Introduction
Donor cell leukemia (DCL) is a rare complication of
hematopoietic stem cell transplantation (HSCT), in which normal
donor cells become transformed into aggressive leukemia or
myelodysplastic syndrome in the host environment (1). Common symptoms of DCL include anemia,
neutropenia and thrombocytopenia (2).
The latency between HSCT and DCL ranges between 1 and 193 months
(median, 24 months) (2). Reinduction
chemotherapy or/and a second HSCT are the main treatments for DCL
(3). The majority of case reports
that are concerned with DCL report poor prognosis for patients even
following a second HSCT (2). Wiseman
reported that 53% of patients succumb to the disease, with a median
survival time of 5.5 months after DCL diagnosis (3). Furthermore, the mean overall survival
time for treated patients is 32.8 months (3). To date, >60 cases of DCL have been
reported in the English literature (3), but only two cases of DCL following HSCT
for the treatment of SAA have been described previously (4,5). In the
current study, the case of a 25 year-old male patient who developed
acute myeloid leukemia (AML) with an (8;21)(q22;q22) translocation
and an extra copy of the chromosome 8 in donor cells 2.5 years
following peripheral blood stem cell transplantation (PBSCT) for
SAA is presented. In addition, the onset of AML was preceded by the
development of Graves' disease, which occurred 1 year subsequent to
PBSCT. The patient was successfully treated with chemotherapy and
IL-2 maintenance, which may be a unique treatment for DCL.
Case report
In December 2008, a 25 year-old male patient
presented to the West China Hospital (Chengdu, China) with
petechiae and fatigue. A complete blood count revealed pancytopenia
with hemoglobin levels of 55.00 g/l (normal range: 130~175g/L), a
reticulocyte count of 12.10×109 cells/l (normal range:
24~84×109 cells/l), a platelet count of
6.00×109 cells/l (normal range: 100~300×109
cells/l) and a white cell count of 1.90×109 cells/l
(normal range: 3.5~9.5×109 cells/l). The absolute
neutrophil count was 0.38×109 cells/l (normal range:
1.8~6.3×109 cells/l). Physical examination was normal,
with the exception of pallor and petechiae. Serological tests for
viral infections, including hepatitis B and C, human
immunodeficiency virus, toxoplasmosis, rubella, cytomegalovirus,
herpes simplex virus and Epstein-Barr virus were negative. A bone
marrow smear exhibited marked hypoplasia, and mainly contained
lymphocytes and mature plasma cells. Bone marrow biopsy revealed
that the majority of hematopoietic tissue had been replaced by fat
tissue. Furthermore, no megakaryocytes were identified on the bone
marrow smear or biopsy specimens. Flow cytometry analysis of
cluster of differentiation (CD)55 and CD59 expression was normal.
Humoral and cellular immunity tests indicated no autoimmune
disease. Subsequently, idiopathic SAA was diagnosed.
