Introduction
Liver cancer is one of the most prevalent solid
tumors worldwide, with a poor prognosis and limited treatment
options available (1,2); it is also the fifth most frequently
diagnosed cancer and the third leading cause of cancer mortality
worldwide (3). Liver cancer is well
characterized as a highly refractory disease, with high levels of
tumor progression and recurrence. The prognosis of advanced
hepatocellular carcinoma (HCC) remains poor and no effective
systemic therapy has yet been developed (4). The traditional treatment methods,
including surgery, radiofrequency ablation therapy and
chemotherapy, focus on reducing the bulk of the tumor mass,
however, they are limited by drug resistance, various side effects
and metastasis to other organs (5).
Thus, there is a significant requirement to expand our
understanding of the molecular mechanisms underlying liver cancer
in order to develop novel therapeutic strategies. Currently,
targeted therapy is considered a more effective therapeutic
strategy and is receiving an increasing level of attention.
AKT has a wide range of effects on cellular
signaling and has been accepted as a promising anticancer target,
including for the treatment of liver cancer (6). AKT is a serine/threonine protein kinase
that serves a central role in the signaling network of
phosphoinositide 3-kinase (PI3K) and the mammalian target of
rapamycin (mTOR). Accumulating evidence indicates that this pathway
controls key cellular processes, including glucose metabolism,
apoptosis, survival, cell proliferation, cell migration,
transcription and angiogenesis (7,8). Under
normal conditions, this signaling network may be activated by
numerous receptors, including members of the epidermal growth
factor receptor and vascular endothelial growth factor receptor
families and their ligands. The importance of the PI3K/AKT/mTOR
signaling pathway in liver cancer is underlined by the finding that
mTOR inhibition can suppress HCC growth in vitro and in
xenograft models (9). Additionally,
either the loss of phosphatase and tensin homolog deleted on
chromosome 10 (PTEN) function or the overexpression/activation of
AKT leads to HCC development in mouse models (10,11).
Aberrant mTOR signaling has been detected in ~48% of HCCs (9), with a negative feedback loop, resulting
in the activation of AKT following mTOR inhibition, being observed
in a variety of cancer cell lines and human tumor samples of colon
and breast cancer (12). Suppressor
with morphogenetic effect on genitalia family member-1 (SMG-1) and
mTOR each belong to the PI3K-related kinase (PIKK) family.
Recently, the level of awareness regarding SMG-1 has increased due
to evidence indicating that it is likely to be a potential human
tumor suppressor gene product (13).
Several types of small molecular AKT inhibitors have
been studied, including ATP-competitive protein kinase inhibitors
[A-443654, GSK690693 and GDC0068 (14–16)],
phosphatidylinositol-3,4,5-trisphosphate (PIP3) binding inhibitors
[perifosine (17)] and allosteric
inhibitors [MK-2206 (18)]. In order
to evaluate the mechanism of AZD5363 suppression of cell
proliferation and migration in liver cancer, the present study
investigated the effect of AZD5363, a novel AKT inhibitor, on the
phosphorylation of AKT downstream molecules, and on the SMG-1 and
mTOR pathways.
Materials and methods
Inhibitor preparation
AZD5363
[(S)-4-amino-N-[1-(4-chlorophenyl)-3-hydroxypropyl]-1(7H-pyrrolo[2,3-d]
pyrimidin-4-yl) piperidine-4-carboxamide (#S8019; Selleck
Chemicals, Houston, TX, USA), a novel AKT inhibitor, was prepared
as a 100 mM stock solution in dimethyl sulfoxide (DMSO) and stored
at −80°C. The final concentration of DMSO was <0.5% in all the
assays.
Cell culture reagents
Human liver cancer Hep-G2 and Huh-7 cell lines were
obtained from Shanghai Saiqi Biological Engineering Co., Ltd.
(Shanghai, China). The study protocol was approved by the Ethical
Committee of the Shandong Provincial Hospital Affiliated to
Shandong University. All cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 1%
penicillin-streptomycin solution and 1% non-essential amino acids.
All cells were maintained in a humidified incubator with 5%
CO2 at 37°C. The structure and synthesis of the AKT
inhibitor, AZD5363, has been described previously (19).
Cell counting kit-8 (CCK-8) assay
The cell growth rate was measured using CCK-8
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Briefly,
cells seeded at 1,000–2,000/per well in 96-well plates were
cultured overnight with 90 µl DMEM, containing 10% FBS, and were
subsequently treated with AZD5363 at varying concentrations (5 to
30 µM) for 24, 48 and 72 h. CCK-8 One Solution Reagent (Dojindo
Molecular Technologies) was added to each well according to the
manufacturer's protocols. Following a total of 1.5 h in culture,
the cell viability was determined by measuring the absorbance at
450 nm.
Transwell migration assay
Monolayers of serum-starved adherent cells were
trypsinized (0.25% Trypsin-EDTA; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), and 50,000 cell suspensions [counted using
a cell counting board (Bio-Rad Laboratories, Inc., Hercules, CA,
USA)] were placed in 200 µl serum-free DMEM into the upper well of
Transwell filter apparatus (Corning Inc., Corning, NY, USA). The
filter was suspended within a well of a 24-well plate (NEST
Biotechnology Co., Ltd., Wuxi, China) and the lower reservoir was
filled with 800 µl DMEM, containing 10% FBS. The cells were then
incubated under normal conditions for 24 h. Migration assays were
terminated by retrieving the filter and rubbing off non-migrated
cells from the top surface, which was then treated with
formaldehyde (Far Eastern Group, Laiyang, China), methanol (Far
Eastern Group) and Giemsa (Beijing Seajet Scientific, Inc.,
Beijing, China) staining. The cells that were identified on the
underside of the filter were fixed, stained with Giemsa and
captured under a microscope (model no. BX51; Olympus Corporation,
Tokyo, Japan) using a digital camera (Microshot MC55, Guangzhou
Mingmei Photoelectric Technology Co. Ltd., Guangzhou, China). The
cells were counted in 3 randomly selected fields for each
chamber.
