Introduction
The majority of adrenal lesions are benign adrenal
cortical adenomas. However, nodular lymphocyte-predominant Hodgkin
lymphoma (NLPHL) is a rare disease manifestation in the adrenal
gland. NLPHL is a subtype of Hodgkin lymphoma (HL), which
represents ~5% of HLs and involves peripheral lymph nodes. NLPHL is
a ‘benign’ disease which is characterized by an indolent course
(1,2). Mortalities due to NLPHL are rare,
however treatment toxicities and varsious secondary malignancies
may contribute to the overall morality rate. Secondary aggressive
non-Hodgkin lymphoma may occur in ~7–14% of NLPHL cases, but the
majority of NLPHL patients present at an early stage and have a
favourable prognosis (3–6). The peripheral and most commonly the
cervical regions are involved (1,2). NLPHL
has a peak incidence in the fourth decade of life with a
significant male predominance (75%). B symptoms, extranodal
involvement and bulky disease are particularly uncommon (7). We report a unique case of NLPHL
affecting only the adrenal gland in a patient who was successfully
diagnosed by histological examination, and treated with surgery and
a chemotherapy regimen of doxorubicin, bleomycin, vinblastine and
dacarbazine. This study was approved by the ethics committee of the
Second Xiangya Hospital of Central South University (Changsha,
China). Patient provided written informed consent.
Case report
Case presentation
A 36-year-old male presented at The Second Xiangya
Hospital of Central South University (Changsha, China) with high
fever and weight loss lasting for 10 days, as well as a left
adrenal mass that had been identified one week previously. The
patient had no history of hypertension and physical examination
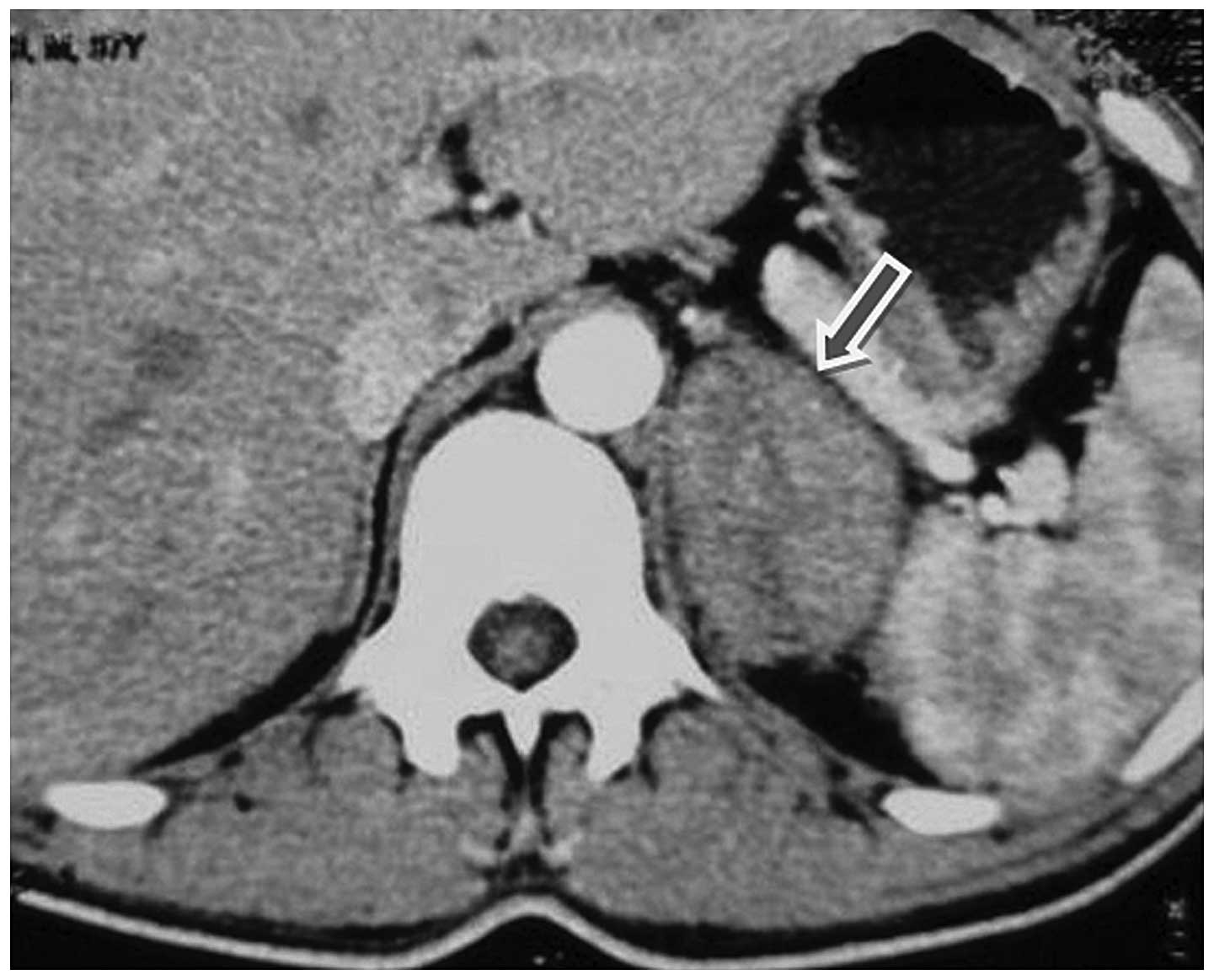
showed no abnormalities. A total body computed tomography (CT) scan
