the right neck, and a needle biopsy of the
mass revealed a papillary thyroid carcinoma. On positron emission
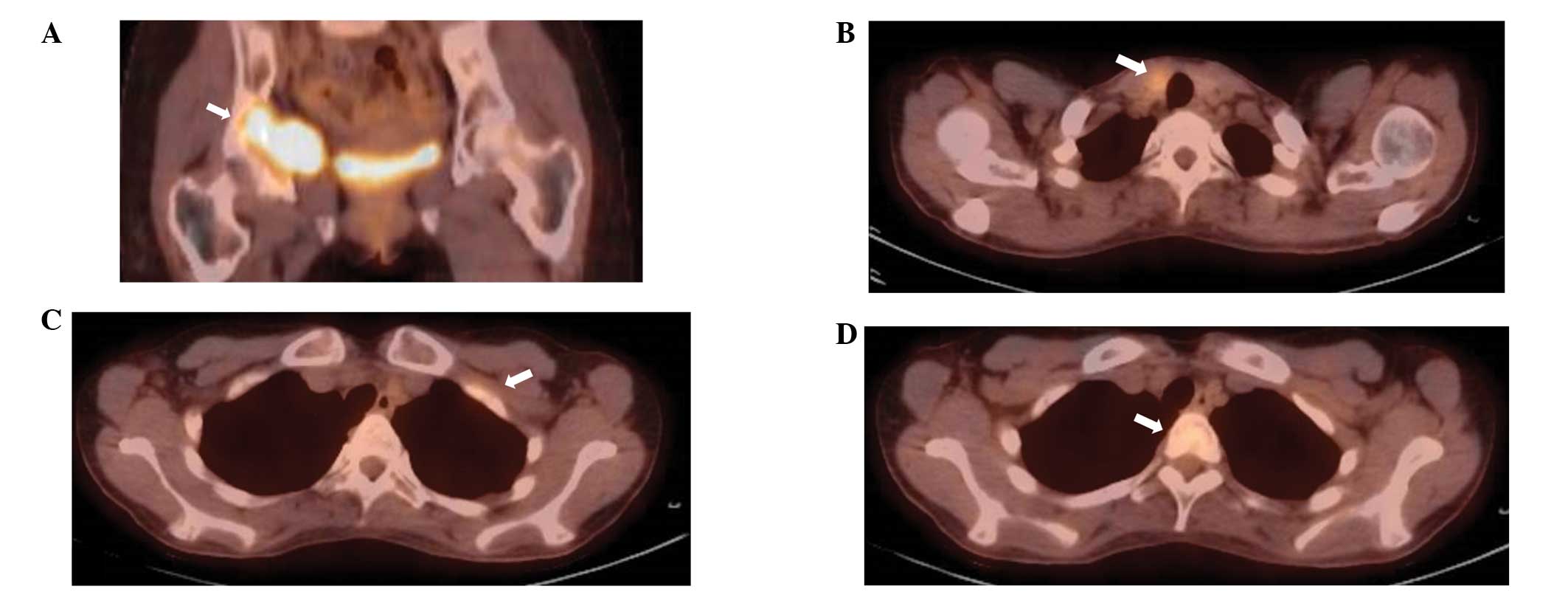
tomography with 18F-fluorodeoxyglucose (FDG-PET),
abnormal uptake was observed in the right pelvis (maximum
standardized uptake value of 7.0), left first rib, T2 vertebra and
right lobe of the thyroid gland (Fig.