The patient was administered cyclosporine (300 mg
orally, daily; Huadong Medicine Co., Ltd., Hangzhou, China) for 2
months, however no response was achieved. Allogeneic-PBSCT
(allo-PBSCT) was subsequently performed using cells obtained from
the patient's 28 year-old brother. The patient's sibling was
demonstrated to be clinically healthy and shared an identical human
leukocyte antigen (HLA) with the patient. The patient was
conditioned for 3 days with a total dose of 500 mg rabbit
anti-thymocyte globulin, followed by treatment with
cyclophosphamide (3,000 mg intravenously, daily; Chengdu Suncadia
Pharmaceuticals Co., Ltd., Chengdu, China) for 4 days. Prophylactic
treatment with cyclosporine, methotrexate and γ-globulin was
administered to prevent rejection and graft versus host disease
(GVHD). A total of 2.30×106 peripheral stem cells/kg
were infused. Granulocyte-colony stimulating factor (G-CSF; 300 µg,
daily) was administered 2 weeks preceding and 2 weeks following
stem cell infusion. A bone marrow examination on day 28 following
stem cell infusion indicated trilineage hematopoiesis. In addition,
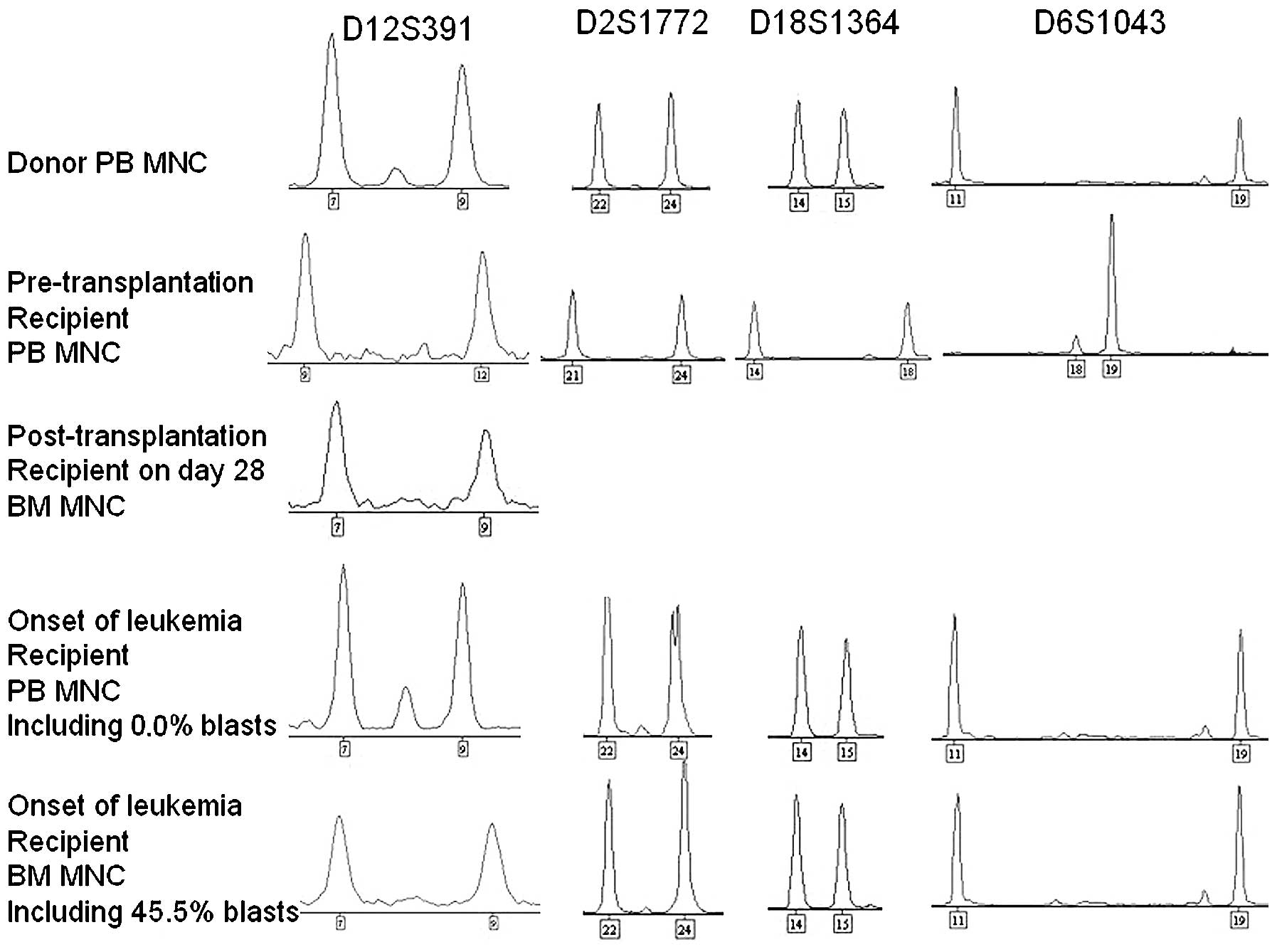
bone marrow engraftment analysis using multiplex polymerase chain
reaction (PCR) of short tandem repeat (STR) markers was performed
using a STR Typer-10G kit (Codon Ltd., Zhuhai, China). The PCR was
performed in an Eppendorf AG 22331 Thermocyler (Eppendorf, Hamburg,
Germany) and GeneMapper® ID version 3.2 (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was used to interpret the
results. PCR was performed under the following conditions: Five
cycles of initial denaturation at 95°C for 2 min and 94°C for 30
sec, annealing at 61°C for 60 sec and extension at 70°C for 60 sec,
followed by 25 cycles of denaturation at 92°C for 30 sec, annealing
at 61°C for 60 sec and extension at 70°C for 60 sec, with a final
extension step at 60°C for 45 min. The results of the PCR revealed
complete engraftment by donor cells (Fig.
