Introduction
Gastric cancer is the fourth most common cancer
worldwide (1). Advances in the
molecular analysis of preneoplastic and neoplastic lesions of the
stomach have revealed a large number of epigenetic and genetic
alterations that determine a multistep process of stomach
carcinogenesis (2). Helicobacter
pylori infection is inherently responsible for the pathogenesis
of intestinal metaplasia, and the epidemiological contribution of
the infection to gastric cancer is similar in European and Japanese
populations (3). In 1994, the
International Agency for Research on Cancer recognized H.
pylori as a definite carcinogen in gastric cancer, based on a
strong epidemiological association between chronic colonization of
H. pylori and gastric cancer (4). In addition, a Mongolian gerbil model
revealed that the inoculation of H. pylori into the stomach
is closely associated with the occurrence of chronic gastritis,
intestinal metastasis and promotion of adenocarcinoma (5).
AKT is a pivotal regulator of cell survival,
proliferation and differentiation, and AKT is also a member of the
phosphatidylinositol-3 kinase (PI3K) signaling pathway (6–9).
Stimulation of receptor tyrosine kinases or G-proteins results in
the activation of PI3K, which in turn activates AKT. AKT
phosphorylation is catalyzed by heat shock protein 90, and the
dephosphorylation of AKT is mediated through protein phosphatase 2A
(9). Subsequently, AKT regulates
signaling through various growth factors and cytokines. Upstream of
AKT, activation of insulin-like growth factor-1 receptor, epidermal
growth factor receptor and human epidermal growth factor receptor
2, which are important in cancer progression, activates AKT
(6,7).
Therefore, AKT has been reported as a biomarker that predicts
metastasis in human gastrointestinal cancer (8).
Phosphorylation of AKT modulates signals from
phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and
the mammalian target of rapamycin (mTOR), resulting in diverse
effects of AKT on cells (9). AKT1 is
recognized as an apoptotic inhibitor that contributes to cancer
progression (9). Phosphorylation
catalyzed by AKT inactivates B-cell lymphoma 2 (Bcl-2) antagonist
of cell death, resulting in the dissociation of the promoter from
Bcl-2. In addition, AKT activates nuclear factor κB, which results
in the upregulation of transcription for numerous survival genes
(10). AKT also promotes angiogenesis
through the upregulation of vascular endothelial growth factor
(VEGF) (11). The AKT-microRNA
regulatory network suggests that microRNA-mediated gene regulation
interacts with the AKT signal pathway (12). Thus, the expression and activation of
AKT promotes tumorigenesis, and AKT is a relevant molecular target
for cancer treatment (7).
AKT activation in the gastric mucosa
To establish the role of oxidative stress and AKT
activation in gastric cancer development, the levels of
phosphorylated AKT (pAKT), inducible nitric oxide synthase (iNOS),
nitrotyrosine (NT) and human telomerase reverse transcriptase
(hTERT), which is the catalytic component of telomerase, have
previously been examined by ELISA in non-cancerous gastric mucosa
and gastric cancers (13). In this
previous study, the levels of pAKT were found to be directly
associated with the levels of iNOS, NT and hTERT. Inflammation of
the gastric mucosa was classified into four categories: Chronic
gastritis without H. pylori (CG); chronic active gastritis
with H. pylori (CAG); chronic metaplastic gastritis without
H. pylori (CMG); and chronic gastritis with atypia without
H. pylori (CGA). Increased levels of pAKT, iNOS and NT were
found in tissues from CG, CAG, CMG and CGA. hTERT protein
expression was detected only in CGA. These previous findings
suggest that oxidative stress may be implicated in AKT activation
and hTERT induction, and also that the presence of CGA in the
mucosa may present a high-risk status for gastric carcinogenesis
(13).
H. pylori infection is the major cause of
chronic persistent inflammation of the gastric mucosa (5,14). In
gastric mucosal pathology, the infection induces chronic
inflammation and increases the production of reactive oxide species
(ROS) (3). H. pylori
stimulates the proliferation of gastric mucosal cells through type
IV secretion of cytotoxin-associated gene A (CagA) followed by CagA
phosphorylation by src homology 2 domain-containing protein
tyrosine phosphatase-2 (SHP2) (15,16). CagA
activates SHP2 phosphatase, which in turn inhibits signal
transducers and activators of transcription-mediated
growth-suppressive signaling and also activates extracellular
signal-regulated kinase-mediated growth signaling (17). The increased growth activity may
enhance the risk of gene alteration (15,16).