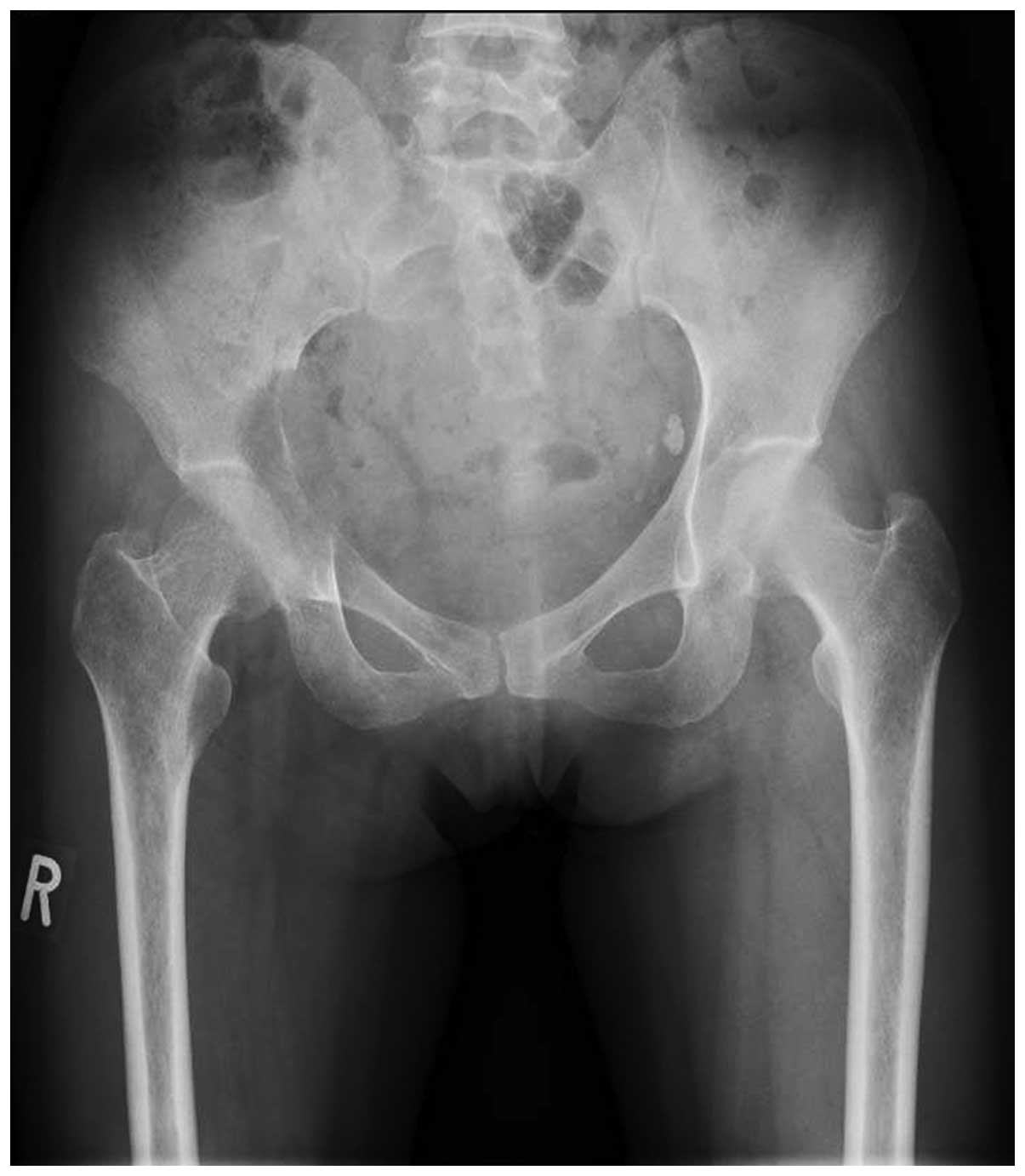
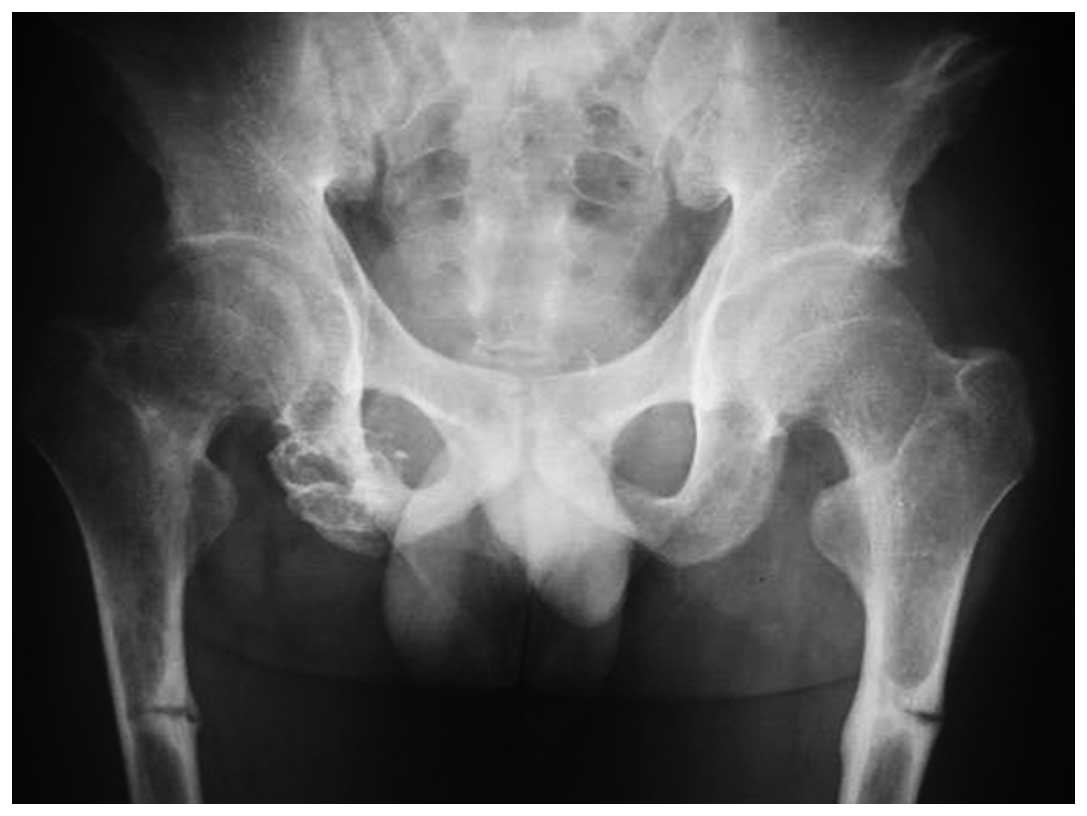
1). X-ray examination revealed an osteolytic lesion in the
right pelvis (Fig. 2), and
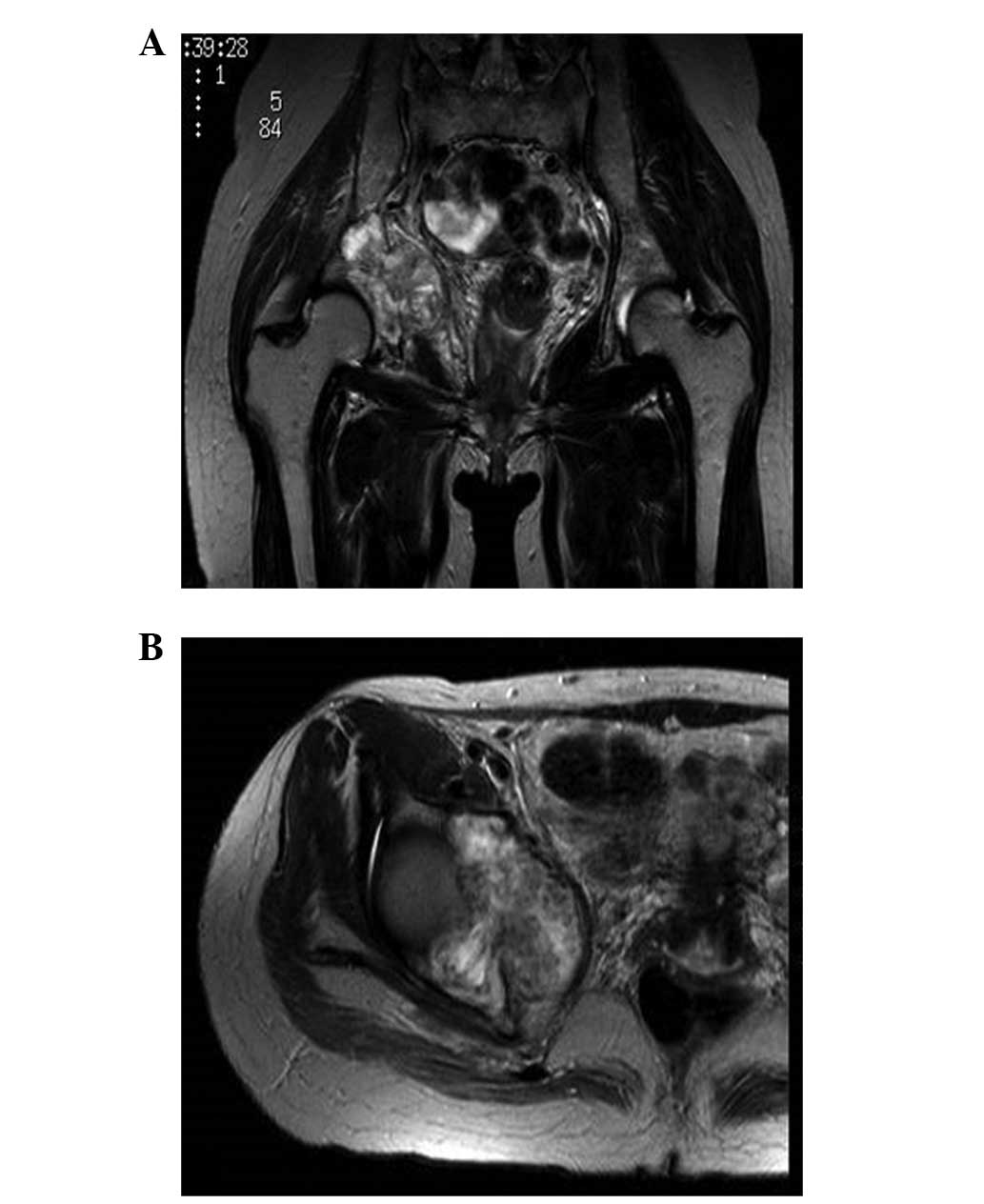
T2-weighted magnetic resonance imaging (MRI) revealed a mass with
inhomogeneous intensity in the right acetabulum (Fig. 3). The diagnosis was of multiple bone
