Introduction
Ovarian cancer is a serious global threat to female
health and is a leading cause of cancer-related mortality in
females, often due to late-stage recognition and aggressive tumor
relapse (1). High patient morbidity
is attributable in part to the recurrent growth of residual ovarian
tumor cells that become resistant to standard chemotherapeutic
treatments, and then aggressively proliferate and spread or
metastasize to multiple sites. In total, 70% of females diagnosed
with ovarian cancer present with advanced malignant disease and
usually undergo surgery followed by a combination of paclitaxel and
platinum-based chemotherapy (2,3).
However, recurrences occur in the majority of patients, and only
~30% of patients with distant metastases survive five years
following diagnosis (4,5). Failure of chemotherapy in recurrent
ovarian cancer is usually due to the development of resistance to
one or the two main classes of chemotherapy agents used to treat
ovarian cancer (6–8). Novel therapeutic approaches are
therefore necessary for the management of advanced and recurrent
ovarian cancer.
Platinating agents, such as carboplatin, are potent
chemotherapeutic agents widely used for the adjuvant treatment of
primary ovarian cancer and metastatic disease. The drug induces the
formation of DNA adducts, G2 phase cell cycle arrest and
the subsequent triggering of apoptosis. However, the efficacy of
carboplatin is limited by drug resistance and side-effects,
including nephrotoxicity, myelosuppression and neurotoxicity
(9,10). The mechanisms underlying the
development of resistance to platinating agents, particularly
carboplatin, include the repair of DNA lesions, translesion DNA
synthesis, altered drug transport, increased antioxidant production
and reduction of apoptosis (11–13).
Altered gene expression affecting cellular transport, DNA repair,
apoptosis and cell-cell adhesion are the mechanisms of platinum
resistance that have been observed in patient samples (14,15).
Therapeutic success may therefore be improved if tumor cells can be
sensitized to carboplatin treatment with a combination therapy.
1α,25-Dihydroxyvitamin D3
[1,25(OH)2D3] is the most active metabolite
of vitamin D3. It is a scarce natural product that is synthesized
predominantly in the skin from 7-dehydrocholesterol by exposure to
ultraviolet sunlight. Although its classical role as the major
regulator of calcium homeostasis and bone formation/resorption has
been recognized for some time (16), more recent findings suggest that
1,25(OH)2D3 is an important modulator of
cellular proliferation and differentiation in a variety of benign
and malignant cells. 1,25(OH)2D3 also
exhibits anti-invasion, antiangiogenesis and antimetastatic
activity in vivo (17–21)
and acts as a chemopreventive agent in animal models of lung,
colorectal and breast cancer (22–24).
The aim of this study was to determine whether
1,25(OH)2D3 enhances the cytostatic effects
of carboplatin in SKOV-3 cells and to characterize the mechanism of
its effect.
Materials and methods
Cell culture and agents
The human ovarian serous papillary
cystadenocarcinoma SKOV-3 cell line was purchased from the Type
Culture Collection of the Chinese Academy of Sciences (TCCCAS;
Shanghai, China) and was verified as mycoplasma free. Authenticity
of the cell line was confirmed by the TCCCAS. The SKOV-3 cells were
maintained in RPMI 1640 medium supplemented with 10% fetal bovine
serum, 100 U/ml penicillin and 5 mg/ml streptomycin. These agents
and trypsin-EDTA solution were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). 1,25(OH)2D3
and carboplatin were purchased from Sigma-Aldrich (St. Louis, MO,
USA) and Qilu Pharmaceutical Co., Ltd. (Jinan, China),
respectively. 1,25(OH)2D3 was dissolved in
ethanol and stored in a concentrated solution (10−5
mol/l) at −80°C. The 1,25(OH)2D3 was freshly
diluted in RPMI 1640 prior to each experiment. The ethanol
concentrations in each experiment were ≤0.1%. The carboplatin
solution was prepared with sterile distilled water and fresh stocks
were prepared on the day of each experiment, and dilutions were
prepared with RPMI 1640.
Cell viability assay
The viability of SKOV-3 cells was determined by Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan).
Briefly, cells at the exponential phase were collected, transferred
to 96-well plates (2,000 cells/well) and cultured overnight. The
plating medium was removed and replaced with a medium containing
the appropriate concentration of vehicle (0.1% ethanol),
1,25(OH)2D3 (0.1, 1, 5, 10, 50, 100, 200 and
500 nM) or carboplatin (0.2, 2, 20, 40, 80 and 160 mg/l). The
combined effects were evaluated by incubation with
1,25(OH)2D3 and carboplatin. Cells were
allowed to grow for an additional 72 h, then 10 μl of CCK-8
solution was added and the cells were incubated for 1 h. Absorbance
(Abs) was measured at 450 nm in a microplate reader (BioTec
Instruments, Inc., Winooski, VT, USA) and growth inhibition was
calculated as the percentage difference of the treated cells versus
the vehicle controls, according to the following formula:
Inhibition rate (%) = [(Abs of vehicle control cells - Abs of
treated cells)/Abs of vehicle control cells] × 100. Each experiment
was performed in triplicate.
Data were analyzed using KaleidaGraph (Synergy
Software, Reading, PA, USA) to determine the drug IC50
value. The combined index (CI) was used to evaluate the drug
combination assays according to the following formula (25): CI = DA/IC50,A
+ DB/IC50,B, where DA is the
IC50 of drug A when A was combined with B, DB
is the IC50 of drug B when A was combined with B,
IC50,A is the IC50 of drug A, and
IC50,B is the IC50 of drug B. Each CI was
calculated from the mean affected fraction at each drug ratio
concentration in triplicate. CI>1, CI=1, and CI<1 indicated
antagonism, additive effect or synergy, respectively (26).