revealed the presence of a large left adrenal mass measuring
4.5×5.5 cm in size (Fig. 1). No
obvious abnormalities in the right adrenal gland, retroperitoneal
area and other parts of the body were identified by CT. Bone marrow
aspiration and biopsy were normal. Plasma metanephrine, plasma
renin activity, serum aldosterone, dehydroepiandrosterone and basic
metabolic panel test results were all normal. Routine hematological
parameters and liver and kidney function tests were normal.
Hepatitis B and C and human immunodeficiency virus serology test
results were negative. In addition, Epstein-Barr virus (EBV)
serology showed negativity for immunoglobulin M (IgM) and IgG
antibodies. The patient subsequently underwent laparoscopic left
adrenalectomy.
Pathology
The adrenalectomy specimen measured 5.5×4.0×4.0 cm.
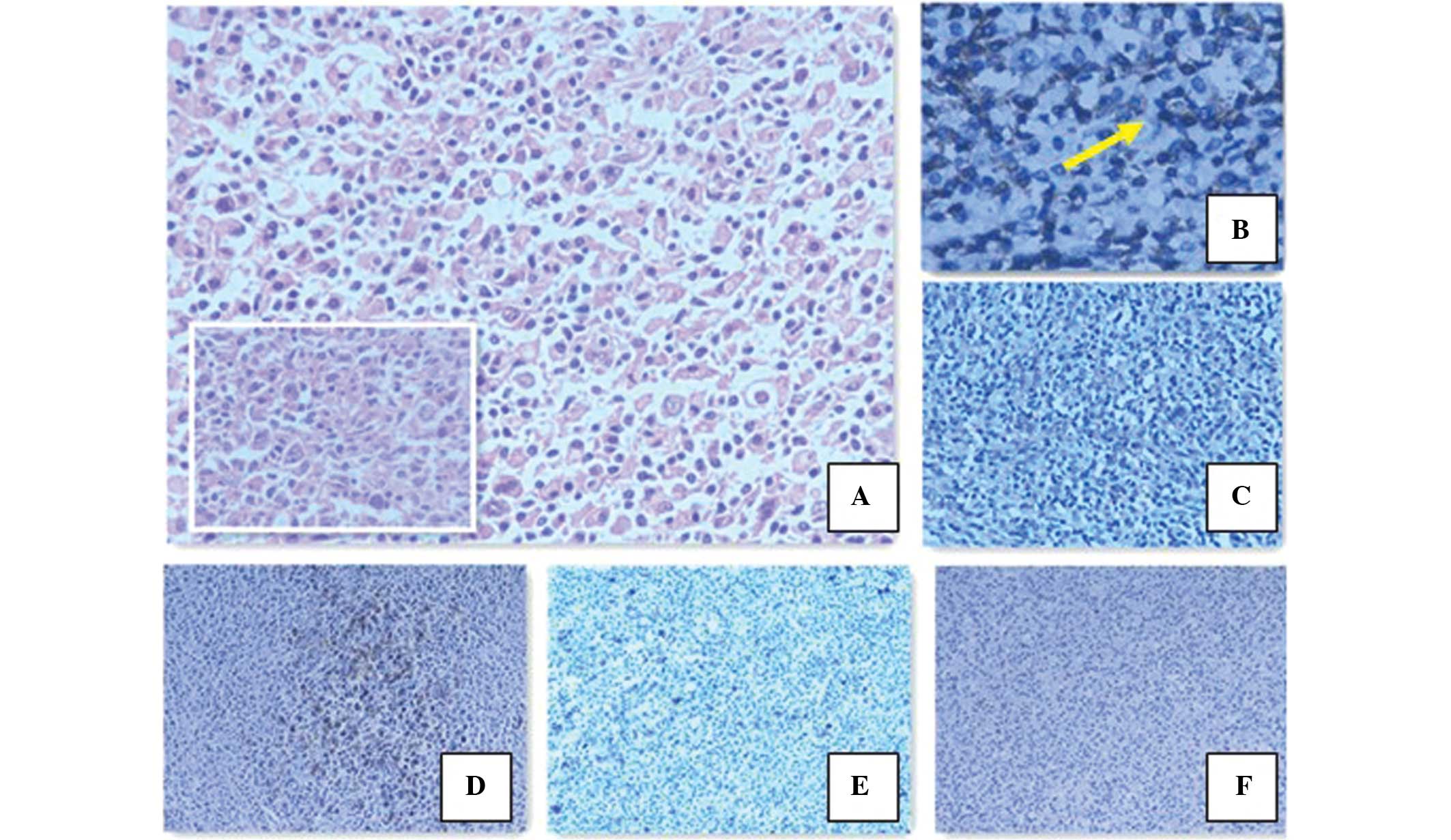
Microscopic examination revealed tumors of lymphoid-hematopoietic
tissues in the adrenal tissue, which destroyed the adrenal gland
and formed a mass in the retroperitoneum. Morphologically, the
majority of atypical cells resembled lymphocyte-predominant (LP)
cells. Immunohistochemical analysis showed that the tumor cells
were negative for cluster of differentiation 30 (CD30) and
EBV-encoded small RNAs (EBERs), and positive for CD20, paired box
protein 5, CD79a, octamer binding protein 2 and epithelial membrane
antigen (EMA). Additionally, very few cells were positive for CD15.
The tumor cells were surrounded by T cells, which were positive for
CD3 and CD57 (Fig. 2). According to
the immunohistochemistry results, the patient was diagnosed with
primary adrenal NLPHL, stage IB according to the World Health
Organization (WHO) (8).
Postoperative treatment
Systemic positron emission tomography (PET)-CT
examination was conducted following surgery, and there were no