metastases from papillary thyroid carcinoma. Radiation therapy to
the right pelvis (40 Gy/20 fractions) followed a total
thyroidectomy. However, the serum Tg levels normalized completely
following thyroidectomy, despite the presence of multiple lesions
considered to be metastases. Bone scintigraphy revealed multiple
linear hot spots over the ribs, as frequently observed with
pseudofractures in osteomalacia. Subsequently, an open biopsy of
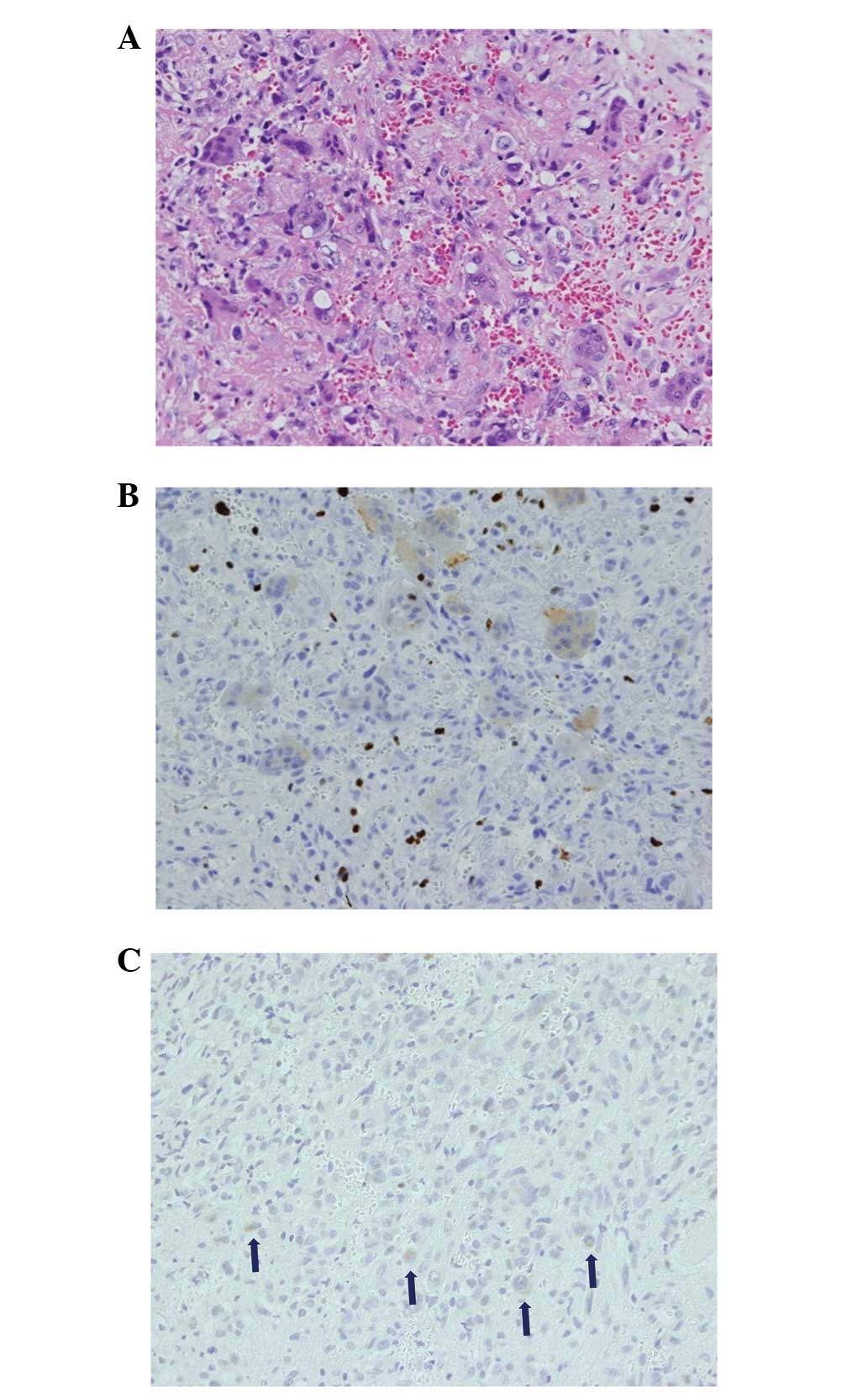
the pelvic lesion was performed. Histopathology showed spindle and
round cells, with multinucleated giant cells in a collagenous
matrix with capillaries (Fig. 4A).
Immunological studies revealed FGF-23-positive tumor cells and a
Ki-67 index of 20% (Fig. 4B and C).