1). Cyclosporine treatment was gradually reduced, and was
discontinued 5 months following allo-PBSCT, as the patient
exhibited no symptoms of GVHD. Periodic monitoring revealed that
the patient's complete blood count was normal.
In April 2009, one year following allo-PBSCT, the
patient presented to the West China Hospital with fatigue,
palpitation and increased appetite. Physical examination indicated
a diffuse goiter and tachycardia. Laboratory findings revealed
elevated levels of free thyroxine (T4; 29.22 pmol/l; normal range,
12–22 pmol/l), decreased levels of thyroid stimulating hormone
(TSH; <0.005 mU/l; normal range, 0.27–4.2 mU/l) and high levels
of antithyrotrophin receptor (1,329 IU/ml; normal range, <115
IU/ml) and antithyroid peroxidase (184.3 IU/ml; normal range,
<34 IU/ml) antibodies. Complete blood count was normal. In
consequence, Graves' disease was diagnosed. Oral propylthiouracil
(200 mg, daily for 3 months; Fosun International, Shanghai, China)
and thiamazole (20 mg, daily for 9 months; Merck KGaA, Darmstadt,
Germany) were administered discontinuously to treat the disease for
1 year. Initially, the symptoms were alleviated, and then recurred.
The levels of free T4 and TSH were not well controlled. Radioactive
iodine therapy was suggested, and treatment with antithyroid drugs
was stopped in preparation for iodine therapy. However, the patient
felt well, and refused further treatment with iodine therapy or
antithyroid drugs.
In October 2011, 6 months after the treatment with
antithyroid drugs had been discontinued, and 2.5 years subsequent
following allo-PBSCT, the patient presented the West China Hospital
with petechiae, hyperhidrosis and low-grade fever. Complete blood
count revealed pancytopenia with hemoglobin levels of 106.00 g/l, a
platelet count of 19.00×109 cells/l and a white cell
count of 2.20×109 cells/l without leukemia cells. High
levels of free T4 and low levels of TSH indicated hyperthyroidism.
