Introduction
Cervical cancer is the second highest cause of
female cancer-associated mortality worldwide, accounting for
288,000 mortalities yearly (1). In
total, ~510,000 cases of cervical cancer are reported each year,
with almost 80% of cases occurring in developing countries.
Persistent infection with high-risk human papillomavirus (HPV) is
regarded as an etiological origin of cervical carcinogenesis
(2,3).
HPV16 is the most common type of high-risk HPV, and accounts for
>50% of all cervical cancers. HPV16 early protein E6 and E7 are
the major oncoproteins that are crucial for host cell
immortalization and transformation. In particular, E6 recruits the
ubiquitin protein ligase E6-associated protein, and the resulting
complex targets the p53 tumor suppressor protein for
proteasome-mediated degradation (4,5). HPV16 E6
also interacts with several other cellular proteins, including
activating transcription factor 3 (6), E6 binding protein (7), human discs large (8), interferon regulatory factor 3 (9), B-cell lymphoma 2-antagonist/killer 1
(10), E6-targeted protein 1
(11) and human telomerase reverse
transcriptase (12). There is also a
switch between mouse double minute 2 homolog (Mdm2) and HPV
E6-mediated degradation of p53 in cervical cancer cells (13). HPV16 E6 regulates cell
differentiation, adhesion, polarity, proliferation, apoptosis, gene
transcription and chromosomal stability through these interactions.
The interactions are not only important for cell carcinogenesis,
but also for the survival of the virus within the host.
Aurora A is a centrosomal serine-threonine kinase
that is responsible for proper mitotic progression. This kinase
plays an essential role in coordinating mitotic events, including
centrosome separation, bipolar spindle assembly, chromosome
segregation and cytokinesis (14).
The expression and kinase activity of Aurora A are regulated by the
cell cycle, peaking when the cells reach M phase. The Aurora A
protein is localized in the centrosomes of interphase cells and the
spindle of mitotic cells (15). The
centrosomes maintain genomic stability through the establishment of
bipolar spindles during cell division, ensuring equal segregation
of replicated chromosomes to the two daughter cells. The abnormal
duplication and distribution of centrosomes in segregation leads to
the aneuploidy observed in numerous cancer cell types. The
expression of Aurora A is upregulated in several human tumors,
including colon, breast, ovarian, gastric and pancreatic cancers,
and hematological malignancies (16).
Aurora A is also involved in the regulation of drug resistance to
several chemotherapeutic agents. The overexpression of Aurora A may
inhibit the cell death induced by Taxol (17), and knock-down of Aurora A increases
the sensitivity of tumor cells to cisplatin toxicity (18). Aurora A abrogates p53 DNA binding and
transactivation activity through the phosphorylation of serine 215
and 315, resulting in the ubiquitylation and proteasomal
degradation of p53 via the Mdm2-mediated pathway (19,20).
Overexpression of Aurora A correlates with an advanced clinical
stage and shortened survival period (21). Although the deregulated expression and
mechanism of the Aurora family involvement in carcinogenesis have
been reported (22), the association
has yet to be elucidated in virus-mediated tumorigenesis. In the
present study, it was demonstrate that HPV16 E6 upregulates the
expression of Aurora A.
Materials and methods
Reagents and antibodies
The E6 DNA fragment of HPV16 was obtained by
polymerase chain reaction (PCR) from SiHa genomic DNA and ligated
to a PCDNA3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) as previously described (23). The p3XFLAG-E6 expression vector was
generated by PCR cloning of the HPV16 PCDNA3-E6 cDNAs, followed by
HindIII and XbaI double digestion and insertion into the HindIII
and XbaI sites of the pA3F vector (Sigma-Aldrich, St. Louis, MO,
USA). Human Aurora A cDNA (a gift from Dr Bingyi Xiao, University
of Pennsylvania, Philadelphia, PA, USA) was subcloned into the
pcDNA3.1HA (Invitrogen; Thermo Fisher Scientific, Inc.) and
pGEX-4T-2 vectors (GE Healthcare Life Science, Chalfont, UK) to
produce hemagglutinin (HA)-tagged and glutathione S-transferase
(GST)-tagged plasmids. The antibodies used in the present study
were the mouse monoclonal anti-FLAG M2 (catalog no. F1804;
Sigma-Aldrich), anti-HA (catalog no. H3663; Sigma-Aldrich),
anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and anti-Aurora A (catalog
no. 610939; BD Biosciences, San Jose, CA, USA) and anti-HPV16 E6
C1P5 (catalog no. sc460; BD Biosciences) antibodies (at 1:1,000
dilution for western blotting).