In addition, the serum FGF-23 levels were elevated to 121 pg/ml
(reference range, 10–50 pg/ml). Based on these findings, the tumor
was diagnosed as a malignant PMT. The patient rejected wide
resection of the pelvic tumor with reconstructive total hip
arthroplasty. Subsequently, transcatheter arterial embolization
(TAE) of the feeding artery of the pelvic tumor was performed, and
the patient was administered disodium phosphate (2 g/day) and
vitamin D (alphacalcidol; 2 μl/day) with monitoring serum phosphate
and 1.25(OH2)D co- ncentrations. The tumor decreased in
size after TAE had been performed twice. The serum phosphate and
ALP levels were gradually normalized, and the multiple uptake on
FDG-PET also disappeared, with the exception of the pelvic lesion,
which indicated that this uptake was due to pseudofractures as
opposed to malignancies. However, regrowth of the pelvic tumor and
multiple metastases in the lung and bones were observed 32 months
after the second TAE. In addition, leukocytosis (24,950 cells/ml),
without C-reactive protein elevation, and a high level of
granulocyte colony-stimulating factor (G-CSF; 713 pg/ml) were
observed. It is possible that the tumor had been transformed from
an FGF-23-producing tumor to a G-CSF-producing tumor. Chemotherapy
consisting of combined Adriamycin (55 mg/m2) and
ifosfamide (8 g/m2), and combined gemcitabine (900
mg/m2) and docetaxel (75 mg/m2), was
administered, however no effect was observed and the patient
succumbed to rapidly progressive lung metastases.
Case two
In 1982, a 10-year-old male was originally diagnosed
with hypophosphatemic osteomalacia of unknown cause. At 25 years
old, an intrapelvic tumor was incidentally found, however, the
patient did not undergo a detailed examination due to a lack of
enlargement in the subsequent two years. In December 2003, at 31
years old, the patient visited the Osaka Koseinen-kin Hospital
(Osaka, Japan) due to worsening bilateral thigh pain and gait
disturbance. A TIO was suspected and as a result, the patient was
referred to the Osaka Medical Center for Cancer and Cardiovascular
Diseases (Osaka, Japan). The laboratory analysis revealed severe
hypophosphatemia (1.3 mg/ml), accompanied by high ALP (1,648 mg/ml)
and FGF-23 (3,319 ng/ml) levels. X-ray examination revealed a bony
destructive lesion, with calcification in the right ischium and
ununited fractures in the shaft of bilateral femora, indicating
Looser’s zones (Fig. 5). In
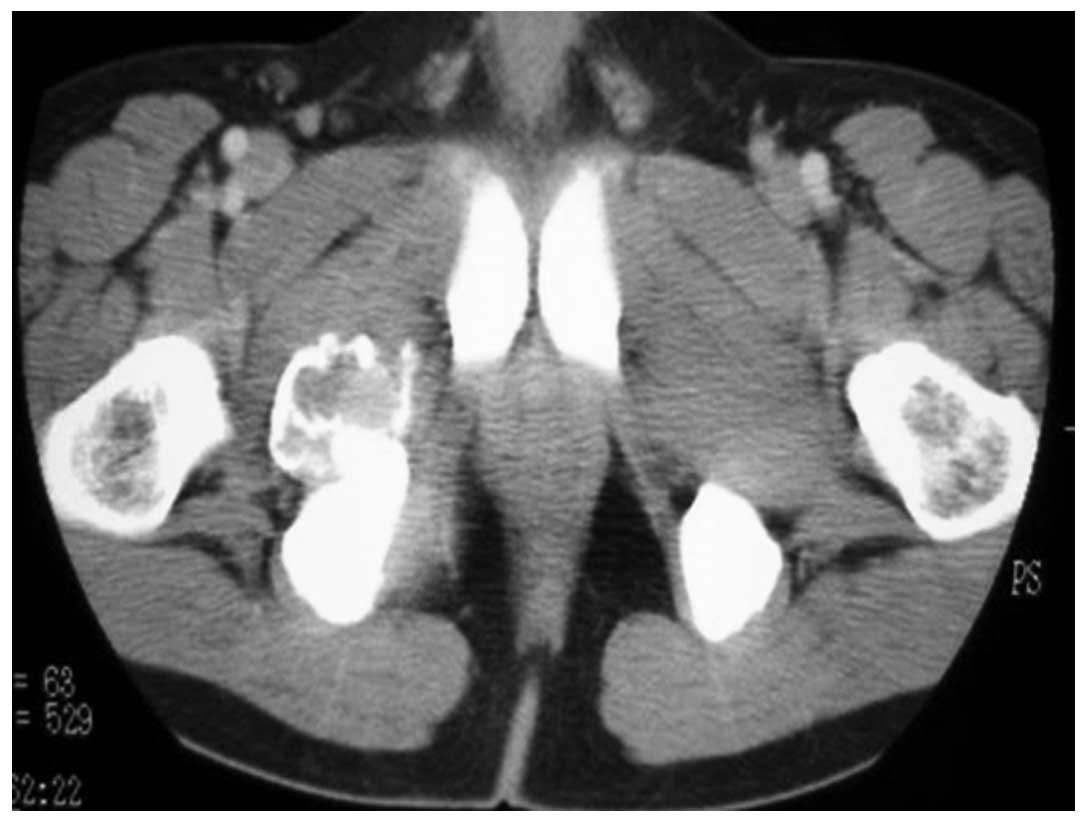
addition, contrast-enhanced computed tomography (CT) revealed an
inhomogeneously enhanced mass in the pelvis (Fig. 6). The tumor had increased in size
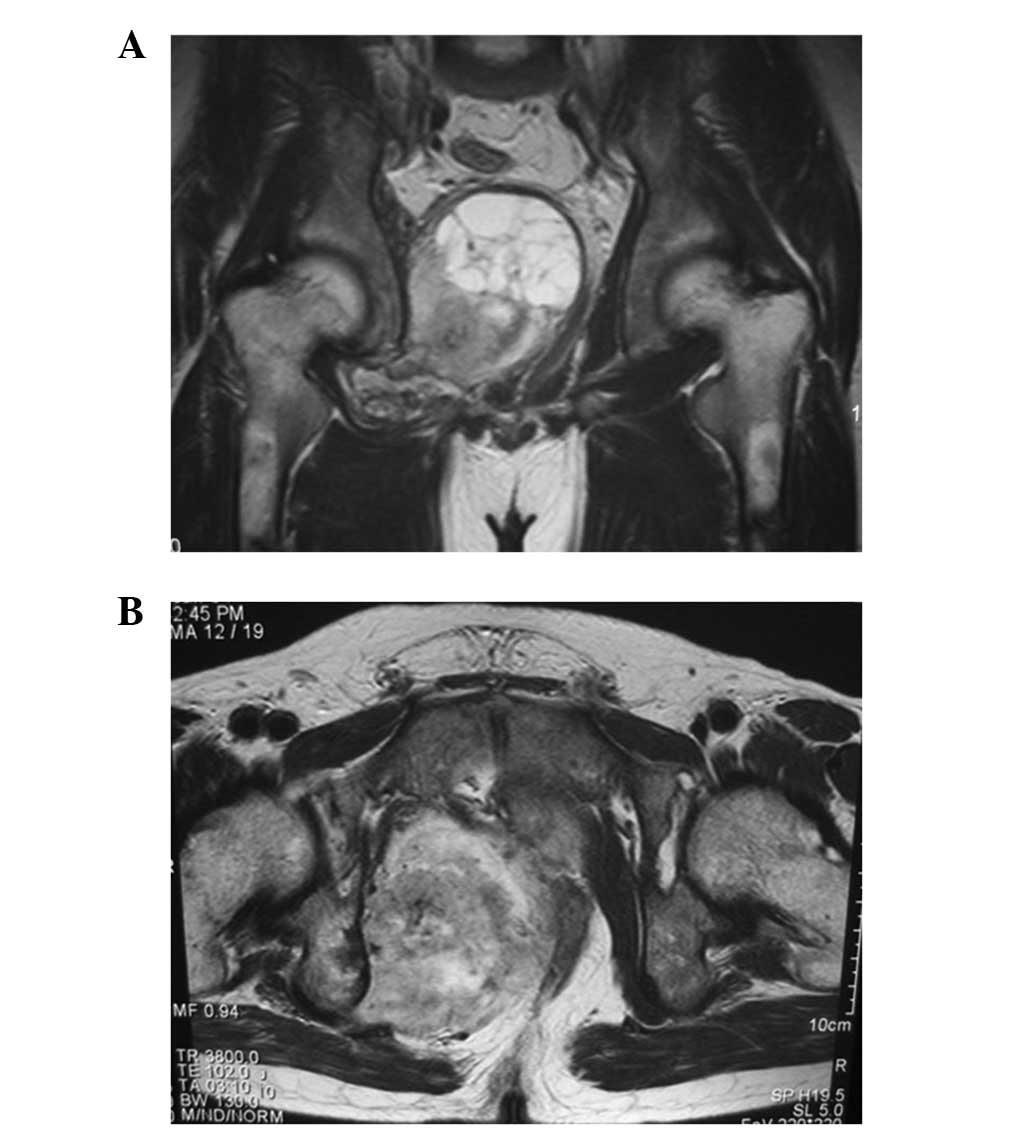
