Introduction
The biological consequence of gene function loss
caused by DNA methylation is analogous to the consequences of gene
mutation (1). Unlike gene mutation,
DNA hypermethylation can be reversed pharmacologically using DNA
demethylating agents. An association between DNA methylation of
tumor suppressor genes and the development of colorectal cancer has
been previously reported (2–5). The re-activation of tumor suppressor
genes that are silenced by DNA hypermethylation is commonly termed
epigenetic therapy, which is a feasible and achievable strategy for
cancer treatment. Previous in vitro experiments identified
that, 5-aza-2′-deoxycytidine (5-aza-CdR) can reactivate
epigenetically silenced tumor suppressor genes, thereby restoring
their inherent anti-cancer effect.
The Ras association domain family 1A
(RASSF1A) gene is located in the short arm of chromosome 3,
originally found as a novel candidate tumor suppressor in lung
cancer (6,7). The aim of the present study was to
examine the effect of 5-aza-CdR on proliferation, cell cycle and
apoptosis in Caco-2 cells in vitro. In addition, a
semi-quantitative analysis of RASSF1A transcripts was
carried out to determine the reactivation of the tumor suppressive
function and whether 5-aza-CdR can be extended to treat colon
cancer.
Materials and methods
Cell lines and culture
Human Caco-2 colon adenocarcinoma cells, purchased
from Shanghai Jiahe Biotechnology Co., Ltd., Shanghai, China, were
cultured in RPMI-1640 medium supplemented with 100 ml/l calf serum
(Wisent, Nanjing, China), 100 kU/l streptomycin (Wisent), and 100
kU/l penicillin (Wisent) at 37°C with 5% CO2.
Subsequently, 5-aza-CdR (Sigma, St. Louis, MO, USA) was dissolved
in tri-distilled water and stored at 70°C. The desired
concentration of 5-aza-CdR was obtained by serial dilution of the
stock solution.
Monoplast suspension was obtained by digesting the
Caco-2 cells in the logarithmic phase using trypsin (2.5 g/l). This
monoplast suspension was cultured and passaged to obtain the
concentration of 2×106/l. The cell suspension was then
treated with 5-aza-CdR at different concentrations of 0.4, 1.6,
6.4, 25.6 and 102.4 µmol/l. At every 24 h, the medium was aspirated
and replaced with fresh RPMI-1640 medium containing the same
concentration of 5-aza-CdR and this process was repeated for 3
days. The RPMI-1640 medium containing the drug was then replaced by
complete culture medium and incubated for 4-days. The same
procedure as described above was performed with the exception of
5-aza-CdR in cultured cells, which served as the control. During
the incubation process, morphological changes in the cells treated
with 5-aza-CdR were observed using phase contrast microscope
(Aipuda, Shanghai, China).
Growth curve using MTT assay
Caco-2 cells were seeded in a 96-well plate at a
density of 3×103 to a final volume of 200 µl. Cell
culture medium containing a concentration of 5 g/l of 5-aza-CdR was
changed regularly. A negative control (without 5-aza-CdR) and a
blank control (without cells) were included in each plate. MTT (20
µl) was added to each well and incubated for 4 h at 37°C. Following
incubation, MTT was aspirated and the cells were rinsed twice with
PBS. This step was followed by the addition of 150 µl of DMSO and
incubation for 15 min. The optical density (OD) was determined at
570 nm in an ELISA reader (Perlong, Beijing, China). Cell
proliferation was calculated according to the formula: Cell
proliferation = (OD of treated - OD of blank)/(OD of the negative
control-OD of the blank) × 100%.
Cell cycle and apoptosis
The 5-aza-CdR-treated cells were collected and
rinsed twice in PBS. The cells were adjusted to contain a cell
density of 1×109/l in a flask. Subsequently, 5 ml of ice
cold hexanol (700 ml/l) was added to immobilize the cells for 24 h.
RNase A (Solarbio, Beijing, China) then was added (1 g/l).
Propidium iodide (Leagene, Beijing, China) was added at a final
concentration of 50 mg/l and incubated for 30 min at 37°C. The cell
cycle and apoptosis were determined in a flow cytometer (Potenov,
Bejing, China).
Reverse transcription-polymerase chain
reaction (RT-PCR)
TRIzol® reagent (Leagene, Beijing, China)
was used to extract total RNA from the treated and untreated cells.
The extracted total RNA was then reverse transcribed. Briefly, 2 µg
of total RNA was added to the pre-existing mixture of 1 µl 10X
reaction buffer (Leagene) with MgCl2 and 1 µl DNase I.
