Introduction
In the 1920s, Warburg observed that cancer cells had
a distinct metabolism that was highly dependent on glycolysis
instead of mitochondrial oxidative phosphorylation, regardless of
oxygen availability (1). This process
was termed the Warburg effect, or aerobic glycolysis (2). Hanahan and Weinberg (3) also revealed that reprogramming energy
metabolism was one of the most common characteristics of cancer
cells. Compared with oxidative phosphorylation, glycolysis is a
less efficient pathway for producing adenosine triphosphate (ATP)
(4). To compensate for the loss of
ATP due to preferential glycolysis, cancer cells upregulate genes
that encode glucose transporters and glycolytic enzymes, which
leads to glucose uptake and an altered metabolism (5). Increased glucose uptake is used in the
clinic for the detection of tumors using fluorodeoxyglucose
positron emission tomography (4). The
metabolic reprogramming of cancer cells provides ATP and allows
cells to survive under hypoxic conditions (6). In addition, metabolic reprogramming
provides cells with biosynthetic building blocks, including
intermediates and substrates for the synthesis of nucleotides,
proteins and membrane components, which are required in
proliferating cells (7).
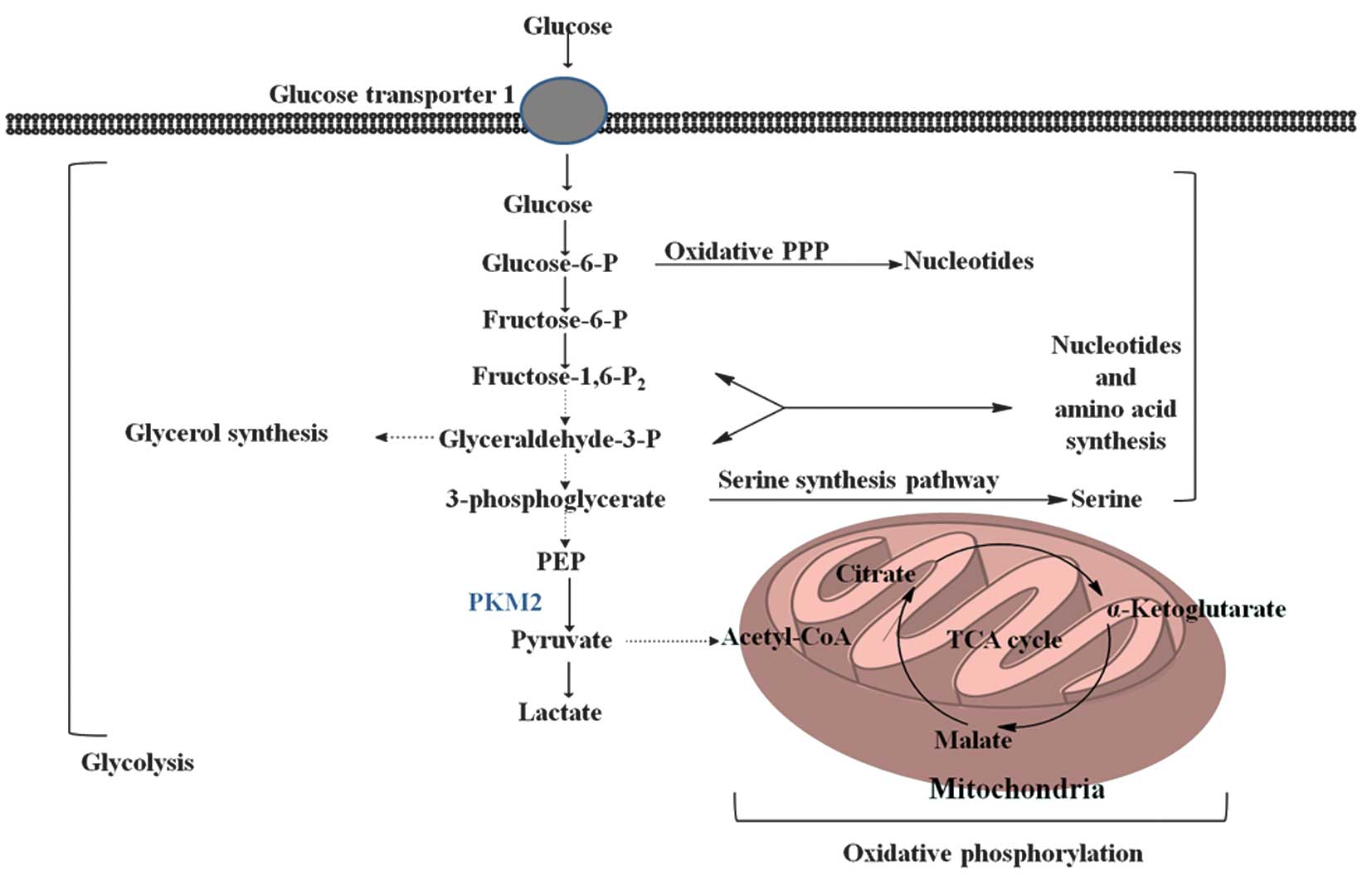
Pyruvate kinase (PK) is a rate-limiting glycolytic
enzyme that catalyzes the irreversible transphosphorylation between
phosphoenolpyruvate (PEP) and adenosine diphosphate, which produces
pyruvate and ATP (8). This reaction
is a committed step that leads to glycolysis or oxidative
phosphorylation of pyruvate (Fig. 1).