since its discovery when the patient was 25 years old, and MRI
revealed a heterogeneous intensity mass with partial cystic change
compressing the rectum and bladder (Fig. 7). A bone scan showed multiple
abnormal accumulations, indicating multiple pseudofractures. An
open biopsy was subsequently performed. The histopathology of the
pelvic lesion revealed spindle cells in a collagenous and
cartilaginous matrix, with vasculature, but without cytological
atypia or mitosis. The diagnosis was of a benign PMT of the mixed
connective tissue (MCT) type. The patient underwent two courses of
TAE of the feeding artery of the pelvic tumor, with only limited
response, and therefore subsequently underwent tumor excision. The
tumor was completely resected with tumor-free margins, and the
serum phosphorus levels were normalized within 10 days after
surgery. Furthermore, on radiographs, the bilateral femoral
pseudofractures were shown to gradually heal. However, two years
after the surgery, follow-up blood tests revealed elevated serum
FGF-23 (230 pg/ml) levels, and CT and MRI showed local recurrence
in the pelvis. In addition, FDG-PET CT showed local recurrence in
the pelvis, multiple coin lesions in the lung and a subcutaneous
mass in the left elbow with FDG uptake. Metastases of the PMT were
diagnosed clinically and the patient underwent chemotherapy,
including two courses of gemcitabine (1,000 mg/m2) and
docetaxel (120 mg/m2), with no effect. The patient then
developed metastases in the bilateral lungs, bones and liver. Upon
pathological analysis, the diagnosis was of metastases from the PMT
with malignant transformation. The patient underwent liver
metastasis resection, followed by two courses of Adriamycin (59
mg/m2) and ifosfamide (7 g/m2), and two
courses of Adriamycin (71 mg/m2) and cisplatin (76
mg/m2). A complete response of the lung lesion was
achieved briefly, whereas the intrapelvic tumor continued to grow
despite radiation therapy and TAE. In addition, the skin of the
buttocks became thinner, ultimately forming a malignant ulcer. The
patient succumbed to respiratory failure due to relapsing lung
metastases and disseminated intravascular coagulation.
Discussion
The first case presented in the current study was
difficult to diagnose, as the malignant pelvic PMT presented as a
synchronous double cancer with thyroid carcinoma. TAE was selected,
as the patient was unable to accept the functional impairment that
would be a result of surgery. Although TAE safely achieved a
temporary cytoreductive effect without massive release of FGF-23,
32 months after the second course of TAE, regrowth of the pelvic
tumor and multiple metastases emerged. The patient succumbed to the