H. pylori-induced inflammation generates
morphological changes, including intestinal metaplasia, which is
formed by transdifferentiation of the gastric epithelium to the
intestinal phenotype (18). In
intestinal metaplasia, the antral or fundic mucosa of the stomach
is replaced with mucosa that resembles intestinal mucosa (19) through the ectopic expression of caudal
type homeobox 2 (20). Intestinal
metaplasia differs from normal mucosa in that cell division is
accelerated (21,22). Intestinal metaplasia demonstrates
several genetic and epigenetic alterations that are also present in
gastric cancer (3). It has been
reported that transcripts of mutated adenomatous polyposis coli and
abnormal cluster of differentiation 44 are present in intestinal
metaplasia and gastric cancer (23).
Furthermore, a previous study has identified that microsatellite
instability and p53 mutation (24)
are detected in 33% of intestinal metaplasias (25). Thus, it is hypothsized that H.
pylori induces iNOS and the synthesis of ROS, which activates
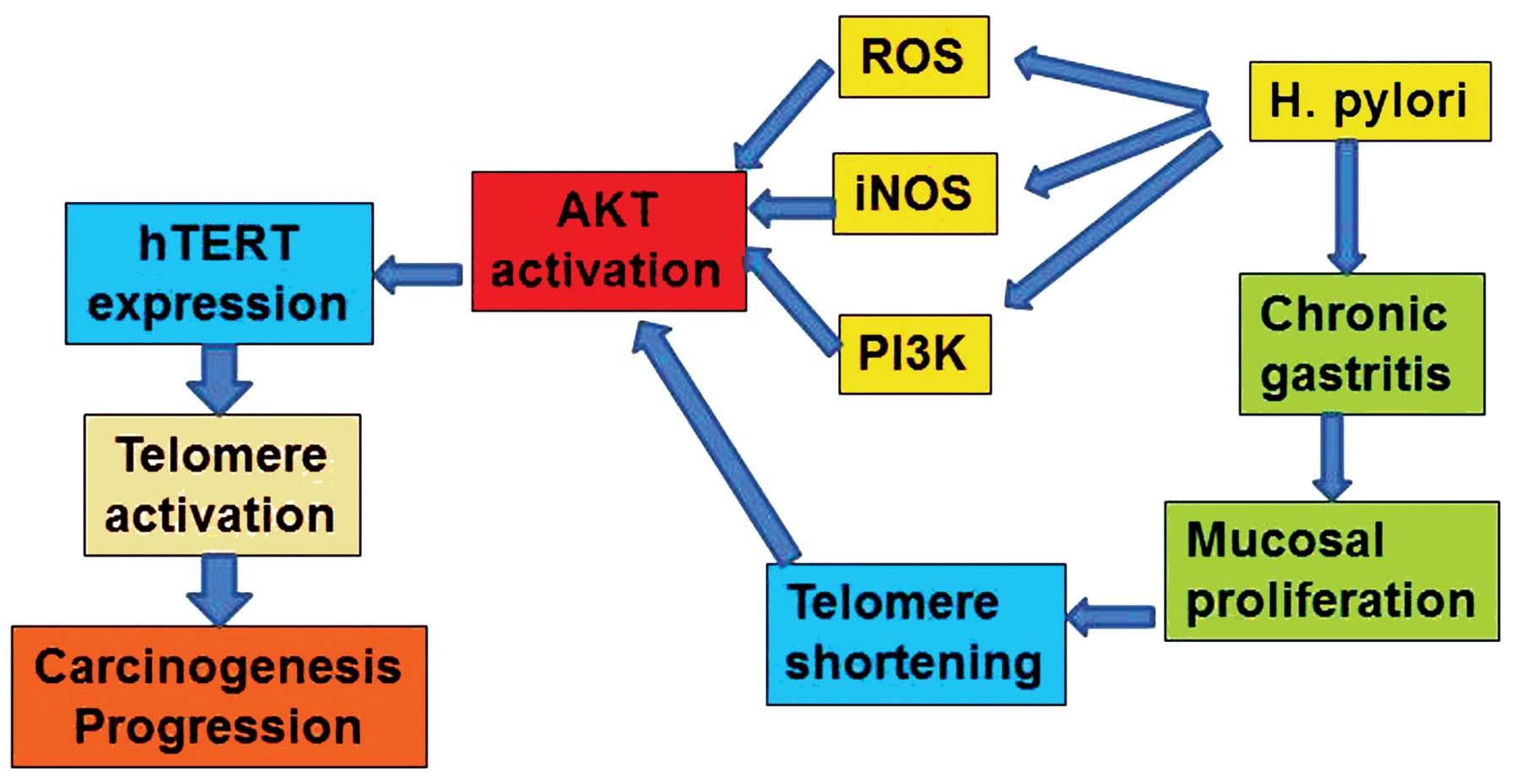
AKT (Fig. 1).