Cell cycle analysis
SKOV-3 cells were grown to 50% confluence in 35-mm
dishes and treated with the vehicle control, 10 nM
1,25(OH)2D3, 40 mg/l carboplatin, or a
combination of the two drugs for 72 h. The cells were harvested by
pooling the floating cells with the trypsinized monolayers and were
pelleted by centrifugation at 179 × g for 5 min. Following fixation
with cold 75% ethanol, the cells were resuspended in a solution of
phosphate-buffered saline (PBS; pH 7.4) containing 25 mg/ml
propidium iodide (PI; Sigma-Aldrich), 0.1 mM EDTA (Invitrogen Life
Technologies) and 0.01 mg/ml DNase-free RNase (Invitrogen Molecular
Probes, Inc., Eugene, OR, USA). The samples were incubated for 15
min at room temperature prior to cell cycle analysis using a FC500
flow cytometer (Beckman Coulter, Fullerton, CA, USA). Statistics
were performed on 20,000 events per sample using MultiCycle DNA
Content and DNA cell cycle analysis software (MutiCycle AV for
Windows; Phoenix Flow System, Inc., San Diego, CA, USA).
Apoptosis assay
The number of apoptotic cells was determined using
the Alexa Fluor 488 Annexin V/Dead Cell apoptosis kit (Invitrogen
Life Technologies). Following treatment, the cells were harvested
and washed with PBS, then suspended in PBS with PI and Annexin V.
The cell suspensions were incubated in the dark for 15 min at 37°C
and then analyzed on a FC500 flow cytometer.
Confocal laser-scanning microscopy was performed
using an SP-2 confocal laser-scanning microscope (Leica, Wetzlar,
Germany) equipped with an oil immersion objective (63X). Nuclear
images were obtained at an excitation wavelength of 405 nm of
4′,6-diamidino-2-phenylindole (DAPI).
Measurement of mitochondrial membrane
potential (MMP)
MMP was measured using JC-1 dye (Invitrogen Life
Technologies), a cationic dye that aggregates in the mitochondria
of healthy cells; at high concentrations, JC-1 monomers (green
fluorescence) form aggregates (red fluorescence). The ratio of the
green/red fluorescence is independent of mitochondrial shape,
density or size, and depends only on the membrane potential. MMP
analysis was performed as previously described (27). Briefly, SKOV-3 cells were treated
for 72 h, then harvested and stained with 10 μM JC-1 at 4°C for 1 h
prior to flow cytometry analysis. JC-1 was excited with the 488-nm
argon laser (Beckman Coulter) and JC-1 green and red fluorescence
was recorded using 530 nm ± 15 nm and a 590 nm ± 15 nm band pass
filter channels. A minimum of 20,000 cells within the gated region
were analyzed. The cell sorting gates used were FL-2 versus FL-1
blotting (28). The ratio of the
fluorescence intensity at 590 nm to that at 530 nm (FL-2:FL-1
ratios) was considered the relative MMP value. Data are presented
as the mean of three experiments.
Measurement of intracellular reactive
oxygen species (ROS)
Intracellular ROS was measured by a cell-permeating
probe, 5-[and-6]-chloromethyl-2′,7′-dichlorodihydrofluorescein
diacetate, acetyl ester (CM-H2DCFDA, Invitrogen
Molecular Probes), as previously described (29). CM-H2DCFDA is a non-polar
compound that is hydrolyzed upon cell entry, forming a
non-fluorescent derivative that can be converted into a fluorescent
product in the presence of a true oxidant. Mean fluorescence
intensity was used as measure of ROS level as determined by flow