The non-specific inhibitor diethylpyrocarbonate (DEPC)-treated
water was added to increase the volume to 10 µl, followed by
incubation for 30 min at 37°C. Subsequently, 1 µl Oligo dT18 was
added and mixed gently. A centrifugal separation step at 1,000 × g
was performed after 5 min incubation at 70°C. The tube was kept on
ice. For reverse transcription, 5 µl 5X Moloney Murine Leukemia
Virus (M-MLV) buffer, 1.25 µl deoxyribonucleotide (dNTP) mixture, 1
µl M-MLV, 0.5 µl RNasin and DEPC-treated water were added until a
total volume of 25 µl was achieved. Incubation was performed again
for 15 min at 72°C. RT-PCR was performed in a total volume of 25 µl
and the constituents used were: 2.5 µl 10X PCR buffer, 0.5 µl dNTP
mixture, 0.625 µl MBI TaqDNA polymerase, 1 µl primer 1 (10 µmol/l),
1 µl primer 2 (10 µmol/l), 1.5 µl MgCl2, 1 µl cDNA and
sterile distilled water (final volume of 25 µl).
RASSF1A-specific primers were used to achieve PCR
amplification. GAPDH was selected as a reference owing to its
stable expression (8). RASSF1A
primers were selected from a previously published study (9). The primers used were: forward:
5′-GGCGTCGTGCGCAAAGGCC-3′ and reverse: 5′-GGGTGGCTTCTTGCTGGAGGG-3′.
The primer sequences for GAPDH were: forward
5′-ACCACAGTCCATGCCATCAC-3′ and reverse 5′-TCCACCACCCTGTTGCTGTA-3′.
The PCR amplification step consisted of initial denaturation at
95°C for 5 min, 35 cycles of denaturation at 94°C for 30 sec,
annealing at 56°C for 30 sec, extension at 72°C for 60 sec and a
final extension at 72°C for 5 min. Thus the PCR amplicons were
visualized on 2% agarose gel (Novelab, Shanghai, China).
Statistical analysis
Experimental data were processed using SPSS software
(IBM, Armonk, NY, USA) and F-test and T-tests were performed.