Telomerase
The telomere is a repetitive ‘TTAGGG’ sequence that
is present at the ends of eukaryotic chromosomes to maintain and
protect the integrity of the chromosomes (26). As cells divide, the telomere is
shortened in length. Therefore, the length of the telomere acts as
a marker of the division limit for cells or for cell death
(27). In stem, cancer and cancer
stem cells, the telomere is elongated by the activity of
telomerase, a ribonucleoprotein enzyme that enables cells to divide
endlessly (28). Thus, through the
activation of the enzyme, telomerase is a key enzyme for the
induction of immortality and malignant properties in somatic cells.
Telomerase activity is detected in 85% of gastric cancer lesions,
regardless of tumor stage and histological types (29). In addition, telomerase activity is
detected in 23% of intestinal metaplasia tissues and 50% of gastric
adenoma tissues, whereas no activity in the corresponding gastric
normal mucosa has been reported (29).
Telomerase RNA
Human telomerase RNA (hTR), which contains 11
nucleotides complementary to the telomere TTAGGG sequence, is
generated to function as the template RNA component of telomerase
(30). hTR is expressed in
pre-crisis cell lines, non-neoplastic tissues, immortalized cell
lines and tumor specimens, but the expression level is not
associated with the level of telomerase activity (31). Notably, Blasco et al reported
that the initial upregulation of hTR is an early event in
carcinogenesis and that telomerase is activated only in end-stage
tumors during multi-stage carcinogenesis (32).
The expression of hTR and telomerase activity
in gastric cancer and corresponding non-cancerous mucosa tissues
has been determined previously (33).
In this study, telomerase activity was detected in 88% of carcinoma
tissues. Although tumor specimens and non-cancerous mucosa tissues
express various levels of hTR, 81% of tumor specimens were
found to express hTR at an increased level compared with the
corresponding mucosa. Gastric carcinoma cell lines were also found
to express hTR at high levels (33). Overall, 35% of non-cancerous mucosa
tissues were found to demonstrate telomerase activity, and all
non-cancerous mucosa tissues infected with H. pylori
contained intestinal metaplasia. The incidence of telomerase
expression in the mucosa with moderate intestinal metaplasia was
revealed to be significantly increased compared with the incidence
in mild intestinal metaplasia, whereas hTR overexpression
was detected in mild intestinal metaplasia and moderate intestinal
metaplasia. Notably, the degree of metaplasia in H.
pylori-infected mucosa increased concordantly with the level of
hTR expression and telomerase activity. These results
indicated that hTR overexpression is caused by H.
pylori, which plays a role upstream of telomerase activity as
an early event in stomach carcinogenesis (33).
In the stomach, various types of inflammation affect
the growth of the mucosa. In particular, intestinal metaplasia and
chronic atrophic gastritis exhibit accelerated cell growth and cell
cycling compared with H. pylori-negative gastric mucosa
(22,34). Although the factors that upregulate
hTR expression remain unknown, continuous inflammation and
regenerative processes may stimulate hTR expression by
affecting stem cells, and subsequently, these processes may enhance
the activity of telomerase in the non-cancerous mucosa of the
stomach (3). Therefore, it is likely
that hTR overexpression in non-cancerous mucosa may reflect
an earlier process of multistep carcinogenesis in the stomach
(33).
Telomere shortening
The shortening of telomeres in non-cancerous tissues
is determined by regenerative processes and is specific to the
individual organs or tissue components, since the regeneration of
different tissues requires various degrees of cell division, which
also occurs in chronic gastritis (21,22). The
difference in telomere shortening of the gastric mucosal epithelium
has been investigated using an in situ telomere
quantification technique (35). The
association between telomere length and H. pylori infection
was assessed, and the difference between the telomere length that
characterizes intestinal metaplasia cells in cancer patients and
the telomere length in cells obtained from non-cancer patients was
also examined (35).