cytometer. Cells were treated for 72 h, washed and loaded with 10
μM CM-H2DCFDA for 1 h. Green fluorescence intensity was
used as a measure of relative intracellular ROS by flow cytometry
at 530 nm. A total of 20,000 cells within the gated region were
analyzed. Data are presented as the mean of three experiments
Statistical analysis
Statistical analysis was performed using SPSS,
version 13.0 for Windows (SPSS, Inc., Chicago, IL, USA). Data are
presented as the mean ± standard deviation. One-way analysis of
variance was used to evaluate differences between the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Dose-response of SKOV-3 cells to
1,25(OH)2D3 and carboplatin
Initial experiments were performed to determine the
range of drug concentrations that would elicit growth inhibition in
ovarian cancer cells. SKOV-3 cells were incubated with graded
1,25(OH)2D3 (0.1, 1, 5, 10, 50, 100, 200 and
500 nM) and carboplatin (0.2, 2, 20, 40, 80, and 160 mg/l). In the
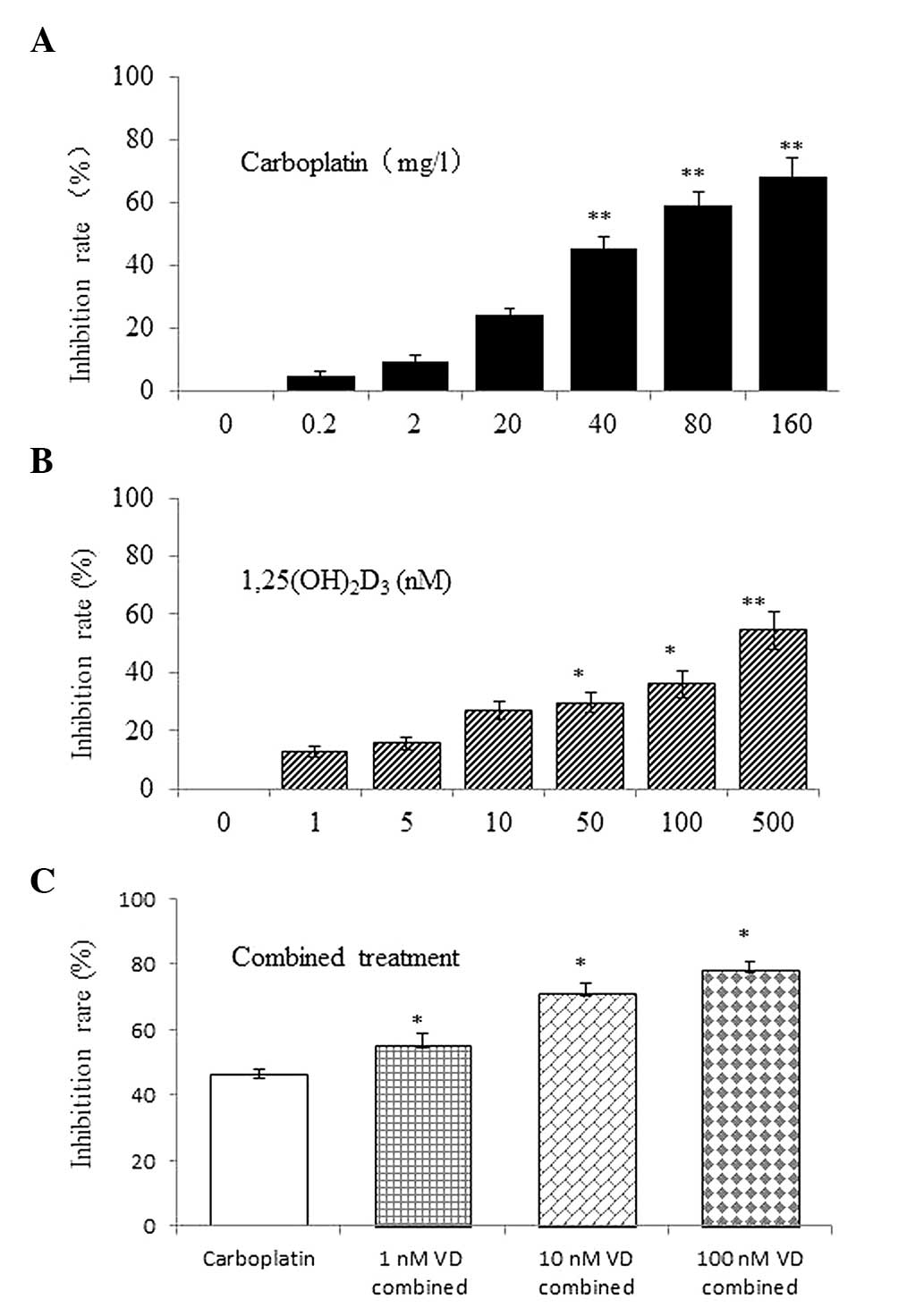
cell viability assay, 1,25(OH)2D3 inhibited
growth in a dose-dependent manner (Fig.
1A). Carboplatin also suppressed the viability of SKOV-3 cells
in a similar manner (Fig. 1B). The
differences between the vehicle control and test groups were
statistically significant (P<0.05). Based on 10–90% inhibition
rates of cell growth in the KaleidaGraph program, the
IC50 values of 1,25(OH)2D3 and
carboplatin were 420.45 nM (R2=0.9904) and 54.6 mg/l
(R2=0.9923), respectively.
Combined administration of
1,25(OH)2D3 and carboplatin
To determine whether the drugs work synergistically,
SKOV-3 cells were treated with carboplatin in the presence of
1,25(OH)2D3. The growth inhibition was
significantly greater with the combined treatment (Fig. 1C) and greater synergy was achieved
at 40 mg/l carboplatin in combination with 10 nM
1,25(OH)2D3 (CI=0.57). The IC50 of
carboplatin evidently decreased with increasing
1,25(OH)2D3 concentrations (Table I).
 | Table IEffect of
1,25(OH)2D3 and carboplatin combination on
the growth of SKOV-3 cells. |
Table I
Effect of
1,25(OH)2D3 and carboplatin combination on
the growth of SKOV-3 cells.
|
1,25(OH)2D3, nM |
IC50a of carboplatin, mg/l | CIb |
|---|
| 0 | 54.6 | NAc |
| 1 | 35.0 | 0.78 |
| 10 | 26.7 | 0.57 |
| 100 | 24.4 | 0.76 |
Cell cycle analysis
To determine whether
1,25(OH)2D3 enhancement of the
antiproliferative activity of carboplatin was due to alterations in
the cell cycle, the cell cycle distribution of SKOV-3 cells treated
with the vehicle control, 10 nM 1,25(OH)2D3,
40 mg/l of carboplatin and the drugs in combination were compared.
Compared with the vehicle control,
1,25(OH)2D3 significantly increased the
percentage of cells in G0/G1 phase,
accompanied by a reduction of cells in G2/M phase. The
reverse effect occurred with carboplatin; a decrease in cells in
G0/G1 phase and an increase in cells in
G2/M phase was observed. However, the percentage of
cells in S phase changed very little. Following treatment with 10
nM 1,25(OH)2D3 and 40 mg/l carboplatin, the
distribution of G0/G1-phase cells in SKOV-3
cells was further reduced, while cells in G2/M phase
evidently increased (Fig. 2A and
B). Therefore, it was concluded that the combined treatment had
a similar effect as carboplatin treatment alone, but this
observation regarding cell cycle arrest requires more
consideration.