obvious abnormalities. Postoperative adjuvant chemotherapy was
administered; the patient received four cycles of doxorubicin,
bleomycin, vinblastine and darcarbazine (ABVD) regimen. This
comprised 25 mg/m2 doxorubicin (day 1 and 14), 10
mg/m2 bleomycin (day 1 and 14), 6 mg/m2
vinblastine (day 1 and 14) and 375 mg/m2 dacarbazine
(day 1 and 14), to be repeated every four weeks. The patient has
been followed up for 16 months from the end of chemotherapy, with a
stable condition and no recurrence.
Discussion
According to the present WHO classification, HLs
comprise two disease entities: NLPHL and classical HL (cHL)
(8). cHL represents ~95% of HLs,
while NLPHL is rare, representing ~5%. NLPHL has a male
predominance and typically presents in middle-aged individuals.
NLPHL frequently presents as localized disease and has a good
outcome. The ten year overall survival rate for early-stage
patients is ~85–100% (3,4). Approximately 20–25% of NLPHL patients
present with an advanced stage of the disease. Xing et al
(9)demonstrated that the ten year
overall survival for patients with advanced NLPHL is 86%. The most
common sites of involvement are the peripheral lymph nodes, and
disease manifestation in the adrenal gland is rare. To the best of
our knowledge, only three cases of primary adrenal HL have been
reported worldwide, two of which were adrenal cHL (10,11)
and one of which was composite lymphoma of NLPHL and cHL
concurrently affecting the same lymph nodes (12).
Ultrasound, CT and magnetic resonance imaging have
become the preferred methods for identifying adrenal tumors. PET-CT
is not only able to distinguish between benign and malignant tumors
of the adrenal gland in order to make a diagnosis, but also plays a
significant role in the staging, evaluation of therapeutic effects
and prediction of patient prognosis in HL (13).