In mammals, the PK family consists of four isoforms: liver-type PK
(PKL); red blood cell PK (PKR); and PK muscle isozyme M1 and M2
(PKM1 and PKM2, respectively). These isoforms are encoded by two
genes, PK, liver and red blood cell (PKLR) and PK, muscle (PKM)
(9,10). The expression of pyruvates is
tissue-specific and regulated by various promoters and alternative
splicing. PKL and PKR are products of the PKLR gene. PKL is
expressed in the liver, kidneys and intestine and has the lowest
affinity to PEP of the isoforms, while PKR is expressed in red
blood cells, inhibited by ATP and activated by
fructose-1,6-bisphosphate (FBP). PKM1 and PKM2 are produced by
alternative splicing of the primary RNA transcript of the PKM gene,
which contains sequences encoded by exons 9 and 10, respectively.
The non-allosteric isoform PKM1 is constitutively active, and
expressed in terminally differentiated tissues, including the
muscle and brain, which require a large supply of ATP. By contrast,
PKM2 is expressed in tissues with anabolic functions, including
proliferating cells and cancer cells, and is subject to complex
allosteric regulation. In the majority of cancer cells, the
expression of PKM2 is increased, which suggests that PKM2 may be an
attractive target for cancer therapy (11). The identification of a novel protein
that interacts with PKM2 has confirmed the key function of PKM2 in
tumor metabolism and cancer growth (12), revealing novel functions of PKM2
beyond the classical concept of glycolysis.
The PKM2 gene and its regulation
The PKM2 gene is expressed by alternative splicing
of PKM pre-messenger (m)RNA (13). In
cancer cells, PKM2 is overexpressed, and overexpression is
controlled by the oncoprotein c-Myc. c-Myc activates the
transcription of heterogeneous nuclear ribonucleoproteins (hnRNPs)
I, A1 and A2, which bind and repress exon 9 encoding RNA sequences
(13). This results in the inhibition
of PKM1 mRNA splicing, which allows the synchronous expression of
the PKM2 isoform (13). A previous
study demonstrated that PKM was regulated by the reciprocal effects
of the mutually exclusive exons 9 and 10, which lead to the
repression of exon 9 and the activation of exon 10 in cancer cells
(14). In addition, knockdown of the
serine/arginine-rich family of pre-mRNA splicing factor, which
reduces lactate production, triggers the expression of exon 10 and
promotes the proliferation of cells and aerobic glycolysis. This
study suggested that the exonic elements are the actual
determinants of PKM2 splicing, as opposed to classical intronic
regions.
In the last decade, studies have focused on the
metabolism and signal transduction of cells, and have revealed that
oncogenes may have direct or indirect associations with metabolic
reprogramming (15,16). Mammalian target of rapamycin (mTOR)
was demonstrated to be a key activator of the Warbug effect, as it
induces PKM2 and other glycolytic enzymes under normoxic conditions
(17). Sun et al (17) demonstrated that PKM2 was a crucial
glycolytic enzyme in the oncogenic mTOR-induced Warbug effect, in
which hypoxia inducible factor-1α (HIF-1α) and c-Myc-hnRNP cascades
are the transducers of mTOR regulation of PKM2. Additionally,
insulin is closely associated with cancer progression, and also
upregulates PKM2 expression through phosphatidylinositide
3-kinase/mTOR mediated HIF-1α induction (18). Notably, the reduction in the activity
of PKM2 is independent of this pathway; insulin-induced reactive
oxygen species (ROS) was revealed to be responsible (18). Under hypoxic conditions, the PKM2 gene
interacts directly with HIF-1α, which activates the hypoxia
response element that is required for HIF-1α binding (19).
A previous study demonstrated that the transcription
of PKM2 was upregulated by the epidermal growth factor receptor
(EGFR) under normoxic conditions (20). EGFR activation induces phospholipase C
γ1-dependent protein kinase C (PKC)ε monoubiquitination at
lysine-321, which is mediated by RING-finger protein that interacts
with C kinase 1 (21,22). Monoubiquitinated PKCε interacts with
an ubiquitin-binding domain in the zinc finger domain of IκB kinase
(IKK)γ, which leads to the recruitment of cytosolic IKK to the
plasma membrane. PKCε phosphorylates IKKβ at serine-177, which
activates IKKβ. Activated v-rel avian reticuloendotheliosis viral
oncogene homolog A (RelA) interacts with HIF-1α, which is required
for the PKM promoter to bind to RelA (21,22).
EGFR-promoted glycolysis and tumorigenesis requires PKCε- and
nuclear factor κ enhancer binding protein (NFκB)-dependent PKM2
upregulation (21,22). These molecular interactions reveal the
importance of the association between EGFR and NFκB pathways in the
upregulation of PKM2 and tumorigenesis of cells.