Western blot analysis
Protein was extracted in a 6-well plate, using a
mixture of radioimmunoprecipitation assay (RIPA) and
phenylmethylsulfonyl fluoride (PMSF) (RIPA:PMSF, 100:1; Beyotime
Institute of Biotechnology, Haimen, China), and protein
concentrations were quantified by the Pierce bicinchoninic acid
assay kit (Thermo Fisher Scientific, Inc.). The soluble proteins
(50 µg) were then subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (PAGE; Bio-Rad
Laboratories, Inc.), followed by immunoblotting. The proteins were
separated electrophoretically in SDS-polyacrylamide gels and
transferred to negative control membranes. Subsequently, the
proteins were incubated at 4°C overnight with a primary antibody
and incubated with HRP-conjugated secondary antibody at room
temperature for a total of 2 h. Immunoreactivity was detected using
the FluorChem E system (ProteinSimple, Santa Clara, CA, USA)
according to the manufacturer's protocols. The majority of
antibodies used were obtained from Cell Signaling Technology Inc.
(Danvers, MA, USA), including rabbit monoclonal phosphor-mTOR
(ser2448; D9C2; catalog no., 5536S; dilution, 1:1,000), rabbit
monoclonal phosphor-Akt (Thr450; D5G4; dilution, 1:1,000; catalog
no., 12178), rabbit monoclonal phosphor-glycogen synthase kinase 3β
(GSK-3β; ser9; 5B3; catalog no., 9323; dilution, 1:1,000), rabbit
monoclonal mTOR (7C10; catalog no., 2983S; dilution, 1:1,000),
mouse polyclonal AKT (catalog. no., 9272; dilution, 1:1,000),
rabbit monoclonal GSK-3β (27C10; catalog no., 9315; dilution,
1:1,000) and rabbit monoclonal SMG-1 (Q25; catalog. no., 4993s;
dilution, 1:1,000). Glyceraldehyde 3-phosphate dehydrogenase
(catalog no., TA-08; dilution, 1:1,000) was purchased from ZSGB-BIO
(Beijing, China), as was the secondary monoclonal antibodies:
Peroxidase-conjugated AffiniPure goat anti-mouse immunoglobulin
(Ig)G (H+L; catalog no., ZB-2305; dilution, 1:1,5000) and
peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H+L; catalog
no., ZB-2301; dilution, 1:5,000).
Statistical analysis
Statistical analysis was performed using SPSS
software, version 18.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad
Prism software, version 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA). The data are presented as the mean ± standard error of the
mean. The differences between groups were analyzed using one-way
analysis of variance, followed by Student-Newman-Keuls post hoc
test for pairwise comparison. P<0.05 was considered to indicate
a statistically significant difference.
Results
AZD5363 inhibits the proliferation of
liver cancer cells in a dose- and time-dependent manner
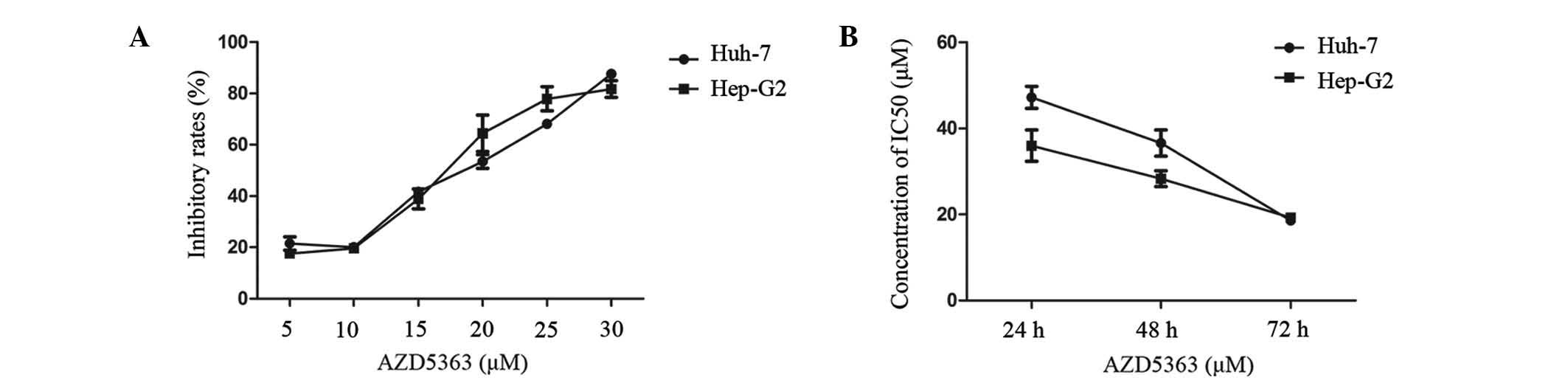
To determine the effect of AZD5363 on cell
proliferation, a panel of two liver cancer cell lines was tested
for anti-proliferative sensitivity using an in vitro cell
growth assay. The Hep-G2 and Huh-7 cells were exposed to AZD5363 at
different concentrations, ranging from 5 to 30 µM. The cells seeded
in 96-well plates were cultured overnight, and were subsequently
treated with AZD5363 at varying concentrations for 24, 48 and 72 h.
The cell growth rate was measured using a CCK-8 assay. A
dose-dependent inhibition of cell viability was observed in each
cell line (Fig. 1A and B). The drug
concentration required for inhibition of growth in the two liver
cancer cell lines was similar, functioning in a dose- and
time-dependent manner. The drug concentration of the half maximal
inhibitory concentration (IC50) was lower when
prolonging the cell exposure; when the exposure time reached 72 h,
the IC50 of the Hep-G2 cells was 18.476 µM and the
IC50 of the Huh-7 cells was 17.80 µM. In order to obtain
a better inhibition curve, a higher density of Huh-7 than Hep-G2
was required. These results indicated that the AZD5363 activity was
selective for the liver cancer cells.