A bone marrow smear revealed an active proliferation of nucleated
cells with numerous myeloblasts, accounting for 45.5% of the total
blood cell count. Peroxidase stain was positive in 97.0% of
myeloblasts, whereas Periodic acid-Schiff and non-specific esterase
stains were negative. Flow cytometry of leukemic cells revealed
positivity for CD34, HLA-DR, CD117, CD13 and cytoplasmic
myeloperoxidase with partial expression of CD56. These results
indicated a typical cell surface antigen expression pattern for AML
with maturation [previously designated as AML M2, according to the
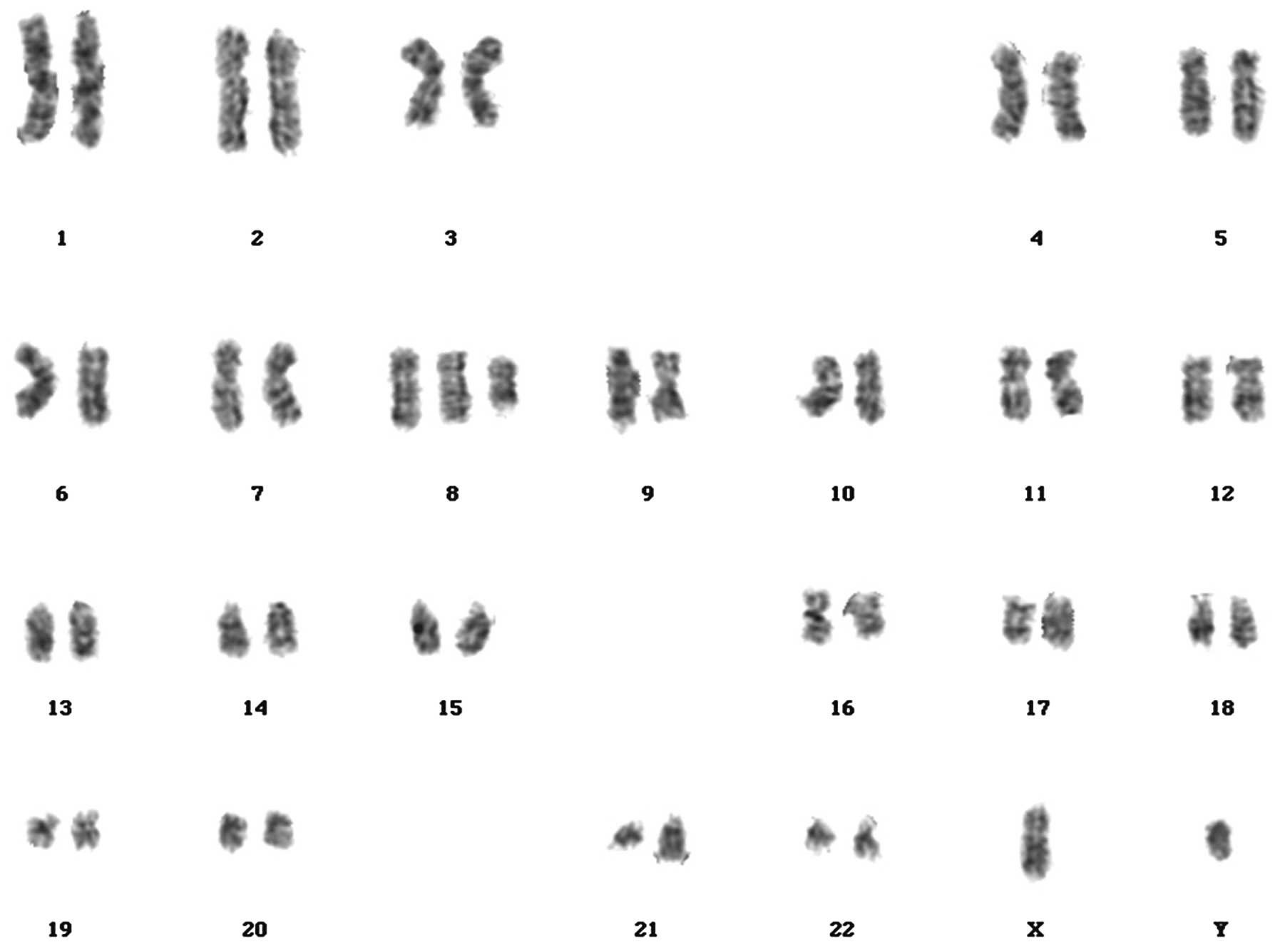
French-American-British (FAB) classification of AML (6)]. Cytogenetic analysis identified an
(8;21)(q22;q22) translocation and an extra copy of the chromosome 8
(Fig. 2). Screening for fusion genes
revealed positivity for AML1/eight twenty one fusion protein and
negativity for core binding factor β/myosin, heavy chain 11, smooth
muscle. No mutations were identified in the feline sarcoma-related
tyrosine kinase 3/internal tandem duplication, c-Kit, nucleophosmin
and CCAAT/enhancer-binding protein alpha genes. Engraftment
analysis of the bone marrow and peripheral blood revealed complete
engraftment by donor cells (Fig. 1).