NLPHL is characterized by a nodular or nodular and
diffuse growth pattern. The neoplastic cells in NLPHL have been
known as lymphocytic and histiocytic cells, LP cells or ‘popcorn’
cells, which are embedded in a nodular non-neoplastic background
mainly composed of small B lymphocytes and some CD57+ T
cells. The immunophenotype of NLPHL is useful for the diagnosis. LP
cells are positive for B-cell markers including CD19, CD20, CD22
and CD79a, in addition to CD45 and EMA; however, they lack
expression of CD15 and CD30, which are the characteristic markers
for cHL. In the background, CD3+ or CD57+ T
cells typically surround the LP cells, forming what are commonly
referred to as T-cell rosettes (14). NLPHL is not associated with EBV
infection, so EBERs are negative in LP cells. The tumor cells of
NLPHL originate from B cells characterized by somatic mutations in
the variable domain of the rearranged Ig heavy chain genes and the
expression of B-cell markers. The morphological and
immunohistochemical features in the present case were consistent
with the diagnosis of NLPHL.
Patients with NLPHL are mainly treated with
radiotherapy and chemotherapy (15,16).
Surgery is mainly used to obtain the pathological tissue and
diagnosis, and does not affect the prognosis (6).
In early-stage, limited NLPHL cases (I and II stage)
treatment with radiotherapy alone has been recommended in the past.
Clinical studies on 202 patients in Australasia showed that the
15-year overall survival (OS) rate was 83% and the freedom from
progression rate was 82% in patients with I-II stage LPHL treated
with radiotherapy alone (17).
Another study, conducted in Germany, showed that of 131 patients,
98% of patients with stage IA LPHL achieved complete remission
following radiotherapy (18). The
suggested cumulative dose is 30–36 Gy (19). However, more recent studies have
found that treating limited-stage NLPHL with ABVD-like chemotherapy
for two cycles followed by radiotherapy may improve outcome
compared with the use of radiation alone (3,20). A
retrospective study analyzed 88 patients with limited stage NLPHL
and the results showed a marked improvement in the 10-year time to
progression (98 vs. 76%; P=0.0074), progression-free survival (91
vs. 65%; P=0.0024) and OS (93 vs. 84%; P=0.074) for patients
treated with ABVD followed by radiotherapy compared with those
treated with radiotherapy alone (3).
Patients with early-stage NLPHL who have received
ABVD chemotherapy and/or radiotherapy have a good prognosis;
studies have shown that the 10-year overall survival rate is
>90%, even up to 100% (1,2).
Patients who seek to achieve long-term survival must consider the
side effects of treatment and treatment-related mortality. A recent
study of 405 patients showed that the 12-year overall survival rate
was 94% among those receiving treatment with ABVD alone, as
compared with 87% among those receiving radiation therapy (P=0.04).
It was suggested that among patients with early-stage HL, ABVD
therapy alone was associated with a higher rate of overall survival
compared with radiation therapy, owing to a lower rate of death
from other causes (21). However, a
number of studies have suggested a watch-and-wait strategy as a
potentially curative approach after initial lymph node surgery,
particularly for children (2,22,23).
The optimal treatment for limited-stage NLPHL is unclear, and very
few cases of adrenal NLPHL have been reported. As a result, there
is no standard treatment for adrenal NLPHL and a suitable treatment
can only be determined based on that of other types of NLPHL. In
order to reduce treatment-related toxicity, the patient in the
present case was only treated with adjuvant chemotherapy after
surgery. The invasion site of the tumor presented in this case was
the kidney. In order to prevent damage to the kideny from
radiotherapy and to reduce treatment-related toxicity, the patient
was treated only with ABVD adjuvant chemotherapy following a
laparoscopic left adrenalectomy.
The current recommendation for advanced-stage NLPHL
consists of standard cHL protocols, with ABVD chemotherapy and
combined therapy including radiotherapy (3).
The LP cells in NLPHL express high levels of CD20
antigen, rituximab, which has emerged as a potential treatment
option for NLPHL. Ekstrand et al (24) conducted a phase 2 trial where 22
patients with LPHL (untreated and previously treated, stages I-III)
were treated with rituximab. The overall response rate was 100%.
The German Hodgkin Lymphoma Study Group reported an overall
response rate (ORR) of 94% in NLPHL patients administered with
rituximab, including eight patients who achieved complete remission
(CR) and six who achieved partial remission (PR) (25). It was suggested that rituximab was
highly effective in relapsed and refractory NLPHL. In another phase
2 trial, newly diagnosed stage IA NLPHL patients treated with
rituximab were investigated. Among the 28 evaluable patients, the
ORR was 100%, 85.7% patients achieved CR and 14.3% achieved PR, and
the 43-month OS rate was 100% (14). Treatment results with rituximab
appear inferior compared with radiotherapy and combined-modality
approaches in early-stage patients. The 2012 National Comprehensive
Cancer Network guidelines recommend rituximab for the treatment of
NLPHL of stage Ib or greater (26).