AZD5363 suppresses liver cancer cell
migration
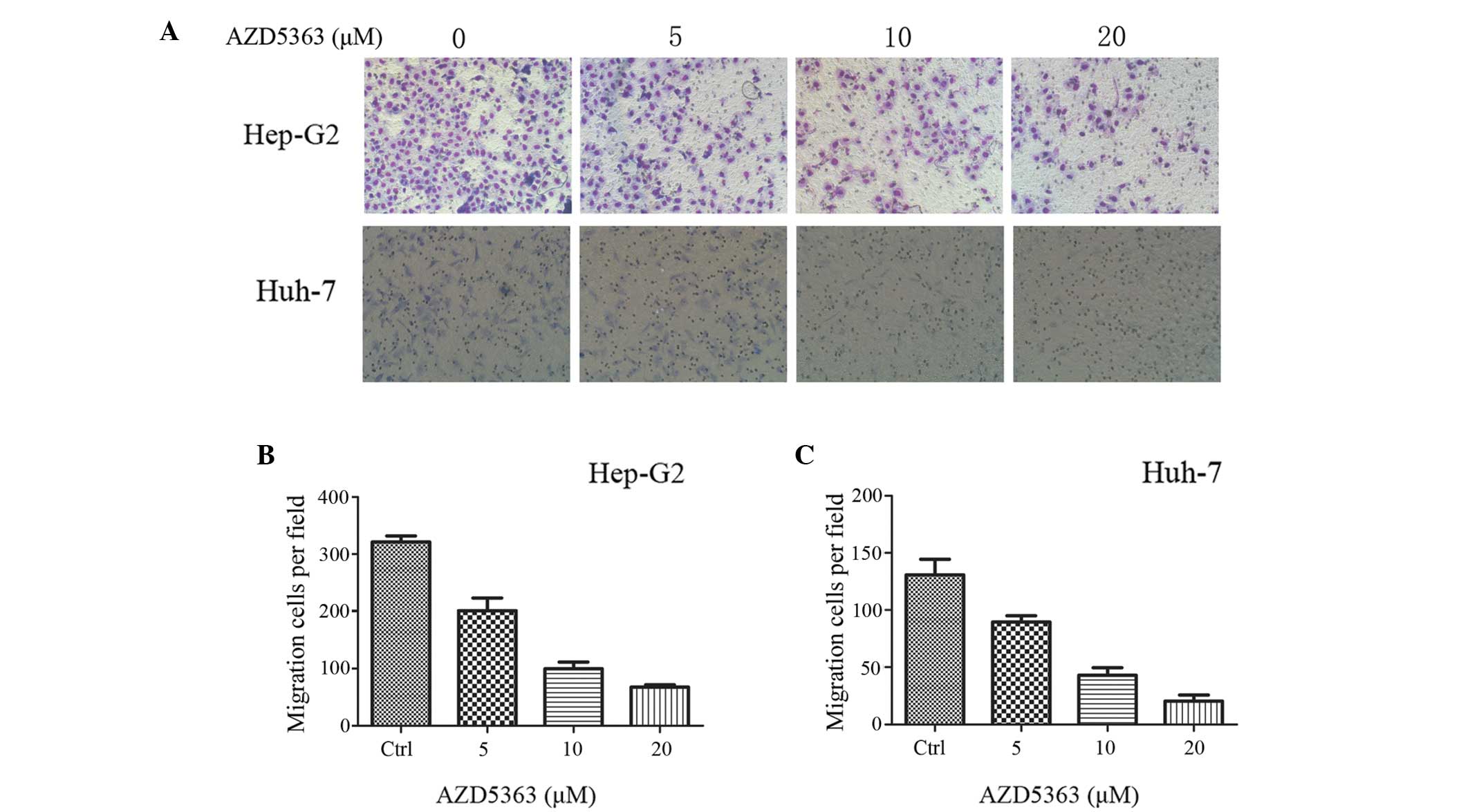
To investigate the effect of AZD5363 on liver cancer
cell migration, a Transwell migration assay was performed. The
treated Hep-G2 and Huh-7 cells, were inoculated in the upper
chamber with serum-free medium, whilst the lower chamber held
medium-containing serum. The cells were cultured for 24 h, and were
subsequently treated with formaldehyde, methanol and Giemsa
staining. As presented in Fig. 2,
AZD5363 significantly reduced the activity of the Hep-G2 and Huh-7
cells.
AZD5363 inhibits phosphorylation of
AKT substrates
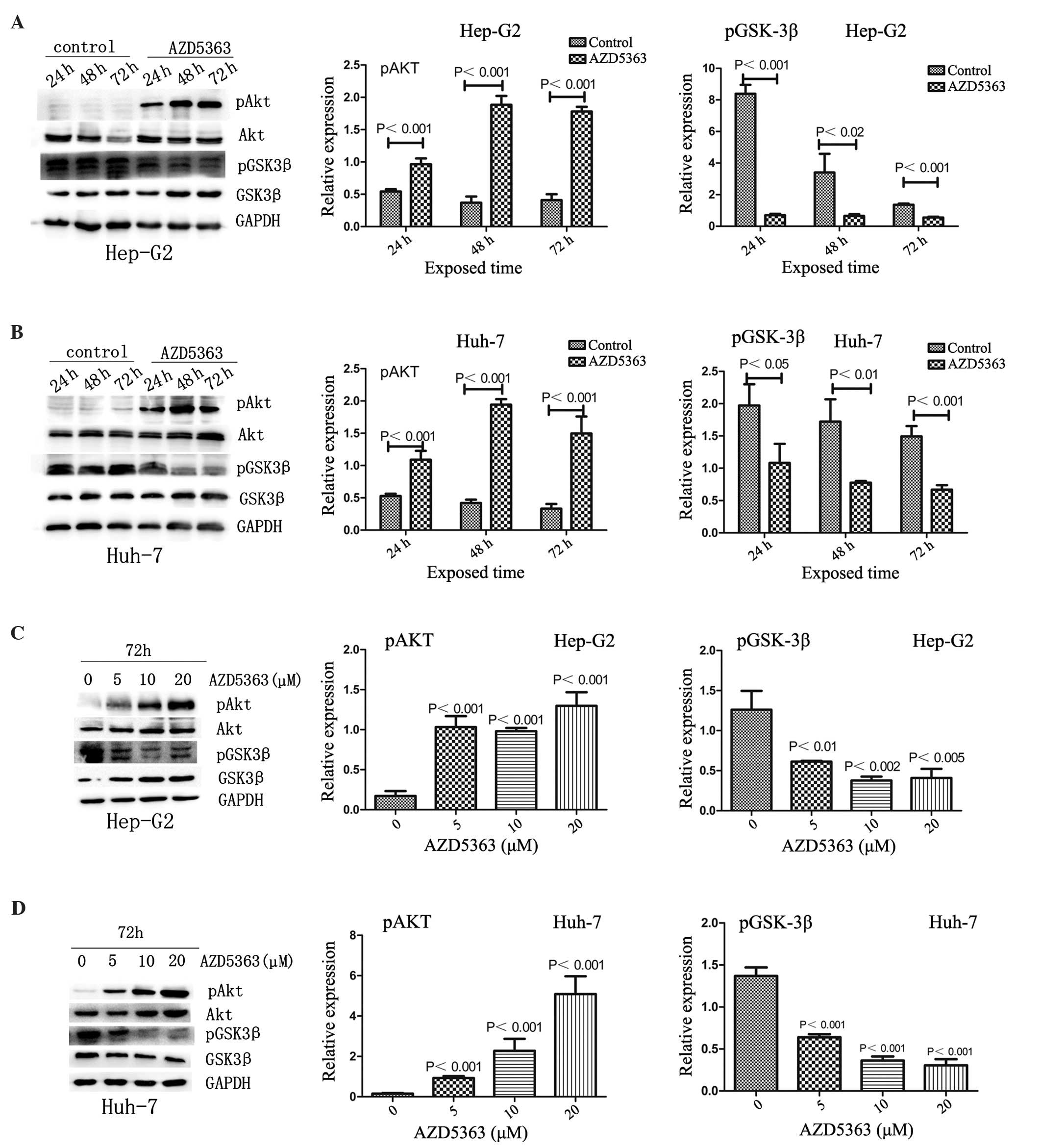
AKT functions in cell survival signaling by
phosphorylating downstream targets, with dephosphorylation of these
substrates indicating the inhibition of AKT activity (19). AKT serves a key role in glucose
metabolism; its substrate, GSK3β, modulates glycogen synthesis and
glucose transporter function. The present study therefore
investigated whether AZD5363 inhibits the phosphorylation of AKT
substrates. As expected, AZD5363 inhibited the phosphorylation of
GSK3β, but increased the phosphorylation of AKT in a dose- and
time-dependent manner in the Hep-G2 and Huh-7 cells (Fig. 3; P<0.05).