rapid progression of the lung metastases.
The second case remained undiagnosed for a long
period, as TIO was not commonly recognized at the time. Therefore,
when the patient was eventually diagnosed with TIO, the tumor was
too large to perform a wide resection with a curative margin. Upon
analysis of a biopsy, the initial diagnosis was of a benign
PMT-MCT, however, two years after surgery, multiple metastases
appeared and resection of the liver metastasis revealed malignant
PMT-MCT.
Ogose et al (9) reported local recurrence and malignant
transformation from benign PMT in the course of long-term
follow-up. Therefore, TIO patients must be followed up even if
diagnosed with a benign tumor. In certain cases of TIO, it is
difficult and time-consuming to detect the tumor inducing the
osteomalacia (9–15). However, it is important to identify
the tumor, as it may be malignant or change from benign to
malignant. Previous studies have demonstrated the usefulness of
FDG-PET CT in detecting the tumor inducing the osteomalacia
(16–20). However, FDG accumulation may also
occur in a pseudofracture, as observed in case one of the current
study. Therefore, it is important to distinguish multiple
metastases and pseudofractures from TIO.
With regard to chemotherapy, few studies have
investigated the chemotherapeutic treatment of malignant PMT
(21,22). Seijas et al (21) reported second distant metastases,
however, the use of six cycles of Adriamycin stabilized the
metastasis sites for two years. Sidell et al (22) reported only a limited response to
chemotherapy consisting of doxorubicin, docetaxel and gemcitabine,
however, significant tumor destruction was observed histologically.
In case two of the current study, a complete response of the
metastatic lung lesion was temporarily achieved using an
Adriamycin-based regimen. Therefore, Adriamycin may exhibit tumor
suppressive activity, however, additional evaluations are
required.
References
|
1
|
Folpe AL, Fanburg-Smith JC, Billings SD,
Bisceglia M, Bertoni F, Cho JY, Econs MJ, Inwards CY, Jan de Beur
SM, Mentzel T, et al: Most osteomalacia-associated mesenchymal
tumors are a single histopathologic entity: an analysis of 32 cases
and a comprehensive review of the literature. Am J Surg Pathol.
28:1–30. 2004.
|
|
2
|
McCance RA: Osteomalacia with Looser’s
nodes (Milkman’s syndrome) due to a raised resistance to vitamin D
acquired about the age of 15 years. Q J Med. 16:33–46. 1947.
|
|
3
|
Weidner N: Review and update: oncogenic
osteomalacia-rickets. Ultrastruct Pathol. 15:317–333. 1991.
|
|
4
|
Econs MJ and Drezner MK: Tumor-induced
osteomalacia - unveiling a new hormone. N Engl J Med.