The donor remained healthy, without evidence of viral infection. A
bone marrow smear, cytogenetic analysis and screening for the
aforementioned fusion genes and mutations in the donor indicated
normality. Subsequently, the patient was diagnosed with AML with an
(8;21)(q22;q22) translocation and an extra copy of the chromosome 8
of donor origin. Complete hematological and molecular remission was
achieved following a single cycle of priming therapy. Two cycles of
consolidation chemotherapy with DA [45 mg/m2 intravenous
daunorubicin (Zheijiang Hisun Chemical Co., Ltd., Taizhou, China),
days 1–3; and 100 mg/m2 intravenous cytarabine
(Sinopharm A-Think, Changchun, China), days 1–7] were administered,
followed by maintenance therapy with recombinant interleukin-2
(IL-2; Shandong Quangang Pharmaceutical Co., Ltd., Jinan, China).
The IL-2 was injected subcutaneously at a dose of 1,000,000 U daily
for 10 days every month, and then tapered every 6 months. In
October 2014 the patient stopped receiving IL-2. A bone marrow
examination is performed on the patient every year at the West
China Hosptial, and DCL has been in molecular remission for 3
years. Notably, hyperthyroidism was relieved following treatment;
however, hypothyroidism subsequently developed, and consequently,
continuous treatment with oral Euthyrox® (50 µg, daily;
Merck KGaA) was administered.
Written informed consent was obtained from the
patient for the use of patient information and accompanying images
in this study.
Discussion
The first case of DCL was reported in 1971 (7), and to date, >60 cases have been
reported (3). The European Group for
Blood and Marrow Transplantation reported that the incidence of DCL
in allo-HSCT recipients is ~0.1% (8).
Recently, Wiseman (3) proposed that,
in a number of cases, DCL may remain undiagnosed for several years,
and may account for ≤5% of all leukemia relapses post-HSCT. The
primary diagnosis in the majority of DCL cases is leukemia
(3). Prior to the present case, only
2 cases of DCL and 2 cases of donor cell-derived myelodysplastic
syndrome (DCM) following HSCT for SAA have been reported in the
English literature to date (4,5,9,10). The 2
cases of DCL were diagnosed as AML (M5a and M0, respectively,
according to the FAB classification of AML) on days 319 and 208,
respectively, following bone marrow transplantation. The 2 cases of
DCM occurred 2 and 13 years following HSCT. Of these cases, ≥3
patients have succumbed to the disease, whereas all donors were
healthy at the time of publication of the present report.
Currently, a number of methods are available to
confirm the origin of donor cells, including conventional
cytogenetics, fluorescent in situ hybridization and molecular
DNA markers, such as variable number tandem repeats, STRs and
restriction fragment length polymorphisms (11). Due to the high sensitivity and
availability of commercial multiplex kits, STRs amplified by PCR
are considered the gold standard technique for analyzing DCL cases
(2,11). Due to the extensive use of molecular
analysis of donor/host chimerism, an increasing number of DCL cases
have been reported; since 2004, more cases of DCL have been
reported than the total number of cases reported in the previous 34
years (3). In the current case, using
this highly sensitive technique, 45.5% blast cells were identified
in the bone marrow of the patient, and the complete blood count of
donor origin was demonstrated to be identical to that from
peripheral blood, which confirmed the diagnosis of DCL following
allo-PBSCT for SAA.