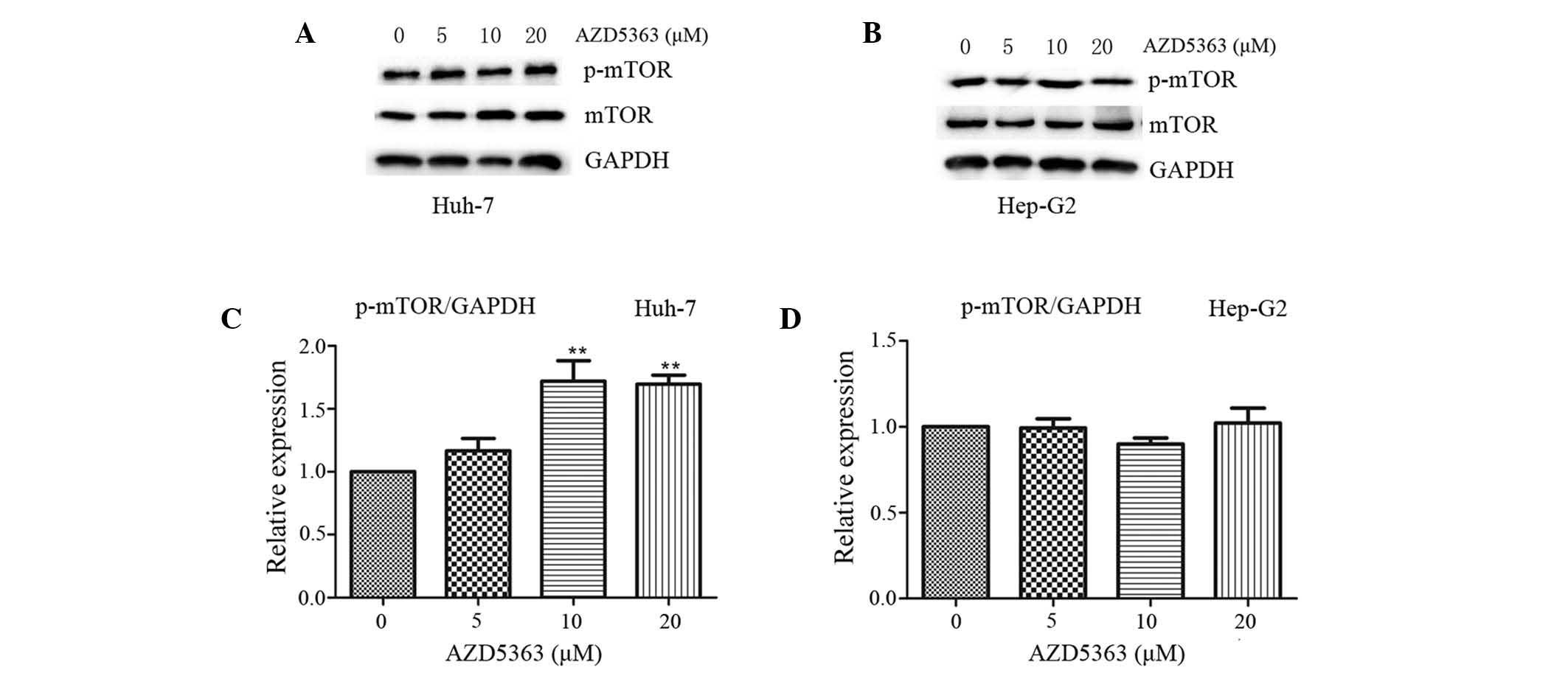
AZD5363 activates the phosphorylation
of mTOR, dependent on liver cancer cell type
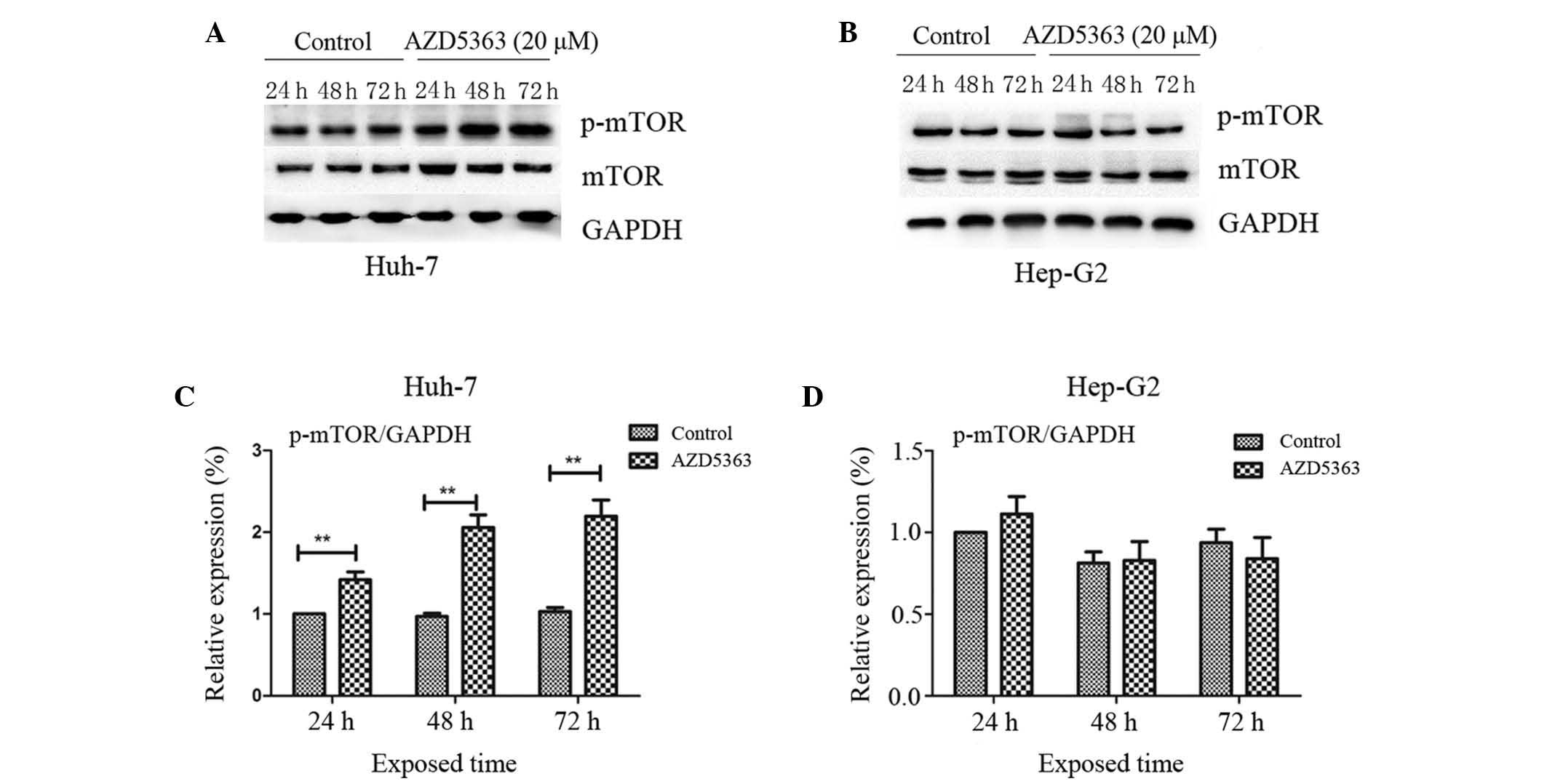
mTOR, a 289-kDa serine/threonine protein kinase,
belongs to the PIKK family and is activated through the PI3K and
AKT signaling pathways via phosphorylation of specific residues;
once activated, mTOR mediates transcription, cytoskeleton
organization, cell growth and cell survival (20,21). To
investigate the effect of AZD5363 on the mTOR pathway, the
phosphorylation levels of mTOR were analyzed. In contrast to the
inhibited phosphorylation of AKT substrates, AZD5363 exhibited
reduced activity in the mTOR pathway, as presented in panels of
tumor cell lines in vitro. AZD5363 enhanced the
phosphorylation of mTOR, however, this was only observed in the
Huh-7 cells. This indicated that AZD5363 significantly stimulated
mTOR signaling, but that this was dependent on liver cancer cell
type (Fig.4 and 5; P<0.01).
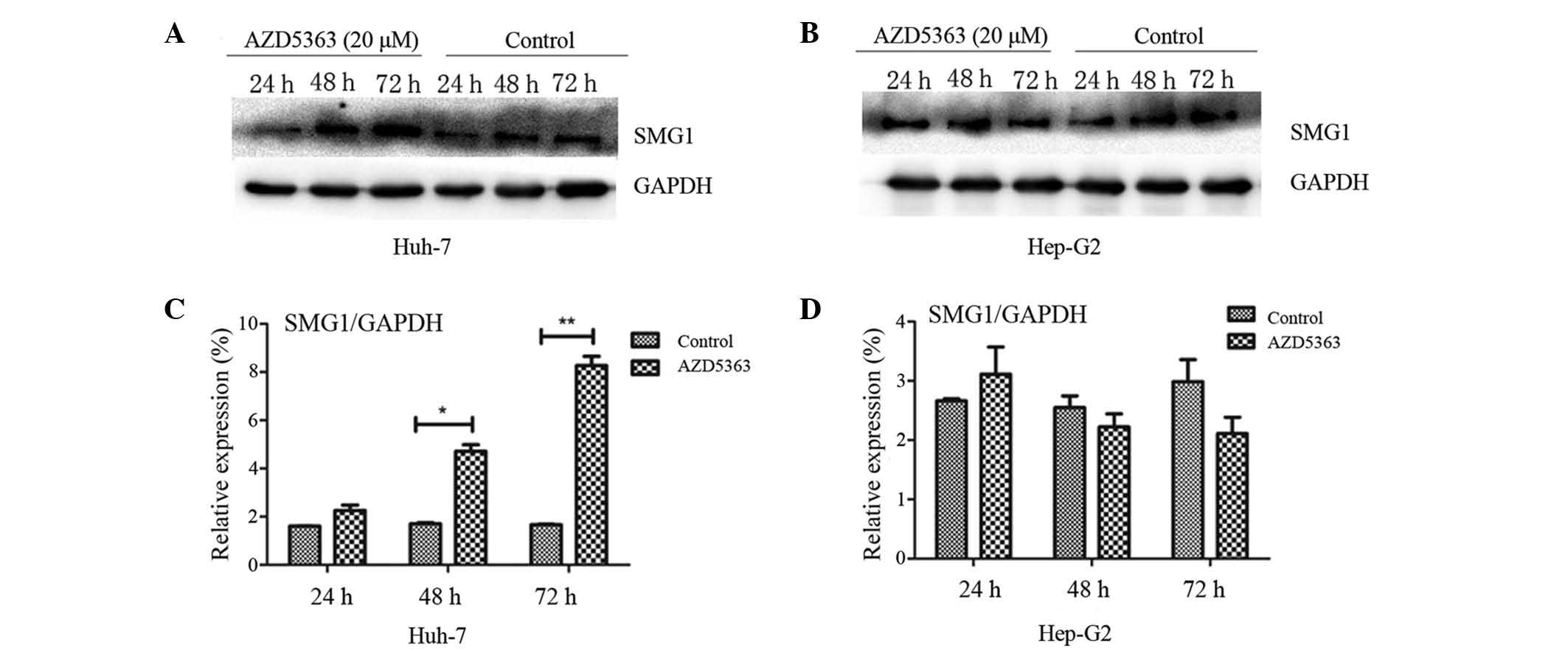
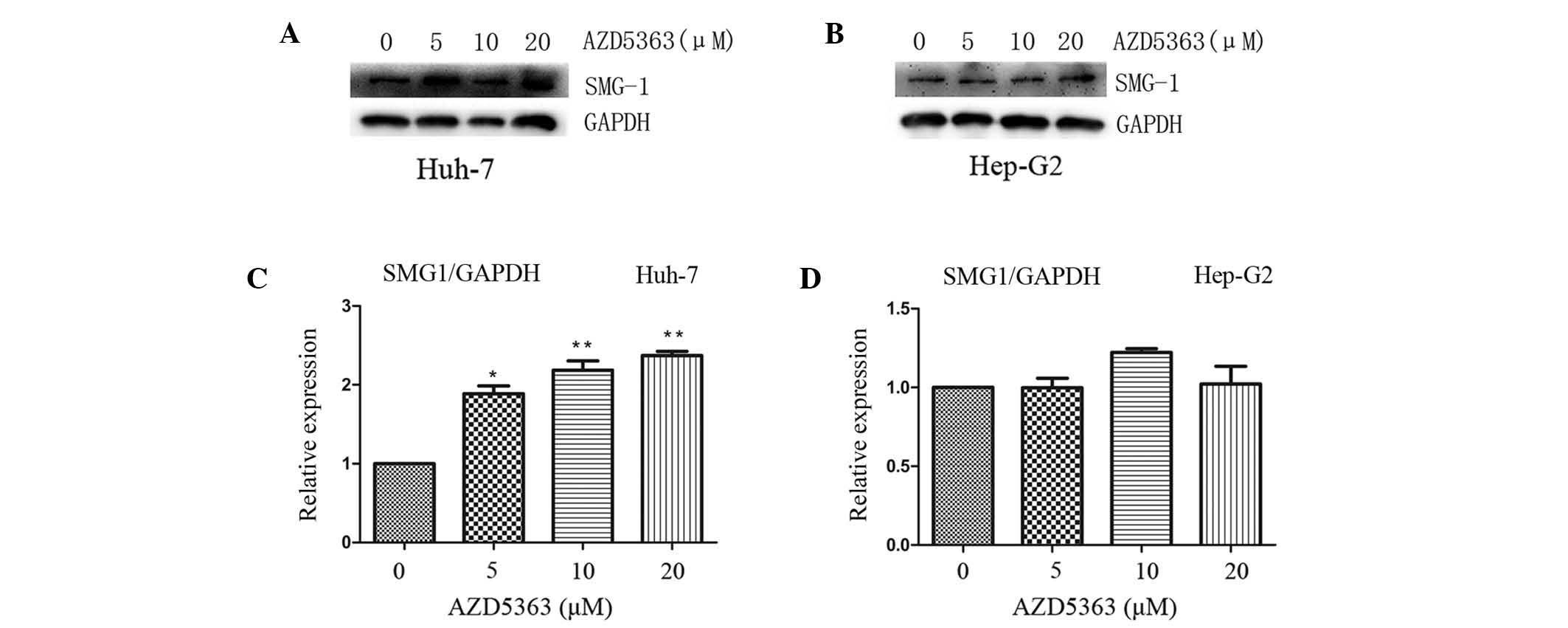
AZD5363 activates SMG-1, dependent on
liver cancer cell type
SMG-1 and mTOR each belong to the PIKK family.
Yamashita et al (22) reported
that the nonsense-mediated mRNA decay (NMD) pathway was suppressed
following inhibition of SMG-1. A follow-up study confirmed that the