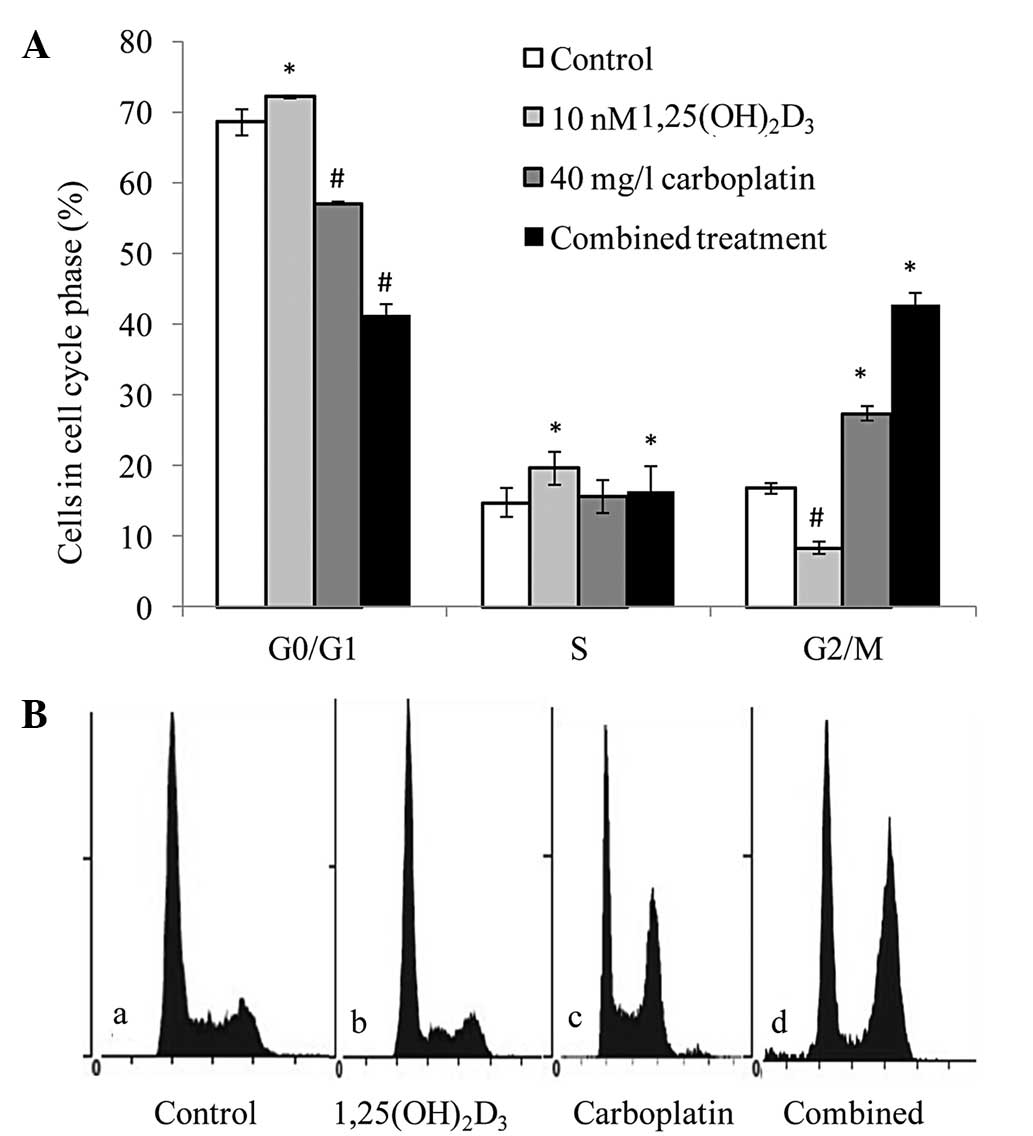 | Figure 2Combined effect of
1,25(OH)2D3 and carboplatin on cell cycle
distribution. (A) G2/M cell cycle arrest in SKOV-3 cells
treated with 1,25(OH)2D3 and carboplatin
versus the untreated cells. */#P<0.05, vs. the
vehicle controls. (B) Flow cytometric analysis: a, control group;
b, 10-nM 1,25(OH)2D3 group; c, 40-mg/l
carboplatin group; and d, combined treatment group. Similar results
were obtained in all three experiments.
1,25(OH)2D3, 1,25-dihydroxyvitamin
D3. |
Apoptosis in combination treatment of
1,25(OH)2D3 and carboplatin
Apoptosis was assessed to identify the mechanism of
growth inhibition by the combined treatment of
1,25(OH)2D3 and carboplatin in ovarian cancer
cells. Apoptosis was increased in SKOV-3 cells treated with 40 mg/l
carboplatin, but the increase was not significant in cells treated
with 10 nM 1,25(OH)2D3 (a dose of 100 nM did
increase apoptosis, data not shown) compared with the control
group. However, apoptosis was evidently increased by combined
treatment (Fig. 3A). Furthermore,
confocal laser-scanning microscopy with DAPI staining demonstrated
that cells treated with the two drugs exhibited apoptotic nuclei
with DNA fragmentation, chromatin condensation and formation of
apoptotic bodies (Fig. 3B), and
that cells treated with single drugs contained fewer apoptotic
nuclei than cells treated with both drugs.