In healthy individuals without H. pylori
infection, the telomere volumes in gastric epithelial tissues were
found to be 70–79% of the volume of the telomeres in intramucosal
lymphocytes, which acted as an internal control (35). In patients without gastric cancer, the
telomere volume in H. pylori-infected mucosa cells was found
to be significantly decreased compared with H.
pylori-negative mucosa cells in metaplastic and non-metaplastic
tissues (P<0.0001) (35). In
gastric cancer patients, the telomere volume in intestinal
metaplasia cells adjacent to a cancerous lesion was identified as
75% of the telomere volume in intestinal metaplasias in patients
without cancer (P=0.0001) (35).
Therefore, H. pylori infection is strongly associated with
the shortening of telomeres, which subsequently stimulates
necessary elongation of the telomeres by telomerase activity,
particularly in the gastric epithelium (35).
Telomeres and AKT
The catalytic subunit of hTERT, which is responsible
for telomerase activity and telomere elongation, is suppressed in
differentiated cells (36).
Shortening of the telomeres has been identified as a significant
factor for the induction of hTERT expression in the gastric
mucosa (35). hTERT expression
is detected in 7% of the corresponding mucosas of cancer-associated
intestinal metaplasias and 3% of cancer-negative intestinal
metaplasias, in which the telomere volume is markedly reduced
(35). The status of gastric mucosa
was classified according to inflammation and immortalization by
hTERT expression. AKT is a key protein that links
inflammation and tumorigenesis. Therefore, AKT became the focus of
previous studies (9–13). iNOS is an important mediator of
inflammation in the gastric mucosa, and increased expression of
iNOS is epigenetically induced upon H. pylori
infection as a host defense (37). NT
is a marker of nitric oxide (NO)-induced protein degradation. In a
previous study, the expression of iNOS and NT was
examined to evaluate inflammation and the expression of
hTERT as a marker of immortality (13). The data revealed that the levels of
pAKT are associated with hTERT expression in CGA (13). In addition, it was also revealed in
this study that pAKT levels are associated with hTERT
expression in gastric adenocarcinomas (13). Overall, these findings indicated that
the immortality of gastric mucosal cells is associated with
inflammation-induced AKT activation.
pAKT levels are elevated in certain types of gastric
mucosa, such as CAG, CMG and CGA (13). Notably, high pAKT levels in CAG are
associated with H. pylori-induced active inflammation. By
contrast, the increased pAKT levels in CMG suggest that persistent
iNOS upregulation develops during the metaplastic process. The
highest pAKT levels in CGA may develop during carcinogenic
processes (38). In vitro
analysis has revealed that only CGA and MKN28 cancer cells
demonstrated upregulation of pAKT in response to brief NO exposure.
In Barrett's esophagus, activation of AKT is associated with
dysplasia-carcinoma sequence (39).
In addition, only CGA has been found to express hTERT
(13). These findings suggest that
CGA is likely to be a distinct category in the process of
carcinogenesis. The activation of telomere shortening induced by
H. pylori infection leads to hTERT expression and
telomerase activation (Fig. 1).
hTERT activity is regulated by phosphorylation, in
addition to the expression of hTERT. Protein kinase C and
AKT each phosphorylate hTERT (40,41).
AKT-catalyzed phosphorylation of hTERT induces intranuclear
translocation of hTERT and subsequently, activates hTERT. By
contrast, ring finger protein 1, which is an E3 ubiquitin ligase,
decreases the activity of hTERT by ubiquitination (42).
AKT is associated with diverse pro-tumor responses,
including hTERT activation (9–13). In a
previous study, the significance of AKT phosphorylation and hTERT
on the prognosis of gastric cancer was examined (43). The activation of AKT by epidermal
growth factor was found to increase hTERT expression and
telomerase activity. By contrast, AKT inactivation by inhibitors
and knockdown decreased hTERT expression and telomerase
activity in MKN28 gastric cancer cells (43). In 40 gastric cancer tissues, a
significant association was found between the levels of pAKT and
hTERT expression and telomere length (43). The levels of pAKT or pAKT/hTERT are
not associated with clinicopathological parameters, such as the
tumor stage and presence of nodal metastasis. However, the survival
rate of the patients with a high level of pAKT or the patients with
a high level of pAKT and hTERT has been found to be significantly
worse compared with other patients (43). These findings suggest that AKT and
hTERT are good molecular targets for the treatment of gastric
cancer.