Results
Morphology and cell proliferation
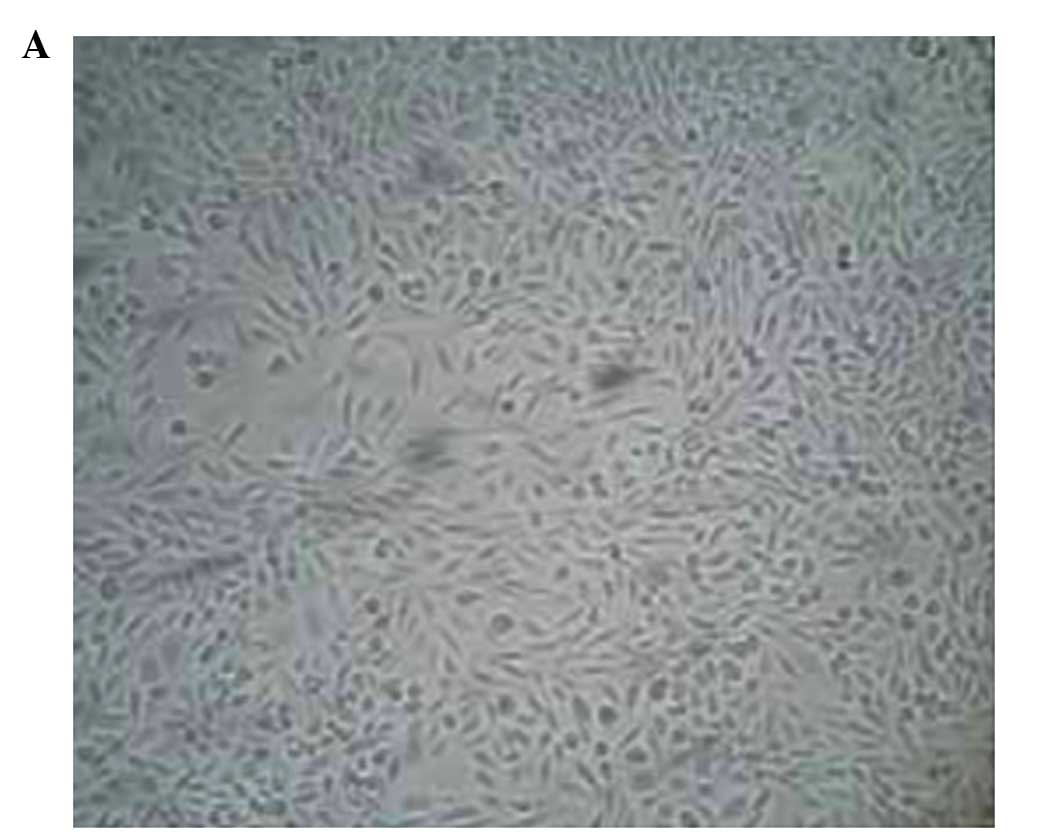
Morphological changes in Caco-2 cells prior to and
following 5-aza-CdR treatment were observed under an inverted
microscope (Dygx, Shanghai, China). The treated cells decreased in
volume and density, and died (Fig. 1A and
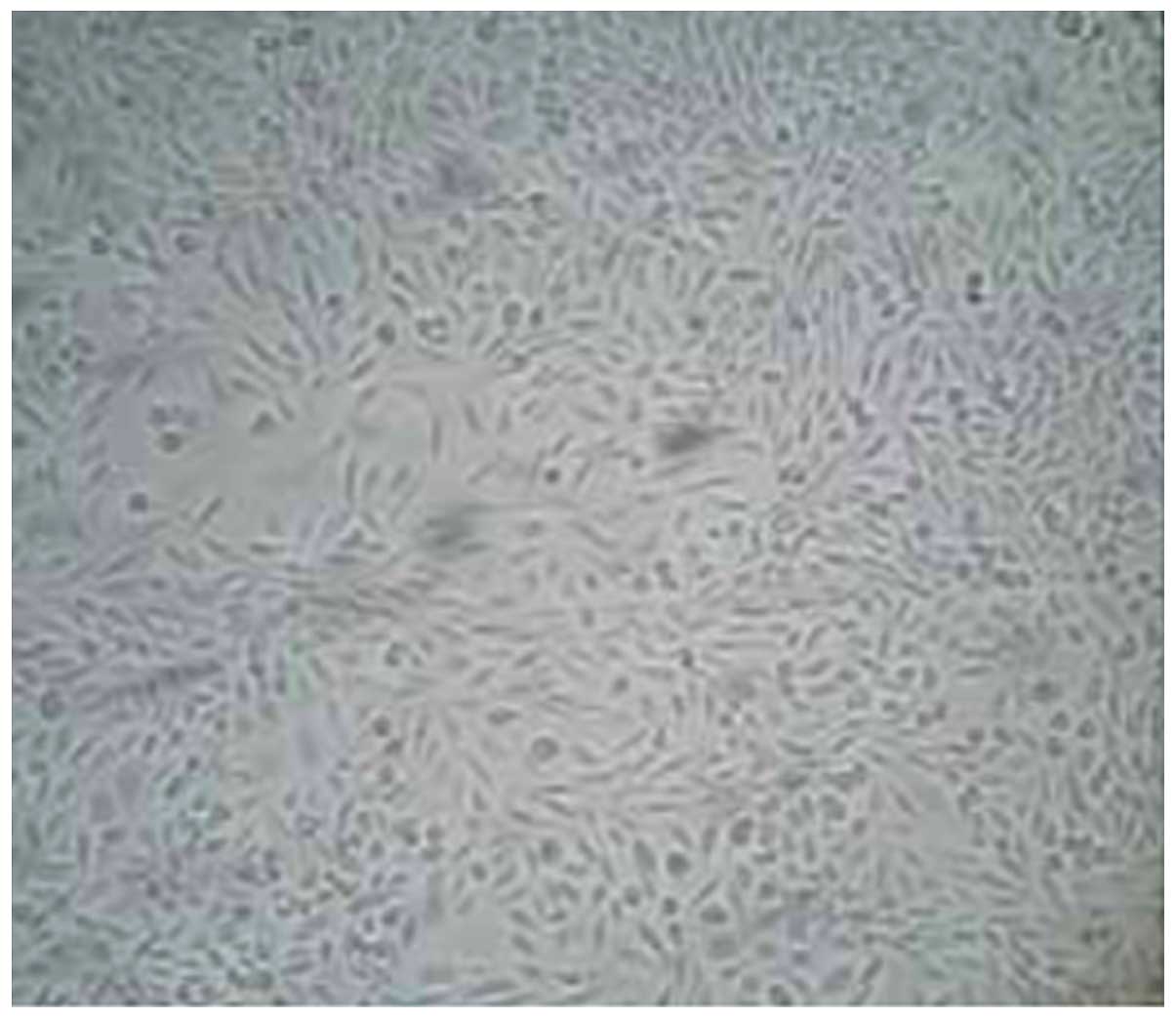
B). No such abnormalities were observed in the untreated cells
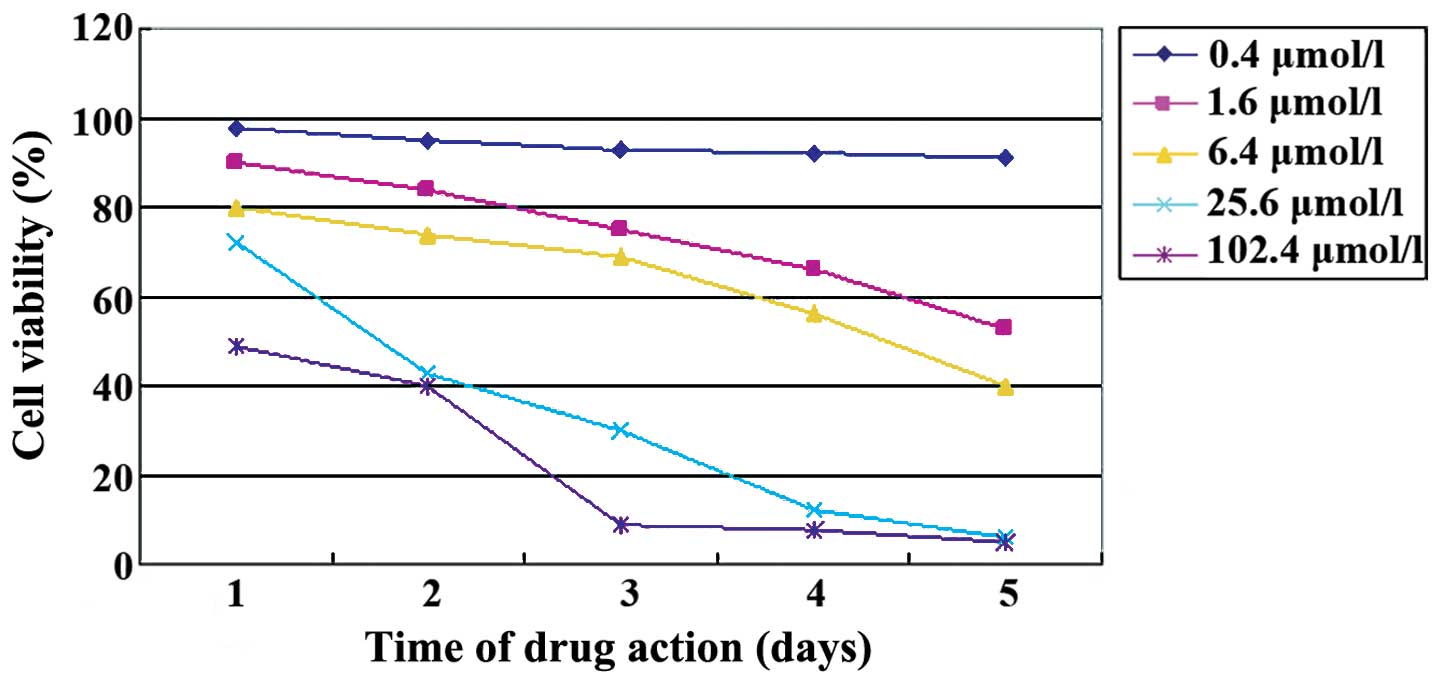
(Fig. 2). The MTT assay showed that
5-aza-CdR inhibited Caco-2 cell proliferation. The number of cells
in which proliferation was inhibited by 5-aza-CdR was elevated with
an increasing concentration of 5-aza-CdR (F=44.079, p<0.01) and
exposure time (F=12.250, p<0.01, Table
I, Fig. 3) was observed.
 | Table I.Inhibition of cell proliferation by
5-aza-CdR in Caco-2 cells. |
Table I.