phosphorylation of up-frameshift protein 1 (Upf1), via SMG-1, is an
important step required to trigger the NMD reaction (23). Therefore, SMG-1 may combine with Upf1
to promote its phosphorylation, subsequently forming the
SMG-1-Upf1-eRF1-eRF3 surveillance complex. The phosphorylation of
Upfl recruits SMG-5/SMG-7 composites and leads to the degradation
of nonsense mRNA (24,25). To investigate the effect of AZD5363 on
SMG-1 in the present study, the levels of SMG-1 were analyzed. It
was observed that AZD5363 promoted the expression of SMG-1 in the
Huh-7 cells, but not in the Hep-G2 cells (Fig. 6 and 7;
P<0.01). However, the mechanism by which AZD5363 induced SMG-1
was different from that of the AKT signaling; furthermore, it was
observed that the effect became more marked with an increasing dose
and time of exposure, which indicated that AZD5363 activated SMG-1
in a dose-and time-dependent manner (Figs. 6 and 7).
Discussion
Emerging evidence supports a crucial role for the
PI3K-AKT-mTOR pathway in tumorigenesis and drug resistance.
Regarding the treatment of solid tumors, including liver cancer, a
number of novel small molecule inhibitors that target PI3K, AKT and
mTOR are currently at varying phases of drug development. However,
the detailed effects that AZD5363 may exert on this signaling