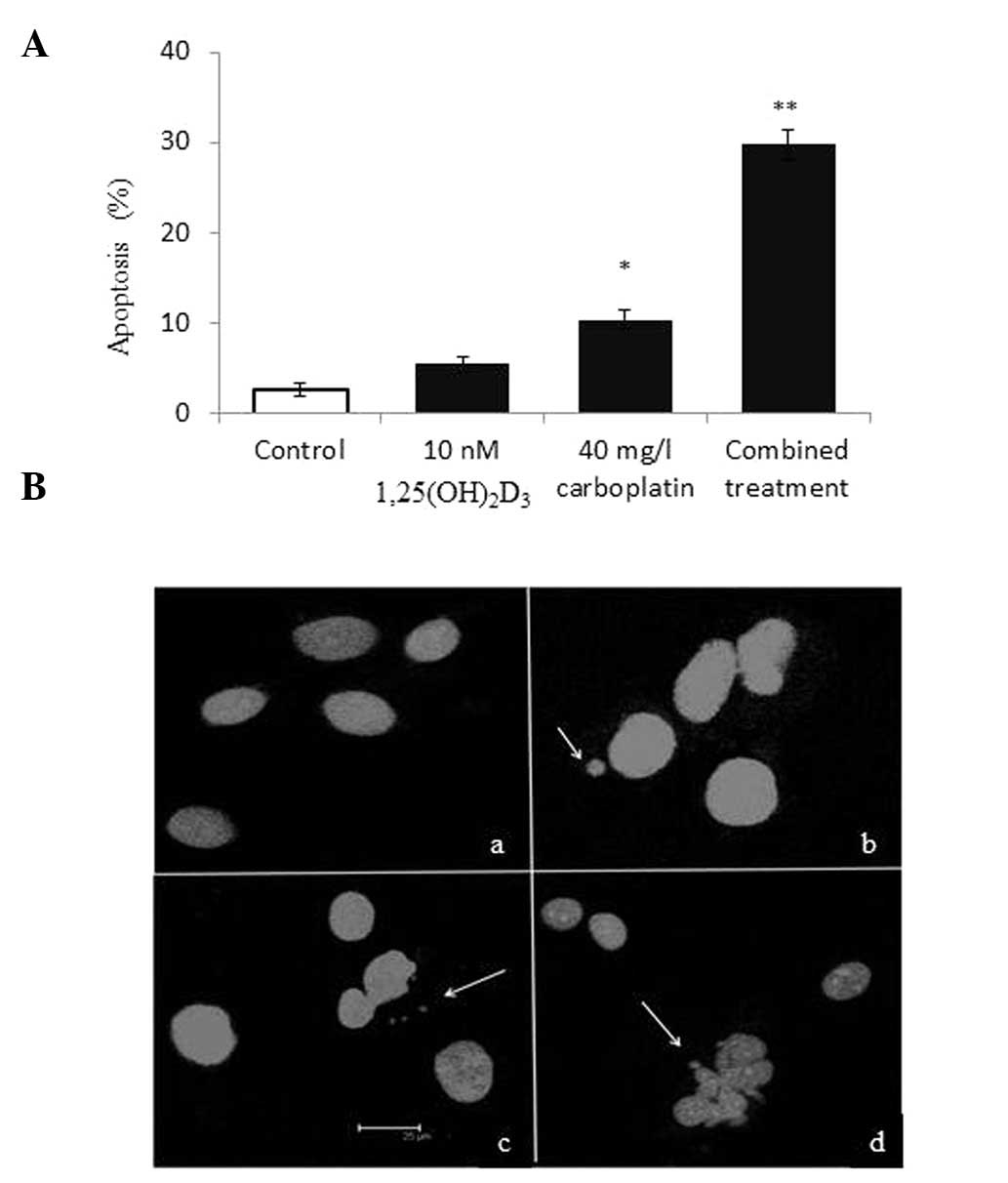 | Figure 3Combined effect of
1,25(OH)2D3 and carboplatin on apoptosis in
SKOV-3 cells. (A) Apoptosis increased following the combined
treatment of 1,25(OH)2D3 and carboplatin
versus the untreated cells. *P<0.05 and
**P<0.01, vs. the vehicle controls. (B) Nuclei of
SKOV-3 cells (stain, 4′6-diamidino-2-phenylindole): a, control
group; b, 10-nM 1,25(OH)2D3 group; c, 40-mg/l
carboplatin group; and d, combined treatment group. Arrows indicate
the fragmented DNA from the nuclei.
1,25(OH)2D3, 1,25-dihydroxyvitamin
D3. |
MMP change following combination
treatment
Depolarization of MMP is a characteristic feature of
apoptosis; hence, treated cells were examined for a drop in MMP.
MMP was not found to significantly reduce with 10 nM of
1,25(OH)2D3 in SKOV-3 cells, but MMP dropped
in cells treated with 40 mg/l carboplatin. The maximal effect was
obtained with combined 1,25(OH)2D3 and
carboplatin treatment (Fig. 4A).
Each drug alone inhibited growth, increased apoptosis and reduced
MMP. Thus, 1,25(OH)2D3 further reduces the
MMP of ovarian cells induced by carboplatin; however, 10 nM of
1,25(OH)2D3 alone did not reduce
MMP.
ROS production following combined
treatment
Oxidative stress appears to be critical for tumor
therapy, as ROS overproduction corresponds to an increase in
apoptosis. In order to assess whether the growth suppression of
1,25(OH)2D3 and carboplatin is due to ROS
production, ROS were measured as detected by CM-H2DCFDA
and expressed as the mean fluorescence by flow cytometry in
comparison with the vehicle controls. The ROS levels in cells
treated with 40 mg/l carboplatin were evidently increased, while
those of cells treated with 10 nm 1,25(OH)2D3
were not found to significantly increase in comparison with the
vehicle control. Although carboplatin treatment alone increases ROS
production, the combined treatment produced a clear increase in ROS
levels compared with the vehicle control (Fig. 4B). Thus, the growth inhibition of
ovarian cancer cells can be induced by the increase of ROS
triggered by the combined treatment.
Discussion
The primary actions of
1,25(OH)2D3 are mediated through the nuclear
vitamin D receptor (VDR), a member of the steroid/thyroid hormone
superfamily of ligand-activated transcription factors. VDR has been
found in rat ovaries by immunohistochemistry (30), as well as hen ovaries by ligand
binding assays (31), indicating
that the ovary is a target organ for vitamin D. Studies have also
shown that VDR is expressed in gynecologic neoplasms, such as
ovarian cancer (32,33), which suggests that
1,25(OH)2D3 may be effective against ovarian
cancer. The correlation between vitamin D and the risk of ovarian
cancer has also received unprecedented attention. Several
ecological studies have reported that ovarian cancer mortality
inversely correlates with sun exposure, which initiates vitamin D
production in the skin (34,35).