Inhibition of cell proliferation by
5-aza-CdR in Caco-2 cells.
|
| Duration of exposure
of cells to 5-aza-CdR |
|---|
|
|
|
|---|
| Concentration of
5-aza-CdR, µmol/l | Day 1 (%) | Day 2 (%) | Day 3 (%) | Day 4 (%) | Day 5 (%) |
|---|
| 0.4 | 97 | 94 | 92 | 90 | 89 |
| 1.6 | 90 | 84 | 75 | 66 | 50 |
| 6.4 | 80 | 74 | 68 | 56 | 40 |
| 25.8 | 71 | 43 | 30 | 11 | 6 |
| 102.4 | 48 | 40 | 9 | 7 | 5 |
Cell cycle and apoptosis
5-aza-CdR treatment induced cell cycle arrest and
caused accumulation of cells in the G0/G1 phase. Accumulation of
the G0/G1 phase cells was enhanced with an increasing dose of
5-aza-CdR (Table II). Flow cytometry
showed that the percentage of apoptotic cells in the absence of
5-aza-CdR was 1.78% while the same increased to 49.25% when they
were treated with 6.4 µmol/l of 5-aza-CdR, and this difference was
statistically significant (t=3.98, p<0.05 vs. 0 µmol/l; Table II). When 5-aza-CdR concentration
reached 102.4 µmol/l, cell necrosis instead of cell apoptosis
occurred.
| Table II.Cell cycle and apoptosis in Caco-2
cells treated with 5-aza-CdR. |
Table II.
Cell cycle and apoptosis in Caco-2
cells treated with 5-aza-CdR.
|
| Cell cycle |
|---|
|
|
|
|---|
| Concentration of
5-aza-CdR, µmol/l | Sub-G1 phase | G1 phase | S phase | G2 phase |
|---|
| 0 | 1.78 | 56.21 | 22.01 | 15.27 |
| 0.4 | 18.90 | 57.21 | 15.99 | 8.94 |
| 1.6 | 37.81 | 45.31 | 9.98 | 7.54 |
| 6.4 | 49.25 | 38.46 | 9.35 | 5.09 |
| 25.6 | 38.21 | 44.67 | 6.72 | 3.75 |
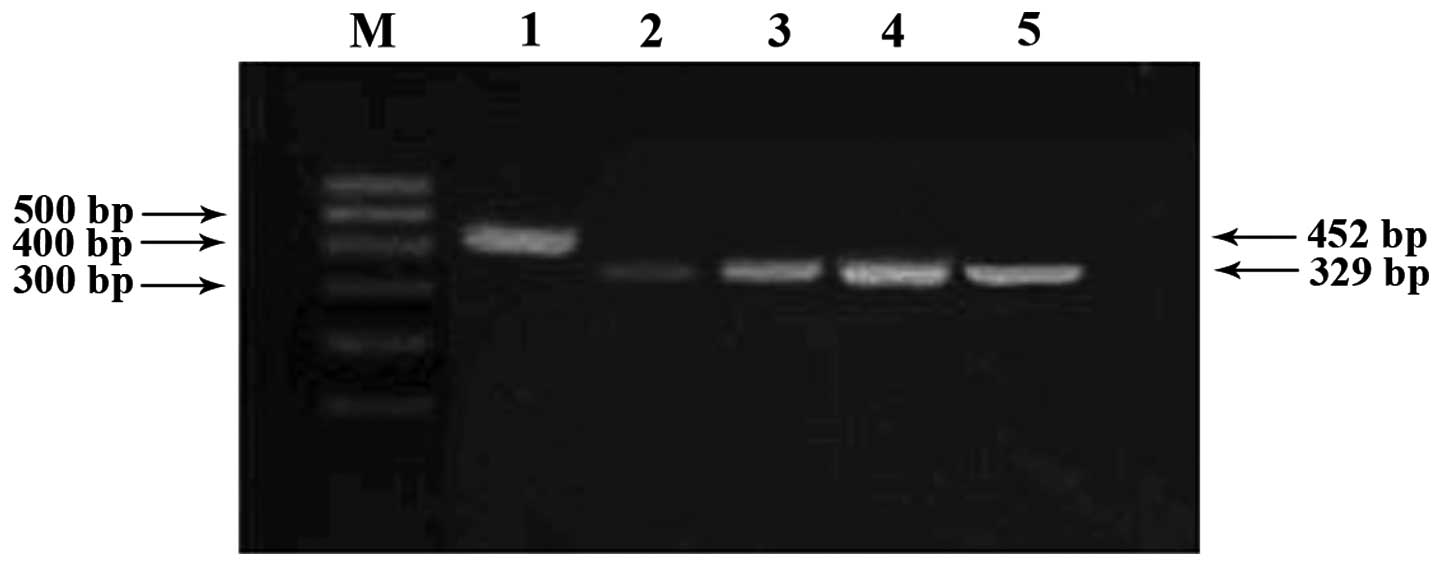
RT-PCR analysis of the RASSF1A
gene
Caco-2 cells originally lacking RASSF1A gene
expression were treated with 5-aza-CdR. Re-expressed RASSF1A
mRNA was dependent on the concentration of 5-aza-CdR as observed in
the 2% agarose gel after RT-PCR analysis (Fig. 4).