AKT is associated with cancer cell survival through
the alteration of the expression of Bcl-2 antagonist of cell death,
p53, forkhead, nuclear factor κB, mTOR and PTEN (10) (44). In
addition, dysregulated PTEN/PI3K/AKT signaling interacts with the
Wnt signaling pathway to induce epithelial-mesenchymal transition
(EMT), which is usually associated with the cancer stem cell
phenotype and a poor prognosis (45).
A previous study has reported that hTERT promotes EMT induced by
transforming growth factor-β and β-catenin by inducing β-catenin
nuclear translocation and its transcriptional activity for
vimentin, a mesenchymal factor, expression (46). Therefore, PTEN/PI3K/AKT signaling
enhances EMT and stem cell phenotypes. In a previous study, the
association between AKT phosphorylation, TERT expression and
telomerase activity was confirmed in MKN28 gastric cancer cells and
gastric cancer tissues (43). These
associations may result in poor prognoses in patients with high
pAKT levels or high pAKT/hTERT levels. Furthermore, multivariate
analysis has revealed that pAKT and pAKT/hTERT levels are reliable
prognostic factors. The examination of additional gastric cancer
cases is required to confirm the hypothesis that the EMT/stem cell
phenotype affects disease progression (43–46).
Angiogenesis is an essential phenotype for cancer
progression, and VEGF expression is associated closely with
neovascularization and cancer progression in numerous malignancies
(47). The PI3K/AKT pathway induces
VEGF response, which includes other downstream inducers, including
mitogen-activated protein kinase (extracellular signal-regulated
kinases or p38), Src, focal adhesion kinase, Rho family GTPases,
and endothelial NO (48). The
PI3K/AKT pathway increases the secretion of VEGF from cancer cells
by hypoxia-inducible factor 1-dependent and independent mechanisms
(49). Therefore, AKT suppression may
result in an anti-angiogenic effect on gastric cancer.
This data revealed that AKT and hTERT are present in
high levels in gastric cancer, and the concurrent synthesis of
these two proteins at high levels is associated with a poor
prognosis (43). These results
suggest that AKT and hTERT are possible molecular targets for the
treatment of gastric cancer.
Conclusion
In gastric carcinogenesis, H. pylori is an
essential factor for the stimulation of transformation in gastric
epithelial cells. As shown in Fig. 1,
H. pylori induces iNOS, synthesis of ROS and telomere
shortening as a result of repetitive destruction/regeneration
cycles in chronic active gastritis. AKT plays a central role in
H. pylori-induced gastric carcinogenesis. H.
pylori-induced oxidative stress activates AKT, and H.
pylori-accelerated cell growth in the gastric mucosa shortens
telomere length. These changes lead to hTERT expression and
telomerase activation. Telomerase activity immortalizes epithelial
cells to enable uncontrolled cancer cell division and instigates
malignant phenotypes, therefore resulting in the progression of
cancer. These findings demonstrated that AKT plays a pivotal role
in gastric cancer development. Thus, the investigation of AKT as a
promising molecular target to prevent the development or
progression of gastric cancer should be considered.
Glossary
Abbreviations
Abbreviations:
|
PI3K
|
phosphatidylinositol-3 kinase
|
|
VEGF
|
vascular endothelial growth factor
|
|
pAKT
|
phosphorylated AKT
|
|
iNOS
|
inducible nitric oxide synthase
|
|
PTEN
|
tensin homolog deleted on chromosome
10
|
|
NT
|
nitrotyrosine
|
|
hTERT
|
human telomerase reverse
transcriptase
|
|
CG
|
chronic gastritis without H.
pylori
|
|
CAG
|
chronic active gastritis with H.
pylori
|
|
CMG
|
chronic metaplastic gastritis without
H. pylori
|
|
CGA
|
chronic gastritis with atypia without
H. pylori
|
|
SHP2
|
src homology 2 domain-containing
protein tyrosine phosphatase-2
|
|
hTR
|
human telomerase RNA
|
|
EMT
|
epithelial-mesenchymal transition
|
|
NO
|
nitric oxide
|
|
ROS
|
reactive oxide species
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yokozaki H, Kuniyasu H, Semba S, Yasui W
and Tahara E: Molecular bases of human stomach carcinogenesisMol
Pathol Gastroenterol Cancer. Tahara E: Springer-Verlag; Tokyo: pp.