330:1679–1681. 1994.
|
|
5
|
Ramon I, Kleynen P, Body JJ and Karmali R:
Fibroblast growth factor 23 and its role in phosphate homeostasis.
Eur J Endocrinol. 162:1–10. 2010.
|
|
6
|
Shimada T, Mizutani S, Muto T, Yoneya T,
Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S and Yamashita T:
Cloning and characterization of FGF23 as a causative factor of
tumor-induced osteomalacia. Proc Natl Acad Sci USA. 98:6500–6505.
2001.
|
|
7
|
Jonsson KB, Zahradnik R, Larsson T, White
KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H,
Ljunggren O, et al: Fibroblast growth factor 23 in oncogenic
osteomalacia and X-linked hypophosphatemia. N Engl J Med.
348:1656–1663. 2003.
|
|
8
|
Weidner N and Santa Cruz D: Phosphaturic
mesenchymal tumors. A polymorphous group causing osteomalacia or
rickets. Cancer. 59:1442–1454. 1987.
|
|
9
|
Ogose A, Hotta T, Emura I, Hatano H, Inoue
Y, Umezu H and Endo N: Recurrent malignant variant of phosphaturic
mesenchymal tumor with oncogenic osteomalacia. Skeletal Radiol.
30:99–103. 2001.
|
|
10
|
Harvey JN, Gray C and Belchetz PE:
Oncogenic osteomalacia and malignancy. Clin Endocrinol (Oxf).
37:379–382. 1992.
|
|
11
|
Schapira D, Ben Izhak O, Nachtigal A,
Burstein A, Shalom RB, Shagrawi I and Best LA: Tumor-induced
osteomalacia. Semin Arthritis Rheum. 25:35–46. 1995.
|
|
12
|
Seufert J, Ebert K, Müller J, Eulert J,
Hendrich C, Werner E, Schuüze N, Schulz G, Kenn W, Richtmann H, et
al: Octreotide therapy for tumor-induced osteomalacia. N Engl J
Med. 345:1883–1888. 2001.
|
|
13
|
Takeuchi Y, Suzuki H, Ogura S, Imai R,
Yamazaki Y, Yamashita T, Miyamoto Y, Okazaki H, Nakamura K,
Nakahara K, et al: Venous sampling for fibroblast growth factor-23
confirms preoperative diagnosis of tumor-induced osteomalacia. J
Clin Endocrinol Metab. 89:3979–3982. 2004.
|
|
14
|
Uno T, Kawai K, Kunii N, Fukumoto S,
Shibahara J, Motoi T and Saito N: Osteomalacia caused by skull base
tumor: report of 2 cases. Neurosurgery. 69:E239–E244. 2011.
|
|
15
|
Yun KI, Kim DH and Pyo SW: A phosphaturic
mesenchymal tumor of the floor of the mouth with oncogenic
osteomalacia: report of a case. J Oral Maxillofac Surg. 67:402–405.
2009.
|
|
16
|
Dupond JL, Mahammedi H, Magy N,
Blagosklonov O, Meaux-Ruault N and Kantelip B: Detection of a
mesenchymal tumor responsible for hypophosphatemic osteomalacia
using FDG-PET. Eur J Intern Med. 16:445–446. 2005.
|
|
17
|
Jagtap VS, Sarathi V, Lila AR, Malhotra G,
Sankhe SS, Bandgar T, Menon P and Shah NS: Tumor-induced
osteomalacia: a single center experience. Endocr Pract. 17:177–184.
2011.
|
|
18
|
Khadgawat R, Singh Y, Kansara S, Tandon N,
Bal C, Seith A and Kotwal P: PET/CT localisation of a scapular
haemangiopericytoma with tumour-induced osteomalacia. Singapore Med
J. 50:e55–e57. 2009.
|
|
19
|
Roarke MC and Nguyen BD: PET/CT
localization of phosphaturic mesenchymal neoplasm causing
tumor-induced osteomalacia. Clin Nucl Med. 32:300–301. 2007.
|
|
20
|
Suryawanshi P, Agarwal M, Dhake R, Desai
S, Rekhi B, Reddy KB and Jambhekar NA: Phosphaturic mesenchymal
tumor with chondromyxoid fibroma-like feature: an unusual
morphological appearance. Skeletal Radiol. 40:1481–1485. 2011.
|
|
21
|
Seijas R, Ares O, Sierra J and
Pérez-Dominguez M: Oncogenic osteomalacia: two case reports with
surprisingly different outcomes. Arch Orthop Trauma Surg.
129:533–539. 2009.
|
|
22
|
Sidell D, Lai C, Bhuta S, Barnes L and
Chhetri DK: Malignant phosphaturic mesenchymal tumor of the larynx.
Laryngoscope. 121:1860–1863. 2011.
|