Active and inactive oligomeric forms of
PKM2: Activity regulation
Cells have evolved complex regulatory mechanisms to
adapt the metabolism to various physiological states. Rapidly
growing cells consume nutrients at a high rate and must maintain a
balance between the utilization of nutrients for ATP synthesis and
anabolic development, including protein, lipid and nucleic acid
synthesis. Cancer cells use glucose at higher rates compared to
non-cancerous cells, but use a smaller fraction for oxidative
phosphorylation, which enables cancer cells to incorporate a
greater fraction of glucose metabolites in macromolecule synthesis
instead of expending it on carbon dioxide production (2,3).
Consequently, metabolic programming of cancer cells is required to
be flexible, which allows the cells to adapt to various
environmental conditions. PKM2 is responsible for the final step of
glycolysis and is key in this process (2,3). The
preferential expression and allosteric enzymatic activity of PKM2
provides the cancer cells with a growth advantage in vivo,
without the accumulation of ROS.
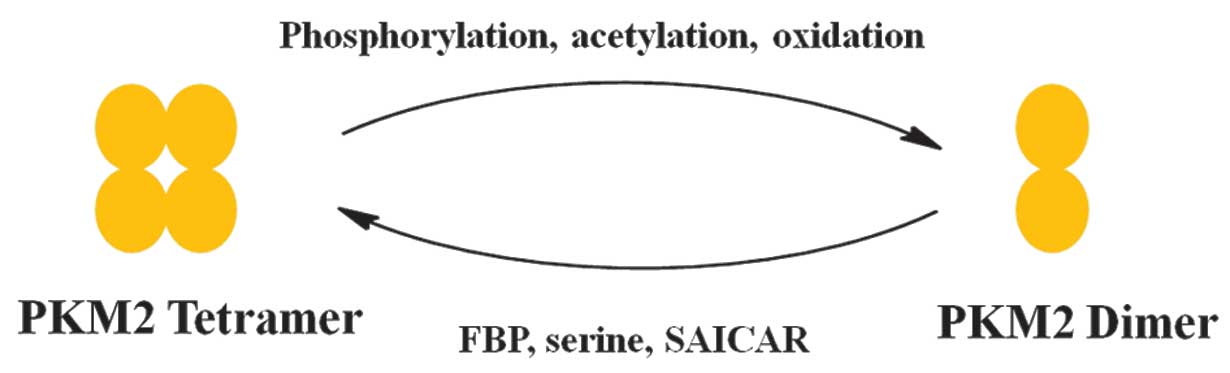
Allosteric regulation of PKM2 allows switching
between a high- and low-activity state. PKM2 exists in the
catalytically distinct tetrameric and dimeric states (2,3).
Tetrameric PKM2 exhibits high catalytic activity, which is
associated with ATP synthesis and catabolic metabolism in cells.
Dimeric PKM2 has low catalytic activity and is the less active
state of PKM2 that facilitates the production of glycolytic
intermediates to enter the glycolysis branch pathways, including
glycerol synthesis and the pentose phosphate pathway, which
produces nicotinamide adenine dinucleotide phosphate-oxidase
(NADPH) to suppress ROS production and is involved in nucleotide
synthesis (2,3).
Various factors have been reported that control the
alteration between the dimer and tetramer of PKM2 (Fig. 2) (23).
FBP is an upstream glycolytic intermediate and is a
well-characterized activator of PKM2, which binds allosterically to
PKM2 and facilitates the formation of the active tetramer (11). In addition, serine, which is a
glycolytic intermediate 3-phosphoglycerate, is a positive regulator
of PKM2 (24). Serine binds and
activates PKM2, and a decrease in serine leads to a reduction in
the PKM2 activity in cells (24).
Phosphoribosylaminoimidazolesuccinocarboxamide (SAICAR) is an
intermediate of the de novo purine nucleotide synthesis
pathway and also stimulates PKM2 activity (25). SAICAR-PKM2 interaction promotes cancer
cell survival in glucose-limited conditions (25–27). This
allosteric regulation may lead to cancer cells coordinating various
metabolic pathways to optimize cell growth in nutrient-limited
conditions. PKM2 activity is also regulated by post-translational
modifications, including phosphorylation, acetylation and
oxidation, which favor the inactive dimeric state of PKM2 (25–27).
Phosphorylation of PKM2 at tyrosine-105 induces the release of FBP,
which causes PKM2 to alter between the tetrameric and dimeric
states. Additionally, acetylation of PKM2 at lysine-305
downregulates PKM2 activity, which promotes glycolytic pooling,
NADPH synthesis and tumor growth (25–27).
Notably, a high glucose concentration induces a lysine-305
acetylation of PKM2, which diverts glucose to anabolic synthesis.
Oxidation of PKM2 at cysteine-358 confers an advantage to cancer
cells, as it allows the cells to withstand ROS (28). Intracellular ROS-induced oxidation of
cysteine-358 decreases the activity of PKM2, which diverts glucose
flux to the anabolic pentose phosphate pathway, thereby producing
sufficient reducing potential for the detoxification of ROS.
Missense mutations in the PKM2 gene, including H391Y
and K422R, are observed within the inter-subunit contact domain of
the PKM2 protein, which may promote cancer metabolism, oxidative
endurance, anchorage independence and tumor growth in a
dominant-negative manner (29). The
two gene mutations have different effects on the activity of PKM2,
which does not affect the stability or expression level of the PKM2
protein. The H391Y mutated isoform of PKM2, resulting from a signal
amino acid residue replacement, leads to the total loss of
allosteric behavior, which affects the dynamic movement of the
protein and results in cell rigidity (29).