55–70. 1997
|
|
3
|
Kuniyasu H, Yasui W, Yokozaki H and Tahara
E: Helicobacter pylori infection and carcinogenesis of the
stomach. Langenbecks Arch Surg. 385:69–74. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
IARC, . Schistosomes Liver Flukes and
Hericobactor pylori. IARC Sci. 1994.
|
|
5
|
Tsukamoto T and Tatematsu M: Role of
Helicobacter pylori in gastric neoplasia. Curr Infect Dis
Rep. 16:4022014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sukawa Y, Yamamoto H, Nosho K, et al:
Alterations in the human epidermal growth factor receptor
2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer.
World J Gastroenterol. 18:6577–6586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berg M and Soreide K: EGFR and downstream
genetic alterations in KRAS/BRAF and PI3K/AKT pathways in
colorectal cancer: implications for targeted therapy. Discov Med.
14:207–214. 2012.PubMed/NCBI
|
|
8
|
Ng L, Poon RT and Pang R: Biomarkers for
predicting future metastasis of human gastrointestinal tumors. Cell
Mol Life Sci. 70:3631–3656. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheung M and Testa JR: Diverse mechanisms
of AKT pathway activation in human malignancy. Curr Cancer Drug
Targets. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Downward J: PI 3-kinase, Akt and cell
survival. Semin Cell Dev Biol. 15:177–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Radisavljevic Z: AKT as locus of cancer
angiogenic robustness and fragility. J Cell Physiol. 228:21–24.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu M and Mo YY: The Akt-associated
microRNAs. Cell Mol Life Sci. 69:3601–3612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sasaki T, Kuniyasu H, Luo Y, et al:
Increased phosphorylation of AKT in high-risk gastric mucosa.
Anticancer Res. 33:3295–3300. 2013.PubMed/NCBI
|
|
14
|
Ho SB: Premalignant lesions of the
stomach. Semin Gastrointest Dis. 7:61–73. 1996.PubMed/NCBI
|
|
15
|
Hatakeyama M: Oncogenic mechanisms of the
Helicobacter pylori CagA protein. Nat Rev Cancer. 4:688–694.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Higashi H, Tsutsumi R, Muto S, et al:
SHP-2 tyrosine phosphatase as an intracellular target of
Helicobacter pylori CagA protein. Science. 295:683–686.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
You M, Yu DH and Feng GS: Shp-2 tyrosine
phosphatase functions as a negative regulator of the
interferon-stimulated Jak/STAT pathway. Mol Cell Biol.
19:2416–2424. 1999.PubMed/NCBI
|
|
18
|
Tosh D and Slack JM: How cells change
their phenotype. Nat Rev Mol Cell Biol. 3:187–194. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ochiai A and Hirohashi S: Genetic
alteration in the precursors of gastric cancerMol Pathol
Gastroenterol Cancer. Tahara E: Springer-Verlag; Tokyo: pp. 43–53.
1997
|
|
20
|
Sugano K: Premalignant conditions of
gastric cancer. J Gastroenterol Hepatol. 28:906–911. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Testino G: Helicobacter pylori,
cell proliferation and carcinogenesis. Am J Gastroenterol.
96:25142001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guerci A, Chambre JF, Franck P, Floquet J,
Gaucher P and Guerci O: Flow cytometric analysis of the cell cycle
in chronic gastritis. Anal Cell Pathol. 4:381–388. 1992.PubMed/NCBI
|
|
23
|
Tahara E, Kuniyasu H, Yasui W and Yokozaki
H: Gene alterations in intestinal metaplasia and gastric cancer.