Discussion
5-aza-CdR has been identified to be effective in
treating recurrent, intractable, acute and chronic myelogenous
leukaemia (10,11). However, its effectiveness against
solid tumors remains unclear. Hypomethylation and hypermethylation
have been observed in various types of cancer (12,13).
Hypomethylation can contribute to genomic instability, activation
of oncogenes, or loss of imprinting. Gene-specific promoter
hypermethylation in tumor suppressor genes cause silencing of tumor
suppressors, which can contribute to many of the hallmarks of
cancer such as evading apoptosis, insensitivity to antigrowth
signals, sustained angiogenesis, limitless replicative potential
and tissue invasion and metastasis. Previous findings have shown
that aberrant gene methylation in cancer renders them resistant to
chemotherapeutics via inhibition of apoptosis. Since methylation
involves changes in gene regulation but not the DNA sequence, the
change is reversible. The silenced tumor suppressor genes can be
re-expressed when hypermethylation in their promoter region is
removed that may ultimately repress tumor growth (14–19).
In the current study, a concentration- and
time-dependent inhibition of proliferation of Caco-2 cells was
identified following treatment with different concentrations of
5-aza-CdR. The percentage of Caco-2 cells in the G0/G1 phase was
enhanced with an increasing the dose of 5-aza-CdR, thereby
increasing the cells that undergo apoptosis. The morphological
changes including decreased cell volume and density and cell death
were observed at low doses of 5-aza-CdR, whereas cell disruption
and necrosis were observed at higher doses of 5-aza-CdR. The
changes were similar to the cytotoxic effects attributed to
chemotherapeutic drugs where apoptosis occurs at lower doses and
necrosis at higher doses. Additionally, the RASSF1A gene,
which is silenced by hypermethylation in Caco-2 cells, was
reactivated by the 5-aza-CdR treatment. The mRNA expression of
RASSF1A gene was identified even after five successive
generations. The reason for this might be the demethylating effect
of 5-aza-CdR. The re-expression therefore contributed to the tumor
suppressive function in Caco-2 cells. It is also possible that the
cytotoxicity of 5-aza-CdR leads to an anticancer effect. However,
5-aza-CdR does not lead to an anticancer effect by exerting
cytotoxity on cancer cells. Studies (20–22) have
been carried out in which Ara-C, an equally cytotoxic drug as
5-aza-CdR, was used to determine whether the anticancer effect
attributed by 5-aza-CdR was due to cytotoxicity in bladder cancer
cells. Ara-C did not possess a demethylating capability. The two
drugs inhibited cell proliferation although the inhibitory effect
of Ara-C was not transmitted, demonstrating that inhibitory effect
of 5-Aza-CdR is not derived from cytotoxicity.
It has been reported that Ras-GTPase is a member of
the superfamily of molecular switches regulating proliferation and
apoptosis (23–27). It performs different functions
dependent on the signal molecule. Ras-GTPase interacts with a
series of different downstream effector molecules to promote cell
growth and differentiation, inducing cell dormancy, terminal
differentiation and apoptosis in order to suppress cell growth.
RASSF1A gene methylation was found to be
present in various types of cancer. Previous studies (28,29) using
methylation-specific PCR examined colorectal cancer and identified
that RASSF1A CpG-island in the neoplastic foci region
methylated more frequently than the periphery of the neoplastic
foci. Kuroki et al (30,31) and
other investigators (32–34) using methylation-specific PCR analyzed
esophageal carcinoma, gastric carcinoma and bladder cancer and
observed that, RASSF1A was hypermethylated and the degree of
methylation correlated closely with the clinical stages of
patients. RASSF1A gene expression was silenced by
hypermethylation of the CPG island in the promoter region of a wide
range of tumors. Müller et al (35) analyzed the aberrant DNA methylation of
RASSF1A in breast cancer and found that patients with
aberrant RASSF1A methylation had a poorer prognosis.