Non-metabolic functions of PKM2
PKM2 acts as a coactivator
PKM2 is not only present in the cytoplasm of cells
as a PK enzyme, but also translocates to the nucleus, which
suggests PKM2 has functions in addition to glycolysis (19,30). There
has been a resurgence of interest in the metabolic effects of PKM2,
and there have been recent studies revealing non-metabolic
functions of PKM2. It has been reported that PKM2 translocates into
the nucleus and interacts directly with the HIF-1α subunit and
promotes transactivation of HIF-1 target genes by enhancing HIF-1
binding and recruitment of p300, a transcriptional coactivator,
which regulates HIF-1 activity (19,30). PKM2
gene transcription is also activated by HIF-1, which creates a
positive feedback loop that promotes HIF-1 transactivation and
alters glucose metabolism in cancer cells (19). The Jumonji C domain-containing
dioxygenase Jumonji domain-containing protein 5 (JMJD5) directly
interacts with PKM2, which induces the translocation of PKM2 into
the nucleus and promotes HIF-1α mediated transactivation (31). The JMJD5-PKM2 interaction occurs at
the intersubunit interface region of PKM2, which hinders PKM2
tetramerization and blocks PK activity.
The translocation of PKM2, and not PKM1, may be in
response to EGFR activation (32). In
PKM2, lysine-433 binds to c-Src-phosphorylated tyrosine-333 on
β-catenin, and this interaction is required for the two proteins to
be recruited to the cyclin D1 (CCND1) promoter (32). This results in the removal of histone
deacetylase 3 (HDAC3) from the promoter, histone H3 acetylation and
CCND1 expression (12).
PKM2-dependent β-catenin transactivation is required for
EGFR-promoted tumor cell proliferation and brain tumor development
(33). In addition, associations have
been identified between c-Src activity, β-catenin tyrosine-333
phosphorylation and PKM2 nuclear accumulation in human glioblastoma
specimens (32). In colorectal
cancer, decrease in the expression of PKM2 stimulates the β-catenin
signaling pathway, which leads to the promotion of c-Myc-mediated
glutamine metabolism (34). PKM2 may
also be acetylated by p300 acetyltransferase at lysine-433, which
prevents PKM2 activation by altering FBP (35). Activation of PKM2 promotes the nuclear
accumulation and protein kinase activity, which is increased by
cell cycle stimulation, EGF and oncoprotein E7. Phosphorylation of
PKM2 at serine-37 promotes the translocation of PKM2 to the nucleus
through the mitogen-activated protein kinase 1 ERK2 docking groove,
which binds directly to PKM2 at isoleucine-429/leucine-431, and the
protein kinase activity of PKM2 (36). Phosphorylation of PKM2 at serine-37
recruits peptidyl-prolyl cis-trans isomerase
NIMA-interacting 1 for the cis-trans isomerization of PKM2
(36). In addition, nuclear PKM2 acts
as a coactivator of β-catenin to induce the expression of c-Myc.
Upregulation of c-Myc expression by PKM2 creates a positive
feedback-loop, since c-Myc elevates the transcription of hnRNPs,
which contributes to an increase in the PKM2/PKM1 ratio and
promotes glycolysis by driving the expression of enzymatic kinases
(36).
Protein kinase activity of PKM2
PKM2 not only acts as a coactivator, but may also
act as a protein kinase that phosphorylates substrates involved in
metabolic reprogramming. Nuclear dimeric PKM2 directly
phosphorylates signal transducer and activator of transcription 3
(STAT3), which is activated in response to inflammatory cytokines,
including interleukin-6 (37). PKM2
uses PEP as a phosphate donor to phosphorylate STAT3 at
tyrosine-705, which activates the transcription of
mitogen-activated protein kinase kinase 5. The activation of STAT3
in malignant cancer cells is possibly one of the most important
molecular signatures for promoting the progression of cancer. PKM2
overexpression facilitates STAT3 nuclear translocation, which
regulates the aggressive progression of colorectal cancer (38). The functional implications of the
study by Yang et al (38)
indicate that PKM2 activates β1-integrin-focal adhesion kinase and
snail-2-E-cadherin signaling, and also upregulates the expression
of matrix metalloproteinase-2 and 9, which induces cell migration
and adhesion. Similarly to STAT3, histone H3 is a substrate for
PKM2 kinase activity (39). PKM2
directly binds and phosphorylates histone H3 at threonine-11, which
leads to the removal of HDAC3 from CCND1 and Myc promoter regions,
subsequent to acetylation at lysine-9 and gene transcription
(39). PKM2-dependent histone H3
modifications are key to EGF stimulation (19). Furthermore, nuclear PKM2 levels are
associated with phosphorylation at proline-3/threonine-11 in
malignant glioma (36). Recently, a
study reported that the PKM2-SAICAR interaction is required and
sufficient to induce robust protein kinase activity in PKM2 in
vitro and in cancer cells (26).