A number of mechanisms have been proposed to explain
the etiology of DCL, including abnormality in donor cells (such as
occult leukemia or preleukemic potential), conditioning or
virus-induced mutagenesis or transformation, impaired immune
surveillance or defective microenvironmental niche in the host
(3,11,12). To
date, multiple factorial processes have been considered to be the
main cause of DCL (3,11,12). In
the present case, the donor was completely healthy prior to PBSCT
and when the recipient developed DCL. Screening tests identified no
evidence of immunological disease, viral infection or genetic
mutations in the donor. These results suggest that the donor cells
exhibited no abnormalities. Certain authors have reported that
long-term use of immunosuppressive agents or G-CSF for the
treatment of SAA are risk factors for the development of
therapy-related AML/myelodysplasia, which is usually a late
complication (13–17). In the current case, the patient
exhibited a rapid hematopoietic recovery following PBSCT, and
exhibited no evidence of GVHD. Furthermore, short-term treatment
with immunosuppressive agents and G-CSF did not appear to affect
donor cells. Previously, it has been hypothesized that malignant
cells are continuously arising in healthy individuals, but the
immune system is able to recognize and eliminate such cells via
complex interactions (3). Therefore,
the development of DCL may be the result of impaired immune
surveillance and an acquired (8;21)(q22;q22) translocation with an
extra copy of the chromosome 8 in donor cells. The latter may
spontaneously arise in a clone or may be induced by the impaired
stem cell niche, which is involved in the regulation of quiescence,
self-renewal, proliferation and differentiation of stem cells
(18,19). The impaired immune surveillance leads
to anergy towards the arising malignant clones, which are favored
by the immunocompromised status following transplantation (3,11).
Consequently, it is possible to hypothesize that the development of
DCL in the present case may be predominantly attributed to impaired
immune surveillance and an abnormal hemopoietic
microenvironment.
The prognosis of DCL is usually poor, with a median
survival time of 5.5 months (range, 1 week-64 months) following DCL
diagnosis. A second HSCT is the main treatment for DCL (3), however, in the present case, only 2
cycles of DA consolidation chemotherapy were administered
subsequent to induction, followed by maintenance therapy with IL-2.
IL-2 is a cytokine signaling molecule within the immune system that
regulates lymphocyte activity. It enhances the anti-tumor effect of
macrophages through the induction of cytokines with anti-neoplastic
activity, including α-tumor necrosis factor and γ-interferon
(20). Recombinant IL-2 binds to IL-2
receptors, and introduces the diphtheria toxin into the cells that
express those receptors, thus killing the malignant cells that
express the IL-2 receptor. This indicates that IL-2 is an effective
drug for DCL maintenance therapy.
In conclusion, the present study highlights DCL as a
rare complication of allo-PBSCT for SAA, and indicates that
impaired immune surveillance is an important mechanism of
leukemogenesis. Furthermore, the current case demonstrates that
treatment with recombinant IL-2 is effective as a maintenance
therapy for DCL.
References
|
1
|
Sala Torra O and Loeb KR: Donor
cell-derived leukemia and myelodysplastic neoplasm: Unique forms of
leukemia. Am J Clin Pathol. 135:501–504. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang E, Hutchison CB, Huang Q, Lu CM, Crow
J, Wang FF, Sebastian S, Rehder C, Lagoo A, Horwitz M, et al: Donor
cell-derived leukemias/myelodysplastic neoplasms in allogeneic
hematopoietic stem cell transplant recipients: A clinicopathologic
study of 10 cases and a comprehensive review of the literature. Am
J Clin Pathol. 135:525–540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wiseman DH: Donor cell leukemia: A review.