pathway remain an unresolved issue. In the present study, the novel
AKT kinase inhibitor, AZD5363, exhibited strong specificity for the
AKT kinase in Hep-G2 and Huh-7 cells. AZD5363 suppressed
proliferation through the inhibition of AKT kinase activity in
vitro.
Recently, numerous studies have demonstrated that
AKT inhibitors may activate varying feedback loops, subsequently
affecting the efficacy of AKT inhibitors, including the MAPK and
HER3 pathways (26,27). A number of feedback loops and layers
of cross-talk have been reported to connect the mTOR complex 1
(mTORC1) and PI3K/AKT pathways. The proximal mTORC1 activator, Ras
homolog enriched in brain, is involved in the direct inhibition of
C-Raf activity and B-Raf/C-Raf heterodimerization, highlighting the
complexity of the connections among the mTORC1 and PI3K/AKT
pathways (28). In the present study,
however, the mechanism by which AZD5363 induced mTOR signaling was
different from that of the AKT depleted situation. It was observed
that while each cell line was sensitive to AZD5363, they exhibited
different reactions to the mTOR pathway. AZD5363 activated
phosphorylation of mTOR only in the Huh-7 cells. Nonetheless, the
explicit mechanism between mTOR and PI3K/AKT pathway activation by
AZD5363 remains elusive.