It was also reported that the PKM2-SAICAR complex phosphorylates
>100 human proteins, the majority of which were previously
unrecognized. In particular, PKM2-SAICAR directly activates ERK1
in vitro; however, activated ERK1/2 phosphorylates PKM2,
creating a positive feedback loop (36). Additionally, when EGFR is activated,
the concentration of cellular SAICAR is increased, which is
required to sustain the activation of ERK1/2 and proliferative
signaling via PKM2 (36).
PKM2 acts as a differentiating
agent
PKM2 acts as a proliferative agent through its
interaction with nuclear proteins, and as a differentiating agent
through its interaction with octamer-binding transcription factor 4
(Oct4), which is a key regulator in cancer stem cell self-renewal
and differentiation (40). In glioma
stem cells, the interaction between PKM2 and Oct4 inhibits the
ability of PKM2 to maintain cell stemness, thereby promoting cell
differentiation and enhancing the sensitivity of cells to cell
death, which is concomitant with an alteration between the dimer
and tetramer states of PKM2 (40).
Notably, dichloroacetate, which is known as an inhibitor of PDK1
and is involved in the mitochondrial tricarboxylic acid cycle,
increases the number of PKM2/Oct4 complexes (41).
PKM2 emergence in blood
circulation
PKM2 detected in the blood of patients with
gastrointestinal, pancreatic, lung and ovarian cancer and renal
cell carcinoma revealed that PKM2 is released into the circulation
(42). The PKM2 levels in the
circulation of patients may be used as a diagnostic marker for
numerous types of cancer (43). A
recent study suggested that PKM2 in the blood circulation
facilitated tumor growth by promoting tumor angiogenesis (44). The study demonstrated that PKM2
promoted tumor angiogenesis by increasing endothelial cell
proliferation, migration and extracellular matrix-cell adhesions;
only dimeric PKM2 was observed to be involved in tumor
angiogenesis.
PKM2 as a therapeutic target
Cancer-specific metabolism, which is closely
associated with tumorigenicity, cancer aggressive progression and
inherent or acquired therapeutic resistance, is an attractive
target for cancer therapy. The role of PKM2 in glycolysis and the
proliferation of cancer cells has been revealed, and has been
demonstrated to balance the production of biomolecular building
blocks and the generation of pyruvate and ATP. Consequently, there
are two therapeutic strategies for targeting PKM2. The first is
inhibitors that block the catalytic activity of PKM2, and the
second is activators that induce tetramerization of PKM2 to
increase glycolysis.
Inhibition of PMK2 activity
Inhibitors of PKM2 appear to rely on the hypothesis
that proliferating cells are highly dependent on energy, and a
reduced activity of PKM2 inhibits energy regeneration. Using a high
throughput screen, small molecules have been identified that
selectively inhibit PKM2 (45). These
molecules target the allosteric FBP binding site of PKM2; however,
since PKL and PKR are also activated by FBP, there may be toxicity
issues for human cancer therapy if PK activity is inhibited in the
liver and red blood cells. Natural products, such as shikonin and
its analog alkannin, which belong to a family of necroptotic
inducers, demonstrated potent and promising selective inhibition of
PKM2 that did not inhibit PKM1 and PKL (46). However, PKM2 inhibition by these drugs
is partially reversed by the addition of FBP to protein
lysates.
RNA interference targeting PKM2 has been observed to
attenuate growth and induce caspase-dependent apoptosis in several
cancer cell lines (47). In addition,
peptide aptamers, which inhibit PKM2 and not the homologous PKM1,
induce a significant decrease in cell proliferation and size under
conditions where glucose metabolism is disrupted (48).
Although the inhibition of PKM2 has been
demonstrated to be successful against cancer cell proliferation,
the majority of cells are characterized by a high glutaminolysis
capacity, which is another source of energy (47). Therefore, an inhibition of PKM2 may
not be sufficient to significantly reduce the cellular
proliferation of tumor cells.
Activators of PKM2
PKM2 activators aim to induce tetramerization of
PKM2, which results in a decrease in glycolytic intermediates that
are used as biosynthetic precursors. A class of quinolone
sulfonamide activators has been reported to possess a distinctive
mode of binding to PKM2 (49). These
activators bind to a site that is distinct from the FBP binding
site, which results in the diversion of glycolytic intermediates
away from the serine biosynthetic pathway, which produces serine
that is required for continued cell proliferation. A library of
13,000,000 drug-like compounds was screened in silico
against the PKM2 structure, and 9 novel scaffolds were identified
as PKM2 activators (50). These
molecules had low nanomolar PKM2 potency, with excellent
selectivity to other PK isoforms and good absorption, distribution,
metabolism and excretion properties. A series of pyridopyrimidine
analogs were also reported as potent activators of PKM2, with good
stability, permeability, solubility and selectivity to PKM2
(51). However, PKM2 activation alone
is not sufficient to alter cancer cell metabolism. Another series
of small molecule PKM2 activators did not affect the growth of
cancer cell lines under normal conditions in vitro, but
strongly inhibited the proliferation of multiple lung cancer cell
lines when serine was removed from the cell culture media (52). Screening of an in-house fragment-like
library of 2,000 other compounds, led to the identification of
another series of small molecules, which caused 50% of the cell
population to undergo apoptosis at a concentration of 36 µM, with
104% maximal response compared with the natural PKM2 ligand and FBP
activator (53). All the reported
small molecule activators of PKM2 bind to a site that is distinct
from the binding site of FBP, which is located on the opposite side
of the tetramer interface compared with the FBP site.