Biol Blood Marrow Transplant. 17:771–789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Browne PV, Lawler M, Humphries P and
McCann SR: Donor-cell leukemia after bone marrow transplantation
for severe aplastic anemia. N Engl J Med. 325:710–713. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lawler M, Locasciulli A, Longoni D, Schiro
R and McCann SR: Leukaemic transformation of donor cells in a
patient receiving a second allogeneic bone marrow transplant for
severe aplastic anaemia. Bone Marrow Transplant. 29:453–456. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Catovsky D, Matutes E, Buccheri V, Shetty
V, Hanslip J, Yoshida N and Morilla R: A classification of acute
leukaemia for the 1990s. Ann Hematol. 62:16–21. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fialkow PJ, Thomas ED, Bryant JI and
Neiman PE: Leukaemic transformation of engrafted human marrow cells
in vivo. Lancet. 1:251–255. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hertenstein B, Hambach L, Bacigalupo A,
Schmitz N, McCann S, Slavin S, Gratwohl A, Ferrant A, Elmaagacli A,
Schwertfeger R, et al: Chronic Leukaemia Working Party of the
European Group for Blood and Marrow Transplantation: Development of
leukemia in donor cells after allogeneic stem cell transplantation
- a survey of the European Group for Blood and Marrow
Transplantation (EBMT). Haematologica. 90:969–975. 2005.PubMed/NCBI
|
|
9
|
Hashino S, Fujisawa F, Kondo T, Imamura M,
Sato K, Torimoto Y, Kohgo Y, Kimura K, Furukawa H, Todo S and Asaka
M: Donor-type myelodysplastic syndrome with t(2;3) and monosomy 7
after allogeneic peripheral blood stem cell transplantation and
liver transplantation in a patient with severe-type aplastic
anemia. Int J Hematol. 84:363–366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haltrich I, Müller J, Szabó J, Kovács G,
Kóos R, Poros A, Dobos M and Fekete G: Donor-cell myelodysplastic
syndrome developing 13 years after marrow grafting for aplastic
anemia. Cancer Genet Cytogenet. 142:124–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruiz-Argüelles GJ, Ruiz-Argüelles A and
Garcés-Eisele J: Donor cell leukemia: A critical review. Leuk
Lymphoma. 48:25–38. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Flynn CM and Kaufman DS: Donor cell
leukemia: Insight into cancer stem cells and the stem cell niche.
Blood. 109:2688–2692. 2007.PubMed/NCBI
|
|
13
|
Socié G, Henry-Amar M, Bacigalupo A, Hows
J, Tichelli A, Ljungman P, McCann SR, Frickhofen N, Van't
Veer-Korthof E and Gluckman E: European Bone Marrow
Transplantation-Severe Aplastic Anaemia Working Party: Malignant
tumors occurring after treatment of aplastic anemia. N Engl J Med.
329:1152–1157. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kojima S, Ohara A, Tsuchida M, Kudoh T,
Hanada R, Okimoto Y, Kaneko T, Takano T, Ikuta K and Tsukimoto I:
Japan Childhood Aplastic Anemia Study Group: Risk factors for
evolution of acquired aplastic anemia into myelodysplastic syndrome
and acute myeloid leukemia after immunosuppressive therapy in
children. Blood. 100:786–790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saracco P, Quarello P, Iori AP, Zecca M,
Longoni D, Svahn J, Varotto S, Del Vecchio GC, Dufour C, Ramenghi
U, et al: Bone Marrow Failure Study Group of the AIEOP (Italian
Association of Paediatric Haematology Oncology): Cyclosporin A
response and dependence in children with acquired aplastic anaemia:
A multicentre retrospective study with long-term observation
follow-up. Br J Haematol. 140:197–205. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaito K, Kobayashi M, Katayama T, Masuoka
H, Shimada T, Nishiwaki K, Sekita T, Otsubo H, Ogasawara Y and
Hosoya T: Long-term administration of G-CSF for aplastic anaemia is
closely related to the early evolution of monosomy 7 MDS in adults.
Br J Haematol. 103:297–303. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Witherspoon RP, Fisher LD, Schoch G,
Martin P, Sullivan KM, Sanders J, Deeg HJ, Doney K, Thomas D, Storb
R and Thomas ED: Secondary cancers after bone marrow
transplantation for leukemia or aplastic anemia. N Engl J Med.
321:784–789. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moore KA and Lemischka IR: Stem cells and
their niches. Science. 311:1880–1885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Z and Li L: Understanding hematopoietic
stem-cell microenvironments. Trends Biochem Sci. 31:589–595. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang SR, Salup RR, Urias PE, Twilley TA,
Talmadge JE, Herberman RB and Wiltrout RH: Augmentation of NK
activity and/or macrophage-mediated cytotoxicity in the liver by
biological response modifiers including human recombinant
interleukin 2. Cancer Immunol Immunother. 21:19–25. 1986.
View Article : Google Scholar : PubMed/NCBI
|