A number of studies focused on the development of
inhibitors of the mTOR signaling pathway are currently in progress
(29). In contrast to inhibitors of
the mTOR kinase (19,30), a recent study has reported that
AZD5363 exerts a reduced range of activity in panels of tumor cell
lines in vitro, for which a drug concentration required to
reduce growth rates to 50% of the maximum rate (GI50)
value of 3 µM was used as a cutoff; only 41 of 182 (23%) of the
cell lines were sensitive to AZD5363 (31). Furthermore, the same study also
observed that these tumor types, which have a high frequency of
phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α
(PIK3CA) mutation and PTEN loss, appear to have more contact with
AKT signaling and are sensitive to monotherapy inhibition by
AZD5363. In the present study, the Hep-G2 and Huh-7 cells also
exhibited differing reactions to mTOR kinase. Therefore, to
determine whether the liver cancer cells have these characteristics
(including HER2 amplification, RAS mutations, PIK3CA mutation and
PTEN loss) is necessary. It follows that certain tumor types may be
enriched for responders to an AKT inhibitor, such as AZD5363,
whereas in cell types that are insensitive to a specific AKT
inhibitor, targeting the AKT pathway alone is not sufficient enough
to eradicate cancer.
SMG-1 is a member of the PIKK family of mammalian
genes that includes mTOR, ataxia telangiectasia mutated (ATM), ATM
and Rad3-related, the DNA-dependent protein kinase catalytic
subunit and transformation/transcription domain-associated protein
(32). The NMD pathway has now been
identified and widely exists within eukaryotic organisms as a
highly-conserved RNA monitoring mechanism (33). Although its role as an NMD effector is
well recognized, SMG-1 also serves well-characterized roles in
other biological aspects, including the maintenance of genomic
integrity, the response to hypoxia and protection against tumor
necrosis factor-α-induced apoptosis, as well as possessing
essential roles in the DNA damage response, embryogenesis, the
regulation of diverse genes, the regulation of lifespan and
oxidative stress resistance, and the activation of p53 in response
to DNA double-strand breaks (34–37).
Recently, an increased level of awareness was focused on the
downregulation of SMG-1 in response to promoter hypermethylation in
human papillomavirus-positive head and neck squamous cell carcinoma
(38). Roberts et al (39) reported that SMG-1 heterozygous mice
exhibited a predisposition to various types of cancer, including
hematopoietic malignancies and lung cancer, and the development of
chronic inflammation. González-Estévez et al (13) reported that SMG-1 and mTORC1 act
antagonistically to regulate response to injury and growth in
planarians; the study also indicated that SMG-1 is likely to be a
potential human tumor suppressor gene product. Based on the studies
discussed, the present study analyzed the expression of SMG-1, with
a significant difference identified between the liver cancer cells.
In the Huh-7 cells, when exposed to AZD5363, SMG-1 was activated in
a time- and dose-dependent manner, but this was not observed in the
Hep-G2 cells. With continued investigation, SMG-1 is expected to
become one of the breakthrough targets in cancer treatment,
possibly providing novel ideas and methods for the diagnosis and
treatment of tumor-associated diseases.
mTOR is a key component of the PIKK/AKT/mTOR
signaling pathway and functions as a kinase-activating molecule
downstream of PIKK/AKT (12). Despite
SMG-1 and mTOR each belonging to the PIKK family, the interactions
between them are not yet fully understood. In the present study, it
was observed that AZD5363 activated the phosphorylation of mTOR in
the Huh-7 cells, but this was not observed in the Hep-G2 cells;
this indicates that AZD5363 activated SMG-1 only in the Huh-7
cells. Therefore, it can be inferred that mTOR expression is
positively correlated with that of SMG-1, possibly suggesting that
SMG-1 may interact with mTOR signaling in a direct or indirect
manner. González-Estévez et al (13) also described near opposite roles for
mTOR and SMG-1 in planarian regeneration. Altman et al
(40) reported that tumor suppressor
molecules may target and inhibit the mTOR pathway, resulting in
regulatory effects on mRNA translation. It has also been noted that
in planarian worms, SMG-1 may function as a regulator of injury and
growth responses, primarily through cross-talk interactions with
mTOR (13). González-Estévez et
al (13) observed that SMG-1 was
essential for the tight control of stem cell proliferation and
differentiation caused by injury or nutrient status in planarian
flatworms (Schmidtea mediterranea). The knockdown of SMG-1
in planarian flatworms, similar to the knockdown of several known
human suppressors, such as PTEN or p53, leads to lethal outgrowths
(13,41,42). Such
findings suggest that SMG-1 may serve a potential role as a tumor
suppressor in human cancer.
In conclusion, extensive genetic and biochemical
research may aid the clarification of the association between mTOR
and SMG-1. Nevertheless, the present study has uncovered novel
roles of AZD5363 that may be targeted in the treatment of liver
cancer, with the investigation of the possible evolutionary
conservation of these roles also likely to be advantageous.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China, 2014 (grant no. 81373172).
References
|
1
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abou-Alfa GK, Schwartz L, Ricci S, Amadori
D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz
B, et al: Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitsui H, Takuwa N, Maruyama T, Maekawa H,
Hirayama M, Sawatari T, Hashimoto N, Takuwa Y and Kimura S: The
MEK1-ERK map kinase pathway and the PI 3-kinase-Akt pathway
independently mediate anti-apoptotic signals in HepG2 liver cancer
cells. Int J Cancer. 92:55–62. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Villanueva A, Chiang DY, Newell P, Peix J,
Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al:
Pivotal role of mTOR signaling in hepatocellular carcinoma.
Gastroenterology. 135:1972–1983, 1983.e1-1983.e11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calvisi DF, Wang C, Ho C, et al: Increased
lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes
development of human hepatocellular carcinoma. Gastroenterology.