By contrast, oleanolic acid (OA) did not interact
with PKM2, which affects the splicing of the PKM gene (54). Decreased expression of c-Myc-dependent
hnRNA1, a subtype of hnRNPs, and mTOR inhibition were responsible
for the OA-induced switch between PKM isoforms.
Overall, there are a number of PKM2 inhibitors and
activators that are in preclinical development; however, to date,
none have advanced to human clinical studies. The present study
suggests that PKM2 activators may be a more promising therapeutic
target for patients with cancer compared with PKM2 inhibitors.
However, the serine auxotrophy induced by PKM2 activators may be
reversed by multiple pathways, including de novo serine
synthesis, conversion from glycine, catabolism from proteins and
serine-containing phospholipids and absorption from the
extracellular environment, which may present a challenge when
investigating activators.
Conclusion
The present review focused on the PKM2 gene and its
regulation, and emphasized the non-metabolic function of PKM2 and
the development of PKM2 as a therapeutic target. In the last
decade, extensive research has clarified the notable involvement of
PKM2 in cancer through its canonical and non-canonical functions.
This allosteric property of PKM2 balances cell growth and oxidative
stress.
However, PKM2 has multiple roles, and the
intracellular mechanisms induced by PKM2 are more complicated than
previously hypothesized. Protein kinases are important regulators
in numerous biological processes. Numerous human proteins have been
identified as potential substrates for PKM2, the majority of which
are involved in the regulation of cell proliferation. Additional
studies concerning the mechanism of PKM2 substrate recognition may
reveal a common mechanism between the proteins involved in
cell-cycle regulation. It has been demonstrated that the cell
microenvironment provides metabolic heterogeneity in cancer
tissues, which may be caused by differences in the supply of
nutrients and oxygen (55). Cancer
cells are located at various distances from blood vessels, in which
PKM2 may play a role. Certain studies suggest that overexpression
of PKM2 may be a biomarker for specific types of cancer (56–58);
however, this is controversial and additional studies are required
for confirmation. The regulation of PKM2 requires additional
clarification. Even by providing a functional readout of the
oxidative and metabolic state of the cell, it is not fully clear
how PKM2 alters the response of the cell to growth factor
stimulation (59); however, the
association between PKM2 and growth factor responses and oxidative
stress responses indicates that this regulation is integrated. It
is also contradictory that certain studies report that knockdown of
PKM2 results in a modest inhibitory effect on in vitro
proliferation of cancer cells and xenograft tumors without signs of
inhibition, which indicates that PKM2 is not a requirement for
tumor growth (45,46).
In conclusion, PKM2 is essential in the metabolic
reprogramming of cancer cells; however, cancer cells reprogram
using several metabolic pathways to support cell growth. An
improved understanding of metabolism transformation in early cancer
cells and how this results in the development and progression of
cancer may provide an understanding of the metabolic and
non-metabolic functions of PKM2. Furthermore, targeting PKM2 as a
treatment for cancer may significantly evolve over the next several
years.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Jiangsu Province (grant no. BK2012482),
National Natural Science Foundation of China (grant no. 81472702)
and Jiangsu Province Special Program of Medical Science (grant no.
BL2012030).
Glossary
Abbreviations
Abbreviations:
|
PKM2
|
pyruvate kinase muscle isozyme M2
|
|
ATP
|
adenosine triphosphate
|
|
ROS
|
reactive oxygen species
|
|
EGFR
|
epidermal growth factor receptor
|
|
PEP
|
phosphoenolpyruvate
|
|
mTOR
|
mammalian target of rapamycin
|
|
HIF-1α
|
hypoxia inducible factor-1α
|
|
NF-κB
|
nuclear factor κ enhancer binding
protein
|
|
PKC
|
protein kinase C
|
|
IKK
|
IκΒ kinase
|
|
FBP
|
fructose-1,6-bisphosphate
|
References
|
1
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gambhir SS: Molecular imaging of cancer
with positron emission tomography. Nat Rev Cancer. 2:683–693. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mathupala SP, Rempel A and Pedersen PL:
Glucose catabolism in cancer cells: identification and
characterization of a marked activation response of the type II
hexokinase gene to hypoxic conditions. J Biol Chem.
276:43407–43412. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van der Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Altenberg B and Greulich KO: Genes of
glycolysis are ubiquitously overexpressed in 24 cancer classes.
Genomics. 84:1014–1020. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noguchi T, Inoue H and Tanaka T: The M1-
and M2-type isozymes of rat pyruvate kinase are produced from the
same gene by alternative RNA splicing. J Biol Chem.