Primary adrenal NLPHL is extremely rare, and
pathological analysis and immunohistochemical examination are key
in achieving a diagnosis. Due to the rarity of NLPHL, there have
been few prospective studies, and as a result, a wide range of
management approaches are available. Thus, the optimal management
strategy for NLPHL is unclear. Patients with limited stage NLPHL
treated with ABVD chemotherapy followed by radiotherapy have more
successful outcome compared with those treated with radiotherapy
alone. The current recommendation for advanced-stage NLPHL consists
of standard cHL protocols, with ABVD chemotherapy and combined
therapy including radiotherapy and rituximab. Rituximab has emerged
as a potential treatment option for NLPHL. The optimal treatment of
NLPHL in the era of targeted therapy should be further explored.
Further studies, particularly long-term, prospective randomized
clinical trials are required to improve the diagnostic techniques
and the optimal treatment of NLPHL. Additionally, the differential
diagnosis of adrenal tumors should be considered.
References
|
1
|
Nogová L, Rudiger T and Engert A: Biology,
clinical course and management of nodular lymphocyte-predominant
hodgkin lymphoma. Hematology Am Soc Hematol Educ Program.
2006:266–272. 2006.
|
|
2
|
Illés A, Simon Z, Tóth E, Rosta A,
Miltényi Z and Molnár Z: Nodular lymphocyte predominant Hodgkin
lymphoma (NLPHL)-clinicopathological features based on the data of
two Hungarian lymphoma centres. Pathol Oncol Res. 14:411–421.
2008.
|
|
3
|
Savage KJ, Skinnider B, Al-Mansour M, Sehn
LH, Gascoyne RD and Connors JM: Treating limited-stage nodular
lymphocyte predominant Hodgkin lymphoma similarly to classical
Hodgkin lymphoma with ABVD may improve outcome. Blood.
118:4585–4590. 2011.
|
|
4
|
Chen RC, Chin MS, Ng AK, et al:
Early-stage, lymphocyte-predominant Hodgkin’s lymphoma: patient
outcomes from a large, single-institution series with long
follow-up. J Clin Oncol. 28:136–141. 2010.
|
|
5
|
Al-Mansour M, Connors JM, Gascoyne RD,
Skinnider B and Savage KJ: Transformation to aggressive lymphoma in
nodular lymphocyte-predominant Hodgkin’s lymphoma. J Clin Oncol.
28:793–799. 2010.
|
|
6
|
Biasoli I, Stamatoullas A, Meignin V, et
al: Nodular, lymphocyte-predominant Hodgkin lymphoma: a long-term
study and analysis of transformation to diffuse large B-cell
lymphoma in a cohort of 164 patients from the Adult Lymphoma Study
Group. Cancer. 116:631–639. 2010.
|
|
7
|
Nogová L, Reineke T, Brillant C, et al:
Lymphocyte-predominant and classical Hodgkin’s lymphoma: a
comprehensive analysis from the German Hodgkin Study Group. J Clin
Oncol. 26:434–439. 2008.
|
|
8
|
Harris NL, Jaffe ES, Diebold J, et al:
World Health Organization Classification of neoplastic diseases of
hematopoetic and lymphoid tissues: Report of the Clinical Advisory
Committee Meeting - Airlie House, Virginia, November 1997. J Clin
Oncol. 17:3835–3849. 1999.
|
|
9
|
Xing KH, Connors JM, Lai A, et al: The
outcome of advanced stage nodular lymphocyte predominant Hodgkin’s
lymphoma (NLPHL) compared to classical Hodgkin’s lymphoma (CHL): a
matched pair analysis. Blood. 123:3567–3573. 2014.
|
|
10
|
Hagspiel KD: Manifestation of Hodgkin’s
lymphoma in an adrenal myelolipoma. Eur Radiol. 15:1757–1759.