Other studies of dietary intake of vitamin D have also observed an
inverse correlation with ovarian cancer risk (36,37).
Therefore, the inverse correlation between vitamin D level and
ovarian cancer-related mortality suggests that the insufficiency of
1,25(OH)2D3 may contribute to ovarian cancer
initiation and/or progression.
In this study, 1,25(OH)2D3 was
demonstrated to inhibit ovarian cancer SKOV-3 cell growth in a
dose-dependent manner. It also markedly enhanced the inhibitory
effects of carboplatin on ovarian cancer cells at a concentration
of 10 nM 1,25(OH)2D3. While this has been
viewed as the major anticancer effect for
1,25(OH)2D3, its mechanism remains
uncertain.
Cell-cycle perturbation is central to
chemotherapy-mediated antiproliferative activity in tumor cells,
and combined treatment with 1,25(OH)2D3 and
carboplatin in the current study led to a significant increase in
the percentage of G2/M-phase cells and an evident
decrease in G0/G1-phase cells. Moffatt et
al (38) also demonstrated
that, over time, combined 1,25(OH)2D3 and
carboplatin increases the percentage of prostate cancer cells in
G2/M phase. The authors found this trend to correlate
with an apparent decrease in the amount of cells in G1
phase. Studies of ovarian cells have suggested that
1,25(OH)2D3 causes cell cycle arrest at the
G1/S and G2/M checkpoints. In addition, these
studies have shown that the proliferation of ovarian cancer OVCAR3
cells is suppressed by 1,25(OH)2D3 (33) and that
1,25(OH)2D3 arrests ovarian cancer cells in
G2/M phase by a mechanism that involves GADD45 (39). They have also indicated that
1,25(OH)2D3 arrests ovarian cancer cells in
the G1 phase by increasing the abundance of p27, an
inhibitor of cyclin-dependent kinase activity (40). 1,25(OH)2D3
enhanced the effects of platinum agents by inhibiting the growth of
the breast cancer cell line, MCF-7 (41). Additionally, in vivo evidence
for the positive interaction between
1,25(OH)2D3 and platinum compounds has been
obtained in a murine squamous cell carcinoma model system (42). These studies have shown that
1,25(OH)2D3 causes cell cycle arrest and
growth suppression in ovarian cancer cells.
The results of the current study indicated that
1,25(OH)2D3 enhances the growth-inhibitory
effect of carboplatin by increasing the rate of apoptosis.
Furthermore, 1,25(OH)2D3 induced apoptosis,
possibly due to increased ROS production, which directly induces
single- and double-strand breaks, abasic sites and DNA
fragmentation, all of which lead to apoptosis. Koren et al
(43) reported that
1,25(OH)2D3 induces ROS production in MCF-7
cells and suggested a compensatory mechanism in which growth arrest
is induced by oxidative stress, while antioxidant activities are
increased. 1,25(OH)2D3 was not found to act
synergistically with anticancer cytokines in the tumor milieu,
which is mediated by ROS (44). In
the present study, 10 nM 1,25(OH)2D3 was not
found to induce ROS production alone, but to enhance ROS production
in ovarian cancer cells treated with carboplatin. These studies
have demonstrated that the anticancer activity of
1,25(OH)2D3 is associated with the
pro-oxidant action of 1,25(OH)2D3 its in
MCF-7 cells, which may be the result of increased intracellular
ROS. However, overproduction of ROS through endogenous or exogenous
sources may induce DNA damage, the accumulation of which may lead
to multistep carcinogenesis (45).
Thus, the antioxidative effects of vitamin D have been suggested by
epidemiological surveys and numerous in vitro and in
vivo laboratory studies (46,47).
The antioxidative effect of vitamin D strengthens its roles in
cancer chemoprevention and adds to a growing list of the beneficial
effects of vitamin D in cancer (48).
One important observation from this study is that
1,25(OH)2D3 enhances the carboplatin-induced
apoptosis of ovarian cells and is associated with the loss of MMP.
Chen et al (49) also
demonstrated that ergocalciferol, vitamin D2, causes HL-60 cell
apoptosis via a drop in MMP. 1,25(OH)2D3 has
also been found to augment the loss of MMP induced by TNF (50). Another finding has suggested that
1,25(OH)2D3 sensitizes breast cancer cells to
ROS-induced death by influencing the caspase-dependent and
-independent modes of cell death, upstream of mitochondrial damage
(51). Therefore,
1,25(OH)2D3 enhances the anticancer effects
of carboplatin through production of ROS and loss of MMP. Other
studies have found that 1,25(OH)2D3 induces
ovarian cancer cell apoptosis by downregulating telomerase, thus
modulating telomere integrity or perhaps via a
telomerase-independent mechanism (52). This result also indicates that
1,25(OH)2D3 may induce ovarian cancer cell
apoptosis through various mechanisms.
In conclusion, the present study demonstrated that
1,25(OH)2D3 is a potent inhibitor of ovarian
cancer cell growth in vitro, and enhances the therapeutic
effects of carboplatin by altering the cell cycle and increasing
apoptosis through changes in ROS and MMP. These findings suggest
the potential utility of combining
1,25(OH)2D3 with cytotoxic agents for the
treatment of ovarian cancer.
Acknowledgements
This study was supported by the National Natural
Scientific Funding of China (grant nos. 81072286 and 81372979), and
in part by the Priority Academic Program Development of Jiangsu
Higher Education Institutions.