261:13807–13812. 1986.PubMed/NCBI
|
|
10
|
Noguchi T, Yamada K, Inoue H, Matsuda T
and Tanaka T: The L- and R-type isozymes of rat pyruvate kinase are
produced from a single gene by use of different promoters. J Biol
Chem. 262:14366–14371. 1987.PubMed/NCBI
|
|
11
|
Christofk HR, Van der Heiden MG, Harris
MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang W and Lu Z: Nuclear PKM2 regulates
the Warburg effect. Cell Cycle. 12:3154–3158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
David CJ, Chen M, Assanah M, Canoll P and
Manley JL: HnRNP proteins controlled by c-Myc deregulate pyruvate
kinase mRNA splicing in cancer. Nature. 463:364–368. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z, Chatterjee D, Jeon HY, Akerman M,
Van der Heiden MG, Cantley LC and Krainer AR: Exon-centric
regulation of pyruvate kinase M alternative splicing via mutually
exclusive exons. J Mol Cell Biol. 4:79–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Semenza GL: HIF-1: Upstream and downstream
of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Q, Chen X, Ma J, Peng H, Wang F, Zha
X, Wang Y, Jing Y, Yang H, Chen R, et al: Mammalian target of
rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is
critical for aerobic glycolysis and tumor growth. Proc Natl Acad
Sci USA. 108:4129–4134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iqbal MA, Siddiqui FA, Gupta V,
Chattopadhyay S, Gopinath P, Kumar B, Manvati S, Chaman N and
Bamezai RN: Insulin enhances metabolic capacities of cancer cells
by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol
Cancer. 12:722013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang
L, You MJ, Koh MY, Cote G, Aldape K, et al: EGFR-induced and PKCε
monoubiquitylation-dependent NF-κB activation upregulates PKM2
expression and promotes tumorigenesis. Mol Cell. 48:771–784. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Breitkreutz D, Braiman-Wiksman L, Daum N,
Denning MF and Tennenbaum T: Protein kinase C family: On the
crossroads of cell signaling in skin and tumor epithelium. J Canc
Res Clin Oncol. 133:793–808. 2007. View Article : Google Scholar
|
|
22
|
Choi JH, Ryu SH and Suh PG:
On/off-regulation of phospholipase C-gamma 1-mediated signal
transduction. Adv Enzyme Regul. 47:104–116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anastasiou D, Yu Y, Israelsen WJ, Jiang
JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al:
Pyruvate kinase M2 activators promote tetramer formation and
suppress tumorigenesis. Nat Chem Biol. 8:839–847. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chaneton B, Hillmann P, Zheng L, Martin
AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding
FP, Vousden KH, et al: Serine is a natural ligand and allosteric
activator of pyruvate kinase M2. Nature. 491:458–462. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Keller KE, Tan IS and Lee YS: SAICAR
stimulates pyruvate kinase isoform M2 and promotes cancer cell
survival in glucose-limited conditions. Science. 338:1069–1072.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Keller KE, Doctor ZM, Dwyer ZW and Lee YS:
SAICAR induces protein kinase activity of PKM2 that is necessary
for sustained proliferative signaling of cancer cells. Mol Cell.
53:700–709. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gui DY, Lewis CA and Van der Heiden MG:
Allosteric regulation of PKM2 allows cellular adaptation to
different physiological states. Sci Signal. 6:pe72013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anastasiou D, Poulogiannis G, Asara JM,
Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW,
Auld DS, et al: Inhibition of pyruvate kinase M2 by reactive oxygen
species contributes to cellular antioxidant responses. Science.
334:1278–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Iqbal MA, Siddiqui FA, Chaman N, Gupta V,
Kumar B, Gopinath P and Bamezai RN: Missense mutations in pyruvate
kinase M2 promote cancer metabolism, oxidative endurance, anchorage
independence, and tumor growth in a dominant negative manner. J
Biol Chem. 289:8098–8105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo W and Semenza GL: Pyruvate kinase M2
regulates glucose metabolism by functioning as a coactivator for
hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2:551–556.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin
YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ and Wang WC: JMJD5
regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated
glucose metabolism. Proc Natl Acad Sci USA. 111:279–284. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu H, Li Z, Yang P, Zhang L and Fan Y:
PKM2 depletion induces the compensation of glutaminolysis through
β-catenin/c-Myc pathway in tumor cells. Cell Signal. 26:2397–2405.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Li X, Wu S, Xue M and Chen W: Long
non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase
2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci.
105:951–955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki
N, Jiang Y, Zhou X, Li TT, Guan KL, et al: Mitogenic and oncogenic
stimulation of K433 acetylation promotes PKM2 protein kinase
activity and nuclear localization. Mol Cell. 52:340–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo
F, Lyssiotis CA, Aldape K, Cantley LC and Lu Z: ERK1/2-dependent
phosphorylation and nuclear translocation of PKM2 promotes the
Warburg effect. Nat Cell Biol. 14:1295–1304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao X, Wang H, Yang JJ, Liu X and Liu ZR:
Pyruvate kinase M2 regulates gene transcription by acting as a
protein kinase. Mol Cell. 45:598–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang P and Li Z, Fu R, Wu H and Li Z:
Pyruvate kinase M2 facilitates colon cancer cell migration via the
modulation of STAT3 signalling. Cell Signal. 26:1853–1862. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Yung Alfred WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Morfouace M, Lalier L, Oliver L, Cheray M,
Pecqueur C, Cartron PF and Vallette FM: Control of glioma cell
death and differentiation by PKM2-Oct4 interaction. Cell Death Dis.