In conclusion, the current findings suggest that
RASSF1A can result in an antitumor effect when Caco-2 cells
are treated with 5-aza-CdR. The demethylating agent 5-aza-CdR
embraces good prospects in antitumor therapy, given the
universality of regional hypermethylation in tumor cells.
References
|
1
|
Fandy TE: Development of DNA
methyltransferase inhibitors for the treatment of neoplastic
diseases. Curr Med Chem. 16:2075–2085. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang Y, Ng HH, Erdjument-Bromage H,
Tempst P, Bird A and Reinberg D: Analysis of the NuRD subunits
reveals a histone deacetylase core complex and a connection with
DNA methylation. Genes Dev. 13:1924–1935. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prokhortchouk A, Hendrich B, Jørgensen H,
Ruzov A, Wilm M, Georgiev G, Bird A and Prokhortchouk E: The p120
catenin partner Kaiso is a DNA methylation-dependent
transcriptional repressor. Genes Dev. 15:1613–1618. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bird AP: The relationship of DNA
methylation to cancer. Cancer Surv. 28:87–101. 1996.PubMed/NCBI
|
|
5
|
Bird A: DNA methylation de novo. Science.
286:2287–2288. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dammann R, Li C, Yoon JH, Chin PL, Bates S
and Pfeifer GP: Epigenetic inactivation of a RAS association domain
family protein from the lung tumour suppressor locus 3p21.3. Nat
Genet. 25:315–319. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dammann R, Takahashi T and Pfeifer GP: The
CpG island of the novel tumor suppressor gene RASSF1A is intensely
methylated in primary small cell lung carcinomas. Oncogene.
20:3563–3567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartling B, Hoffmann J, Holtz J, Schulz R,
Heusch G and Darmer D: Quantification of cardioprotective gene
expression in porcine short-term hibernating myocardium. J Mol Cell
Cardiol. 31:147–158. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burbee DG, Forgacs E, Zöchbauer-Müller S,
Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S,
et al: Epigenetic inactivation of RASSF1A in lung and breast
cancers and malignant phenotype suppression. J Natl Cancer Inst.
93:691–699. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Piekarz RL and Bates SE: Epigenetic
modifiers: basic understanding and clinical development. Clin
Cancer Res. 15:3918–3926. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wijermans P, Lubbert M, Verhoef G, Bosly
A, Ravoet C, Andre M and Ferrant A: Low-dose
5-Aza-2′-deoxycytidine, a DNA hypomethylation agent for the
treatment of high-risk myelodyspastic syndrome: a multicenter phase
II study in elderly patients. J Clin Oncol. 18:956–960.
2000.PubMed/NCBI
|
|
12
|
Xhu XJ and Dai DQ: Epigenetics and
gastrointestinal cancer. Chin J Digest. 14:3215–3256. 2006.
|
|
13
|
Tomita H, Hirata A, Yamada Y, Hata K,
Oyama T, Mori H, Yamashita S, Ushijima T and Hara A: Suppressive
effect of global DNA hypomethylation on gastrc carcinogenesis.
Carcinogenesis. 31:1627–1633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zöchbauer-Müller S, Fong KM, Virmani AK,
Geradts J, Gazdar AF and Minna JD: Aberrant promoter methylation of
multiple genes in non-small cell lung cancers. Cancer Res.
61:249–255. 2001.PubMed/NCBI
|
|
15
|
Daskalakis M, Nguyen TT, Nguyen C,
Guldberg P, Köhler G, Wijermans P, Jones PA and Lübbert M:
Demethylation of a hypermethylated P15/INK4B gene in patients with
myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine)
treatment. Blood. 100:2957–2964. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bae SI, Lee HS, Kim SH and Kim WH:
Inactivation of O6-methylguanine-DNA methyltransferase by promoter
CpG island hypermethylation in gastric cancers. Br J Cancer.
86:1888–1892. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
18
|
Lübbert M, Tobler A and Daskalakis M:
Cytosine demethylation of the proteinase-3/myeloblastin primary
granule protease gene during phagocyte development. Leukemia.
13:1420–1427. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SH, Bae SI, Lee HS and Kim WH:
Alteration of O6-methylguanine-DNA methyltransferase in
colorectal neoplasms in sporadic and familial adenomatous polyposis
patients. Mol Carcinog. 37:32–38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bender CM, Pao MM and Jones PA: Inhibition
of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth
of human tumor cell lines. Cancer Res. 58:95–101. 1998.PubMed/NCBI
|
|
21
|
Gonzalgo ML, Hayashida T, Bender CM, Pao
MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT and Jones PA: The
role of DNA methylation in expression of the p19/p16 locus in human
bladder cancer cell lines. Cancer Res. 58:1245–1252.