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2007. CA Cancer J Clin. 57:43–66. 2007.
|
|
2
|
McGuire WP, Hoskins WJ, Brady MF, et al:
Cyclophosphamide and cisplatin compared with paclitaxel and
cisplatin in patients with stage III and stage IV ovarian cancer. N
Engl J Med. 334:1–6. 1996.
|
|
3
|
Ozols RF, Bundy BN, Greer BE, et al;
Gynecologic Oncology Group. Phase III trial of carboplatin and
paclitaxel compared with cisplatin and paclitaxel in patients with
optimally resected stage III ovarian cancer: a Gynecologic Oncology
Group study. J Clin Oncol. 21:3194–3200. 2003.
|
|
4
|
Mutch DG: Gemcitabine combination
chemotherapy of ovarian cancer. Gynecol Oncol. 90:S16–S20.
2003.
|
|
5
|
Ozols RF: Systemic therapy for ovarian
cancer: current status and new treatments. Semin Oncol. 33(Suppl
6): S3–S11. 2006.
|
|
6
|
Bookman MA, Brady MF, McGuire WP, et al:
Evaluation of new platinum-based treatment regimens in
advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic
Cancer Intergroup. J Clin Oncol. 27:1419–1425. 2009.
|
|
7
|
Stordal B, Pavlakis N and Davey R: A
systematic review of platinum and taxane resistance from bench to
clinic: an inverse relationship. Cancer Treat Rev. 33:688–703.
2007.
|
|
8
|
Markman M, Webster K, Zanotti K, et al:
Survival following the documentation of platinum and taxane
resistance in ovarian cancer: a single institution experience
involving multiple phase 2 clinical trials. Gynecol Oncol.
93:699–701. 2004.
|
|
9
|
Pavelka M, Lucas M and Russo N: On the
hydrolysis mechanism of the second-generation anticancer drug
carboplatin. Chemistry. 13:10108–10116. 2007.
|
|
10
|
Agarwal R and Kaye SB: Ovarian cancer:
strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003.
|
|
11
|
Cheng TC, Manorek G, Samimi G, et al:
Identification of genes whose expression is associated with
cisplatin resistance in human ovarian carcinoma cells. Cancer
Chemother Pharmacol. 58:384–395. 2006.
|
|
12
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007.
|
|
13
|
Roberts D, Schick J, Conway S, et al:
Identification of genes associated with platinum drug sensitivity
and resistance in human ovarian cancer cells. Br J Cancer.
92:1149–1158. 2005.
|
|
14
|
Peters D, Freund J and Ochs RL:
Genome-wide transcriptional analysis of carboplatin response in
chemosensitive and chemoresistant ovarian cancer cells. Mol Cancer
Ther. 4:1605–1616. 2005.
|
|
15
|
Johnatty SE, Beesley J, Paul J, et al:
ABCB1 (MDR 1) polymorphisms and progression-free survival among
women with ovarian cancer following paclitaxel/carboplatin
chemotherapy. Clin Cancer Res. 14:5594–5601. 2008.
|
|
16
|
Walters MR: Newly identified actions of
the vitamin D endocrine system. Endocrinol Rev. 13:719–764.
1992.
|
|
17
|
Saunders DE, Christensen C, Williams JR,
et al: Inhibition of breast and ovarian carcinoma cell growth by
1,25-dihydroxyvitamin D3 combined with retinoic acid or
dexamethasone. Anticancer Drugs. 6:562–569. 1995.
|
|
18
|
Majewski S, Szmurlo A, Marczak M, et al:
Inhibition of tumor cell-induced angiogenesis by retinoids,
1,25-dihydroxyvitamin D3 and their combination. Cancer Lett.
75:35–39. 1993.
|
|
19
|
Koli K and Keski-Oja J:
1alpha,25-dihydroxyvitamin D3 and its analogues down-regulate cell
invasion-associated proteases in cultured malignant cells. Cell
Growth Differ. 11:221–229. 2000.
|
|
20
|
Evans SR, Shchepotin EI, Young H, et al:
1,25-dihydroxyvitamin D3 synthetic analogs inhibit spontaneous
metastases in a 1,2-dimethylhydrazine-induced colon carcinogenesis
model. Int J Oncol. 16:1249–1254. 2000.
|
|
21
|
Peterlik M, Grant WB and Cross HS:
Calcium, vitamin D and cancer. Anticancer Res. 29:3687–3698.
2009.
|
|
22
|
Akhter J, Chen X, Bowrey P, et al: Vitamin
D3 analog, EB1089, inhibits growth of subcutaneous xenographs of
the human colon cancer cell line, LoVo, in nude mouse model. Dis
Colon Rectum. 40:317–321. 1997.
|
|
23
|
VanWeelden KV, Flanagan L, Binderup L, et
al: Apoptotic regression of MCF-7 xenografts in nude mice treated
with the vitamin D3 analog, EB1089. Endocrinology. 139:2102–2110.
1998.
|
|
24
|
Mehta RG and Mehta RR: Vitamin D and
cancer. J Nutr Biochem. 13:252–264. 2002.
|
|
25
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984.
|
|
26
|
Soriano AF, Helfrich B, Chan DC, et al:
Synergistic effects of new chemopreventive agents and conventional
cytotoxic agents against human lung cancer cell lines. Cancer Res.
59:6178–6184. 1999.
|
|
27
|
Smiley ST, Reers M, Mottola-Hartshorn C,
et al: Intracellular heterogeneity in mitochondrial membrane
potentials revealed by a J-aggregate-forming lipophilic cation
JC-1. Proc Natl Acad Sci USA. 88:3671–3675. 1991.
|
|
28
|
Sung DK, Chang YS, Kang S, et al:
Comparative evaluation of hypoxic-ischemic brain injury by flow
cytometric analysis of mitochondrial membrane potential with JC-1
in neonatal rats. J Neurosci Methods. 193:232–238. 2010.
|
|
29
|
Turturro F, Friday E and Welbourne T:
Hyperglycemia regulates thioredoxin-ROS activity through induction
of thioredoxin-interacting protein (TXNIP) in metastatic breast
cancer-derived cells MDA-MB-231. BMC Cancer. 7:962007.
|
|
30
|
Johnson JA, Grande JP, Windebank AJ and
Kumar R: 1,25-Dihydroxyvitamin D(3) receptors in developing dorsal
root ganglia of fetal rats. Brain Res Dev Brain Res. 92:120–124.