Eur J Gastroenterol Hepatol. (6 Suppl 1):1021994.
|
|
24
|
Ochiai A, Yamauchi Y and Hirohashi S: p53
mutations in the non-neoplastic mucosa of the human stomach showing
intestinal metaplasia. Int J Cancer. 69:28–33. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Semba S, Yokozaki H, Yamamoto S, Yasui W
and Tahara E: Microsatellite instability in precancerous lesions
and adenocarcinomas of the stomach. Cancer. 77:1620–1627. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blackburn EH: Switching and signaling at
the telomere. Cell. 106:661–673. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lingner J, Cooper JP and Cech TR:
Telomerase and DNA end replication: no longer a lagging strand
problem? Science. 269:1533–1534. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stewart SA and Weinberg RA: Telomeres:
cancer to human aging. Annu Rev Cell Dev Biol. 22:531–557. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tahara H, Kuniyasu H, Yokozaki H, et al:
Telomerase activity in preneoplastic and neoplastic gastric and
colorectal lesions. Clin Cancer Res. 1:1245–1251. 1995.PubMed/NCBI
|
|
30
|
Feng J, Funk WD, Wang SS, et al: The RNA
component of human telomerase. Science. 269:1236–1241. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Avilion AA, Piatyszek MA, Gupta J, Shay
JW, Bacchetti S and Greider CW: Human telomerase RNA and telomerase
activity in immortal cell lines and tumor tissues. Cancer Res.
56:645–650. 1996.PubMed/NCBI
|
|
32
|
Blasco MA, Rizen M, Greider CW and Hanahan
D: Differential regulation of telomerase activity and telomerase
RNA during multi-stage tumorigenesis. Nat Genet. 12:200–204. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuniyasu H, Domen T, Hamamoto T, et al:
Expression of human telomerase RNA is an early event of stomach
carcinogenesis. Jpn J Cancer Res. 88:103–107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weiss H, Gutz HJ, Schroter J and Wildner
GP: DNA distribution pattern in chronic gastritis. I. DNA ploidy
and cell cycle distribution. Scand J Gastroenterol. 24:643–648.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuniyasu H, Kitadai Y, Mieno H and Yasui
W: Helicobactor pylori infection is closely associated with
telomere reduction in gastric mucosa. Oncology. 65:275–282. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Autexier C and Lue NF: The structure and
function of telomerase reverse transcriptase. Annu Rev Biochem.
75:493–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Angrisano T, Lembo F, Peluso S, Keller S,
Chiariotti L and Pero R: Helicobacter pylori regulates iNOS
promoter by histone modifications in human gastric epithelial
cells. Med Microbiol Immunol. 201:249–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Murata M, Thanan R, Ma N and Kawanishi S:
Role of nitrative and oxidative DNA damage in inflammation-related
carcinogenesis. J Biomed Biotechnol. 2012:6230192012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beales IL, Ogunwobi O, Cameron E, El-Amin
K, Mutungi G and Wilkinson M: Activation of Akt is increased in the
dysplasia-carcinoma sequence in Barrett's oesophagus and
contributes to increased proliferation and inhibition of apoptosis:
a histopathological and functional study. BMC Cancer. 7:972007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, Zhao L, Yang Z, Funder JW and Liu
JP: Telomerase is controlled by protein kinase C alpha in human
breast cancer cells. J Biol Chem. 273:33436–33442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kang SS, Kwon T, Kwon DY and Do SI: Akt
protein kinase enhances human telomerase activity through
phosphorylation of telomerase reverse transcriptase subunit. J Biol
Chem. 274:13085–13090. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim JH, Park SM, Kang MR, et al: Ubiquitin
ligase MKRN1 modulates telomere length homeostasis through a
proteolysis of hTERT. Genes Dev. 19:776–781. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sasaki T, Kuniyasu H, Luo Y, et al: AKT
activation and telomerase reverse transcriptase expression are
concurrently associated with prognosis of gastric cancer.
Pathobiology. 81:36–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Populo H, Lopes JM and Soares P: The mTOR
Signalling Pathway in Human Cancer. Int J Mol Sci. 13:1886–1918.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Karamitopoulou E: Tumor budding cells,
cancer stem cells and epithelial-mesenchymal transition-type cells
in pancreatic cancer. Front Oncol. 2:2092012.PubMed/NCBI
|
|
46
|
Liu Z, Li Q, Li K, et al: Telomerase
reverse transcriptase promotes epithelial-mesenchymal transition
and stem cell-like traits in cancer cells. Oncogene. 32:4203–4213.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fidler IJ: The pathogenesis of cancer
metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Claesson-Welsh L and Welsh M: VEGFA and
tumour angiogenesis. J Intern Med. 273:114–127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Karar J and Maity A: PI3K/AKT/mTOR Pathway
in Angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|