1998.PubMed/NCBI
|
|
22
|
Xiong Z, Wu AH, Bender CM, Tsao JL, Blake
C, Shibata D, Jones PA, Yu MC, Ross RK and Laird PW: Mismatch
repair deficiency and CpG island hypermethylation in sporadic colon
adenocarcinomas. Cancer Epidemiol Biomarkers Prev. 10:799–803.
2001.PubMed/NCBI
|
|
23
|
Dammann R, Schagdarsurengin U, Strunnikova
M, Rastetter M, Seidel C, Liu L, Tommasi S and Pfeifer GP:
Epigenetic inactivation of the Ras-association domain family 1
(RASSF1A) gene and its function in human carcinogenesis. Histol
Histopathol. 18:665–677. 2003.PubMed/NCBI
|
|
24
|
Liu L, Tommasi S, Lee DH, Dammann R and
Pfeifer GP: Control of microtubule stability by the RASSF1A tumor
suppressor. Oncogene. 22:8125–8136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Strunnikova M, Schagdarsurengin U, Kehlen
A, Garbe JC, Stampfer MR and Dammann R: Chromatin inactivation
precedes de novo DNA methylation during the progressive epigenetic
silencing of the RASSF1A promoter. Mol Cell Biol. 25:3923–3933.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chow LS, Lo KW, Kwong J, To KF, Tsang KS,
Lam CW, Dammann R and Huang DP: RASSF1A is a target tumor
suppressor from 3p21.3 in nasopharyngeal carcinoma. Int J Cancer.
109:839–847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tommasi S, Dammann R, Zhang Z, Wang Y, Liu
L, Tsark WM, Wilczynski SP, Li J, You M and Pfeifer GP: Tumor
susceptibility of Rassf1a knockout mice. Cancer Res. 65:92–98.
2005.PubMed/NCBI
|
|
28
|
Lee S, Hwang KS, Lee HJ, Kim JS and Kang
GH: Aberrant CpG island hypermethylation of multiple genes in
colorectal neoplasia. Lab Invest. 87:884–893. 2004. View Article : Google Scholar
|
|
29
|
Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH
and Kim JS: Aberrant CpG island hypermethylation of chronic
gastritis, in relation to aging, gender, intestinal metaplasia, and
chronic inflammation. Am J Pathol. 163:1551–1556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuroki T, Trapasso F, Yendamuri S,
Matsuyama A, Alder H, Mori M and Croce CM: Allele loss and promoter
hypermethylation of VHL, RAR-beta, RASSF1A, and FHIT tumor
suppressor genes on chromosome 3p in esophageal squamous cell
carcinoma. Cancer Res. 63:3724–3728. 2003.PubMed/NCBI
|
|
31
|
Kuroki T, Trapasso F, Yendamuri S,
Matsuyama A, Alder H, Mori M and Croce CM: Promoter
hypermethylation of RASSF1A in esophageal squamous cell carcinoma.
Clin Cancer Res. 9:1441–1445. 2003.PubMed/NCBI
|
|
32
|
Byun DS, Lee MG, Chae KS, Ryu BG and Chi
SG: Frequent epigenetic inactivation of RASSF1A by aberrant
promoter hypermethylation in human gastric adenocarcinoma. Cancer
Res. 61:7034–7038. 2001.PubMed/NCBI
|
|
33
|
Lee MG, Kim HY, Byun DS, Lee SJ, Lee CH,
Kim JI, Chang SG and Chi SG: Frequent epigenetic inactivation of
RASSF1A in human bladder carcinoma. Cancer Res. 61:6688–6692.
2001.PubMed/NCBI
|
|
34
|
Chan MW, Chan LW, Tang NL, Lo KW, Tong JH,
Chan AW, Cheung HY, Wong WS, Chan PS, Lai FM, et al: Frequent
hypermethylation of promoter region of RASSF1A in tumor tissues and
voided urine of urinary bladder cancer patients. Int J Cancer.
104:611–616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Müller HM, Widschwendter A, Fiegl H,
Ivarsson L, Goebel G, Perkmann E, Marth C and Widschwendter M: DNA
methylation in serum of breast cancer patients: an independent
prognostic marker. Cancer Res. 63:7641–7645. 2003.PubMed/NCBI
|