1996.
|
|
31
|
Dokoh S, Donaldson CA, Marion SL, et al:
The ovary: a target organ for 1,25-dihydroxyvitamin D3.
Endocrinology. 112:200–206. 1983.
|
|
32
|
Ahonen MH, Zhuang YH, Aine R, et al:
Androgen receptor and vitamin D receptor in human ovarian cancer:
growth stimulation and inhibition by ligands. Int J Cancer.
86:40–46. 2000.
|
|
33
|
Saunders DE, Christensen C, Lawrence WD,
et al: Receptors for 1,25-dihydroxyvitamin D3 in gynecologic
neoplasms. Gynecol Oncol. 44:131–136. 1992.
|
|
34
|
Garland CF, Garland FC, Gorham ED, et al:
The role of vitamin D in cancer prevention. Am J Public Health.
96:252–261. 2006.
|
|
35
|
Grant WB: An estimate of premature cancer
mortality in the U.S. due to inadequate doses of solar
ultraviolet-B radiation. Cancer. 94:1867–1875. 2002.
|
|
36
|
Bidoli E, La Vecchia C, Talamini R, et al:
Micronutrients and ovarian cancer: a case-control study in Italy.
Ann Oncol. 12:1589–1593. 2001.
|
|
37
|
Salazar-Martinez E, Lazcano-Ponce EC,
Gonzalez Lira-Lira G, et al: Nutritional determinants of epithelial
ovarian cancer risk: a case-control study in Mexico. Oncology.
63:151–157. 2002.
|
|
38
|
Moffatt KA, Johannes W and Miller GJ:
1Alpha,25dihydroxyvitamin D3 and platinum drugs act synergistically
to inhibit the growth of prostate cancer cell lines. Clin Cancer
Res. 5:695–703. 1999.
|
|
39
|
Jiang F, Li P, Fornace AJ Jr, et al: G2/M
arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated
through the induction of GADD45 via an exhonic enhancer. J Biol
Chem. 278:48030–48040. 2003.
|
|
40
|
Li P, Li C, Zhao X, et al: p27(Kip1)
stabilization and G(1) arrest by 1,25-dihydroxyvitamin D(3) in
ovarian cancer cells mediated through down-regulation of cyclin
E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2
ubiquitin ligase. J Biol Chem. 279:25260–25267. 2004.
|
|
41
|
Cho YL, Christensen C, Saunders DE, et al:
Combined effects of 1,25-dihydroxyvitamin D3 and platinum drugs on
the growth of MCF-7 cells. Cancer Res. 51:2848–2853. 1991.
|
|
42
|
Light BW, Yu W, McElwain MC, et al:
Potentiation of cisplatin antitumor activity using a vitamin D
analogue in a murine squamous cell carcinoma system. Cancer Res.
57:3759–3764. 1997.
|
|
43
|
Koren R, Hadari-Naor I, Zuck E, et al:
Vitamin D is a prooxidant in breast cancer cells. Cancer Res.
61:1439–1444. 2001.
|
|
44
|
Wiseman H: Vitamin D is a membrane
antioxidant. Ability to inhibit iron-dependent lipid peroxidation
in liposomes compared to cholesterol, ergosterol and tamoxifen and
relevance to anticancer action. FEBS Lett. 326:285–288. 1993.
|
|
45
|
Poulsen HE, Prieme H and Loft S: Role of
oxidative DNA damage in cancer initiation and promotion. Eur J
Cancer Prev. 7:9–16. 1998.
|
|
46
|
Marchionatti AM, Picotto G, Narvaez CJ, et
al: Antiproliferative action of menadione and
1,25(OH)2D3 on breast cancer cells. J Steroid
Biochem Mol Biol. 113:227–232. 2009.
|
|
47
|
Koren R, Rocker D, Kotestiano O, et al:
Synergistic anticancer activity of 1,25-dihydroxyvitamin D(3) and
immune cytokines: the involvement of reactive oxygen species. J
Steroid Biochem Mol Biol. 73:105–112. 2000.
|
|
48
|
Bao BY, Ting HJ, Hsu JW and Lee YF:
Protective role of 1 alpha, 25-dihydroxyvitamin D3 against
oxidative stress in nonmalignant human prostate epithelial cells.
Int J Cancer. 122:2699–2706. 2008.
|
|
49
|
Chen WJ, Huang YT, Wu ML, et al: Induction
of apoptosis by vitamin D2, ergocalciferol, via reactive oxygen
species generation, glutathione depletion, and caspase activation
in human leukemia cells. J Agric Food Chem. 56:2996–3005. 2008.
|
|
50
|
Weitsman GE, Ravid A, Liberman UA and
Koren R: Vitamin D enhances caspase-dependent and independent
TNF-induced breast cancer cell death: the role of reactive oxygen
species. Ann N Y Acad Sci. 1010:437–440. 2003.
|
|
51
|
Weitsman GE, Koren R, Zuck E, et al:
Vitamin D sensitizes breast cancer cells to the action of H2O2:
mitochondria as a convergence point in the death pathway. Free
Radic Biol Med. 39:266–278. 2005.
|
|
52
|
Jiang F, Bao J, Li P, et al: Induction of
ovarian cancer cell apoptosis by 1,25-Dihydroxyvitamin D3 through
the down-regulation of telomerase. J Biol Chem. 279:53213–53221.
2004.
|