2005.
|
|
11
|
Bourne AE, Bell SW, Wayment RO and
Schwartz BF: Primary Hodgkin lymphoma of the adrenal gland: a
unique case presentation. Cn J Urol. 16:4694–4696. 2009.
|
|
12
|
Szczepanowski M, Masqué-Soler N, Oschlies
I, Schmidt W, Lück A and Klapper W: Composite lymphoma of nodular
lymphocyte-predominant and classical Hodgkin lymphoma-Epstein-Barr
virus association suggests divergent pathogenesis despite clonal
relatedness. Hum Pathol. 44:1434–1439. 2013.
|
|
13
|
Engert A, Haverkamp H, Kobe C, et al:
Reduced-intensity chemotherapy and PET-guided radiotherapy in
patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a
randomised, open-label, phase 3 non-inferiority trial. Lancet.
379:1791–1799. 2012.
|
|
14
|
Anagnostopoulos I, Hansmann ML, Franssila
K, et al: European Task Force on Lymphoma project on lymphocyte
predominance Hodgkin disease: histologic and immunohistologic
analysis of submitted cases reveals 2 types of Hodgkin disease with
a nodular growth pattern and abundant lymphocytes. Blood.
96:1889–1899. 2000.
|
|
15
|
Eichenauer DA, Fuchs M, Pluetschow A,
Klimm B, et al: Phase 2 study of rituximab in newly diagnosed stage
IA nodular lymphocyte-predominant Hodgkin lymphoma: a report from
the German Hodgkin Study Group. Blood. 118:4363–4365. 2011.
|
|
16
|
Eichenauer DA, Engert A and Dreyling M;
ESMO Guidelines Working Group. Hodgkin’s lymphoma: ESMO Clinical
Practice Guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 22(Suppl 6): Svi55–Svi58. 2011.
|
|
17
|
Wirth A, Yuen K and Barton M: Long-term
outcome after radiotherapy alone for lymphocyte-predominant Hodgkin
lymphoma: a retrospective multicenter study of the Australasian
Radiation Oncology Lymphoma Group. Cancer. 104:1221–1229. 2005.
|
|
18
|
Nogova L, Reineke T, Eich HT, et al:
Extended field radiotherapy, combined modality treatment or
involved field radiotherapy for patients with stage IA
lymphocyte-predominant Hodgkin’s lymphoma: a retrospective analysis
from German Hodgkin Study Group (GHSG). Ann Oncol. 16:1683–1687.
2005.
|
|
19
|
Specht L, Yahalom J, Illidge T, et al:
Modern radiation therapy for Hodgkin lymphoma: field and dose
guidelines from the International Lymphoma Radiation Oncology Group
(ILROG). Int J Radiat Oncol Biol Phys. DOI:
10.1016/j.ijrobp.2013.05.005
|
|
20
|
Savage KJ, Hoskins P, Klasa R, et al: ABVD
chemotherapy is essential for optimal treatment of limited stage
nodular lymphocyte predominant Hodgkin lymphoma. Hematologica.
92(Suppl 5): 27–28. 2007.
|
|
21
|
Meyer RM, Gospodarowicz MK, Connors JM, et
al: NCIC Clinical Trials Group; Eastern Cooperative Oncology Group:
ABVD alone versus radiation-based therapy in limited-stage
Hodgkin’s lymphoma. N Engl J Med. 366:399–408. 2012.
|
|
22
|
Pellegrino B, Terrier-Lacombe MJ, Oberlin
O, et al: Lymphocyte-predominant Hodgkin’s lymphoma in children:
therapeutic abstention after initial lymph node resection - a Study
of the French Society of Pediatric Oncology. J Clin Oncol.
21:2948–2952. 2003.
|
|
23
|
Mauz-Körholz C, Gorde-Grosjean S,
Hasenclever D, et al: Resection alone in 58 children with limited
stage, lymphocyte-predominant Hodgkin lymphoma-experience from the
European network group on pediatric Hodgkin lymphoma. Cancer.
110:179–185. 2007.
|
|
24
|
Ekstrand BC, Lucas JB, Horwitz SM, et al:
Rituximab in lymphocyte-predominant Hodgkin disease: results of a
phase 2 trial. Blood. 101:4285–4289. 2003.
|
|
25
|
Schulz H, Rehwald U, Morschhauser F, et
al: Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma:
long-term results of a phase 2 trial by the German Hodgkin Lymphoma
Study Group (GHSG). Blood. 111:109–111. 2008.
|
|
26
|
Hoppe RT, Advani RH, Ai WZ, et al: Hodgkin
lymphoma, version 2.2012 featured updates to the NCCN guidelines. J
Natl Compr Canc Netw. 10:589–597. 2012.
|