140:1071–1083. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stiles B, Wang Y, Stahl A, et al:
Liver-specific deletion of negative regulator Pten results in fatty
liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci
USA. 101:2082–2087. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
González-Estévez C, Felix DA, Smith MD,
Paps J, Morley SJ, James V, Sharp TV and Aboobaker AA: SMG-1 and
mTORC1 act antagonistically to regulate response to injury and
growth in planarians. PLoS Genet. 8:e10026192012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han EK, Leverson JD, McGonigal T, Shah OJ,
Woods KW, Hunter T, Giranda VL and Luo Y: Akt inhibitor A-443654
induces rapid Akt Ser-473 phosphorylation independent of mTORC1
inhibition. Oncogene. 26:5655–5661. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Levy DS, Kahana JA and Kumar R: AKT
inhibitor, GSK690693, induces growth inhibition and apoptosis in
acute lymphoblastic leukemia cell lines. Blood. 113:1723–1729.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin J, Sampath D, Nannini MA, Lee BB,
Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, et al:
Targeting activated Akt with GDC-0068, a novel selective Akt
inhibitor that is efficacious in multiple tumor models. Clin Cancer
Res. 19:1760–1772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Z, Oh DY, Nakamura K and Thiele CJ:
Perifosine-induced inhibition of Akt attenuates brain-derived
neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma
in vivo. Cancer. 117:5412–5422. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu R, Liu D, Trink E, Bojdani E, Ning G
and Xing M: The Akt-specific inhibitor MK2206 selectively inhibits
thyroid cancer cells harboring mutations that can activate the
PI3K/Akt pathway. J Clin Endocrinol Metab. 96:E577–E585. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davies BR, Greenwood H, Dudley P, Crafter
C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, et al: Preclinical
pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics,
antitumor activity, and correlation of monotherapy activity with
genetic background. Mol Cancer Ther. 11:873–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chong ZZ, Shang YC, Zhang L, Wang S and
Maiese K: Mammalian target of rapamycin: Hitting the bull's-eye for
neurological disorders. Oxid Med Cell Longev. 3:374–391. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chong ZZ and Maiese K: Mammalian target of
rapamycin signaling in diabetic cardiovascular disease. Cardiovasc
Diabetol. 11:452012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamashita A, Ohnishi T, Kashima I, Taya Y
and Ohno S: Human SMG-1, a novel phosphatidylinositol
3-kinase-related protein kinase, associates with components of the
mRNA surveillance complex and is involved in the regulation of
nonsense-mediated mRNA decay. Genes Dev. 15:2215–2228. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grimson A, O'Connor S, Newman CL and
Anderson P: SMG-1 is a phosphatidylinositol kinase-related protein
kinase required for nonsense-mediated mRNA decay in
Caenorhabditis elegans. Mol Cell Biol. 24:7483–7490. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hwang J and Maquat LE: Nonsense-mediated
mRNA decay (NMD) in animal embryogenesis: To die or not to die,
that is the question. Curr Opin Genet Dev. 21:422–430. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamashita A: Role of SMG-1-mediated Upf1
phosphorylation in mammalian nonsense-mediated mRNA decay. Genes
Cells. 18:161–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engelman JA, Chen L, Tan X, Crosby K,
Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y,
et al: Effective use of PI3K and MEK inhibitors to treat mutant
Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med.
14:1351–1356. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chandarlapaty S, Sawai A, Scaltriti M,
Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK,
Baselga J and Rosen N: AKT inhibition relieves feedback suppression
of receptor tyrosine kinase expression and activity. Cancer Cell.
19:58–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karbowniczek M, Robertson GP and Henske
EP: Rheb inhibits C-raf activity and B-raf/C-raf
heterodimerization. J Biol Chem. 281:25447–25456. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barrett D, Brown VI, Grupp SA and Teachey
DT: Targeting the PI3K/AKT/mTOR signaling axis in children with
hematologic malignancies. Paediatr Drugs. 14:299–316. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Serra V, Markman B, Scaltriti M, Eichhorn
PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S,
et al: NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K
signaling and inhibits the growth of cancer cells with activating
PI3K mutations. Cancer Res. 68:8022–8030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu K, Shi C, Toral-Barza L, Lucas J, Shor
B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, et al: Beyond
rapalog therapy: Preclinical pharmacology and antitumor activity of
WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and
mTORC2. Cancer Res. 70:621–631. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lloyd JP and Davies B: SMG1 is an ancient
nonsense-mediated mRNA decay effector. Plant J. 76:800–810. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oliveira V, Romanow WJ, Geisen C,
Otterness DM, Mercurio F, Wang HG, Dalton WS and Abraham RT: A
protective role for the human SMG-1 kinase against tumor necrosis
factor-alpha-induced apoptosis. J Biol Chem. 283:13174–13184. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nicholson P, Yepiskoposyan H, Metze S,
Orozco Zamudio R, Kleinschmidt N and Mühlemann O: Nonsense-mediated
mRNA decay in human cells: Mechanistic insights, functions beyond
quality control and the double-life of NMD factors. Cell Mol Life
Sci. 67:677–700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masse I, Molin L, Mouchiroud L, Vanhems P,
Palladino F, Billaud M and Solari F: A novel role for the SMG-1
kinase in lifespan and oxidative stress resistance in
Caenorhabditis elegans. PLoS One. 3:e33542008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McIlwain DR, Pan Q, Reilly PT, Elia AJ,
McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ and Mak TW:
Smg1 is required for embryogenesis and regulates diverse genes via
alternative splicing coupled to nonsense-mediated mRNA decay. Proc
Natl Acad Sci USA. 107:12186–12191. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gewandter JS, Bambara RA and O'Reilly MA:
The RNA surveillance protein SMG1 activates p53 in response to DNA
double-strand breaks but not exogenously oxidized mRNA. Cell Cycle.
10:2561–2567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gubanova E, Brown B, Ivanov SV, Helleday
T, Mills GB, Yarbrough WG and Issaeva N: Downregulation of SMG-1 in
HPV-positive head and neck squamous cell carcinoma due to promoter
hypermethylation correlates with improved survival. Clin Cancer
Res. 18:1257–1267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roberts TL, Ho U, Luff J, Lee CS, Apte SH,
MacDonald KP, Raggat LJ, Pettit AR, Morrow CA, Waters MJ, et al:
Smg1 haploinsufficiency predisposes to tumor formation and
inflammation. Proc Natl Acad Sci USA. 110:E285–E294. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Altman JK, Sassano A and Platanias LC:
Targeting mTOR for the treatment of AML. New agents and new
directions. Oncotarget. 2:510–517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oviedo NJ, Pearson BJ, Levin M and Sánchez
Alvarado A: Planarian PTEN homologs regulate stem cells and
regeneration through TOR signaling. Dis Model Mech. 1:131–143.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pearson BJ and Sánchez Alvarado A: A
planarian p53 homolog regulates proliferation and self-renewal in
adult stem cell lineages. Development. 137:213–221. 2010.
View Article : Google Scholar : PubMed/NCBI
|