5:e10362014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bonnet S, Archer SL, Allalunis-Turner J,
Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta
L, Bonnet S, et al: A mitochondria-K+ channel axis is
suppressed in cancer and its normalization promotes apoptosis and
inhibits cancer growth. Cancer Cell. 11:37–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weinberger R, Appel B, Stein A, Metz Y,
Neheman A and Barak M: The pyruvate kinase isoenzyme M2 (Tu M2-PK)
as a tumour marker for renal cell carcinoma. Eur J Cancer Care
(Engl). 16:333–337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ahmed AS, Dew T, Lawton FG, Papadopoulos
AJ, Devaja O, Raju KS and Sherwood RA: M2-PK as a novel marker in
ovarian cancer. A prospective cohort study. Eur J Gynaecol Oncol.
28:83–88. 2007.PubMed/NCBI
|
|
44
|
Li L, Zhang Y, Qiao J, Yang JJ and Liu ZR:
Pyruvate kinase M2 in blood circulation facilitates tumor growth by
promoting angiogenesis. J Biol Chem. 289:25812–25821. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Van der Heiden MG, Christofk HR, Schuman
E, Subtelny AO, Sharfi H, Harlow EE, Xian J and Cantley LC:
Identification of small molecule inhibitors of pyruvate kinase M2.
Biochem Pharmacol. 79:1118–1124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen J, Xie J, Jiang Z, Wang B, Wang Y and
Hu X: Shikonin and its analogs inhibit cancer cell glycolysis by
targeting tumor pyruvate kinase-M2. Oncogene. 30:4297–4306. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Goldberg MS and Sharp PA: Pyruvate kinase
M2-specific siRNA induces apoptosis and tumor regression. J Exp
Med. 209:217–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Spoden GA, Rostek U, Lechner S,
Mitterberger M, Mazurek S and Zwerschke W: Pyruvate kinase
isoenzyme M2 is a glycolytic sensor differentially regulating cell
proliferation, cell size and apoptotic cell death dependent on
glucose supply. Exp Cell Res. 315:2765–2774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kung C, Hixon J, Choe S, Marks K, Gross S,
Murphy E, DeLaBarre B, Cianchetta G, Sethumadhavan S, Wang X, et
al: Small molecule activation of PKM2 in cancer cells induces
serine auxotrophy. Chem Biol. 19:1187–1198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yacovan A, Ozeri R, Kehat T, Mirilashvili
S, Sherman D, Aizikovich A, Shitrit A, Ben-Zeev E, Schutz N,
Bohana-Kashtan O, et al: 1-(sulfonyl)-5-(arylsulfonyl)indoline as
activators of the tumor cell specific M2 isoform of pyruvate
kinase. Bioorg Med Chem Lett. 22:6460–6468. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guo C, Linton A, Jalaie M, Kephart S,
Ornelas M, Pairish M, Greasley S, Richardson P, Maegley K, Hickey
M, et al: Discovery of
2-((1H-benzo[d]imidazol-1-yl)methyl)-4H-pyrido[1,2-a]pyrimidin-4-ones
as novel PKM2 activators. Bioorg Med Chem Lett. 23:3358–3363. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Parnell KM, Foulks JM, Nix RN, Clifford A,
Bullough J, Luo B, Senina A, Vollmer D, Liu J, McCarthy V, et al:
Pharmacologic activation of PKM2 slows lung tumor xenograft growth.
Mol Cancer Ther. 12:1453–1460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu Y, Liu XH, Saunders M, Pearce S, Foulks
JM, Parnell KM, Clifford A, Nix RN, Bullough J, Hendrickson TF, et
al: Discovery of 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide
activators of the M2 isoform of pyruvate kinase (PKM2). Bioorg Med
Chem Lett. 24:515–519. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu J, Wu N, Ma L, Liu M, Liu G, Zhang Y
and Lin X: Oleanolic acid suppresses aerobic glycolysis in cancer
cells by switching pyruvate kinase type M isoforms. PLoS One.
9:e916062014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vaupel P: Metabolic microenvironment of
tumor cells: A key factor in malignant progression. Exp Oncol.
32:125–127. 2010.PubMed/NCBI
|
|
56
|
Wong N, Yan J, Ojo D, De Melo J, Cutz JC
and Tang D: Changes in PKM2 associate with prostate cancer
progression. Cancer Invest. 32:330–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhan C, Shi Y, Lu C and Wang Q: Pyruvate
kinase M2 is highly correlated with the differentiation and the
prognosis of esophageal squamous cell cancer. Dis Esophagus.
26:746–753. 2013.PubMed/NCBI
|
|
58
|
Zhou CF, Li XB, Sun H, Zhang B, Han YS,
Jiang Y, Zhuang QL, Fang J and Wu GH: Pyruvate kinase type M2 is
upregulated in colorectal cancer and promotes proliferation and
migration of colon cancer cells. IUBMB Life. 64:775–782. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Warner SL, Carpenter KJ and Bearss DJ:
Activators of PKM2 in cancer metabolism. Future Med Chem.
6:1167–1178. 2014. View Article : Google Scholar : PubMed/NCBI
|