Cell culture and transfection
The SiHa, CaSki, C33A and HEK293 cell lines,
purchased from teh American Type Culture Collection (Manassas, VA,
USA) were grown in HyClone Dulbecco's modified Eagle's medium
(DMEM; GE Healthcare Life Sciences) supplemented with 10% fetal
bovine serum (FBS), 50 U/ml penicillin, 50 µg/ml streptomycin and 2
mM L-glutamine. The cells were transfected by electroporation using
a Bio-Rad Gene Pulser II electroporator (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Immunohistochemistry
Slides mounted with sections of paraffin-embedded,
archival, cervical cancer tissue specimens were obtained from the
Department of Pathology, the First Affiliated Hospital of China
Medical University (Shenyang, Liaoning, China) between January 2012
and Dedember 2012. Slides were deparaffinized in xylene and
rehydrated using a graded series of alcohol (70, 80, and 100%
alcohol; 5 min each). Endogenous peroxidase activity was blocked
with 3% hydrogen peroxide for 10 min. Following antigen retrieval
in 10 mM sodium citrate buffer (pH 6.0; Haoran Bio, Shanghai,
China), samples were blocked with 10% normal rabbit/goat serum
(Sangerbio, Shanghai, China) prior to incubation with primary
anti-Aurora antibodies overnight at 4°C. The secondary polyclonal
biotinylated anti-rabbit/goat IgG antibody (1:200 dilution; S-P
kit) and streptavidin-peroxidase conjugate (S-P kit; Dako,
Glostrup, Denmark) were added according to the manufacturer's
instructions. The enzymatic reaction was developed in a freshly
prepared solution of 3,3′-diaminobenzidine (DAB) using Dako Liquid
DAB Color Solution (brown color; Dako). The sections were then
counterstained with hemalum, dehydrated, washed with xylene and
mounted.
Immunoprecipitation and western
blotting
Transfected cells were harvested, washed with
ice-cold phosphate-buffered saline (PBS; Solarbio, Beijing, China),
and lysed in 0.5 ml ice-cold radioimmunoprecipitation (RIPA) buffer
(Novland, Shanghai, China) containing 1% Nonidet P-40 (NP-40), 10
mM Tris (pH 7.5), 2 mM EDTA and 150 mM NaCl, supplemented with
protease inhibitors, which consisted of 1 mM phenylmethylsulfonyl
fluoride, 1 µg/ml aprotinin, 1 µg/ml pepstatin and 1 µg/ml
leupeptin. Cell debris was removed by centrifugation at 21,000 × g
(10 min; 4°C), and the supernatant was transferred to a fresh
microcentrifuge tube. Lysates were then precleared by end-over-end
rotation with normal mouse serum (Sigma-Aldrich) and 30 µl of a 1:1
mixture of protein A-protein G-conjugated Sepharose beads (1 h at
4°C; Yanhuibio, Shanghai, China). Beads were spun out, and the
supernatant was transferred to a fresh microcentrifuge tube. In
total, ~5% of the lysate was saved for input control. The protein
of interest was captured by rotating the remaining lysate with 1 µg
of anti-HA antibody (catalog no. H3663; Sigma-Aldrich) overnight at
4°C. Immune complexes were captured with 30 µl of a 1:1 mixture of
protein A and protein G Sepharose beads, pelleted and washed five
times with ice-cold RIPA buffer. For western blot assays, input
lysates and immunoprecipitated complexes were boiled in Laemmli
buffer (Haoran Bio), fractionated by SDS-PAGE and transferred to a
0.45 µm nitrocellulose membrane. The membranes were then probed
with appropriate antibodies, followed by incubation with
appropriate infrared-tagged secondary antibodies and viewed on an
Odyssey imager (Li-Cor Biosciences, Lincoln, NE, USA).
Purification of GST fusion
proteins
Escherichia coli BL21 (DE3) cells (Biovector,
Beijing, China) were transformed with the plasmid constructs to
obtain the GST and GST-E6 fusion protein. Single colonies were
selected and grown overnight in 3 ml of Luria broth (Seebio,
Shanghai, China) supplemented with 100 µg/ml ampicillin (Solarbio).
In total, 1 ml of the overnight culture was used to inoculate a 500
ml culture. The larger culture was incubated until the optical
density at 600 nm was ~0.6, at which point it was produced by
adding 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 12 h at
30°C. The bacteria were pelleted, washed once with sodium
chloride-Tris-ethylenediaminetetraacetic acid (EDTA) (STE) buffer
(Shenxiangbio, Shanghai, China) consisting of 100 mM NaCl, 10 mM
Tris and 1 mM EDTA (pH 7.5), resuspended in 3 ml NETN buffer
(Helixgen, Guangzhou, China) consisting of 0.5% NP-40, 100 mM NaCl,
20 mM Tris and 1 mM EDTA (pH 8.0), supplemented with protease
inhibitors (Haoran Bio), and incubated on ice for 15 min. A volume
of 150 µl of 1 M dithiothreitol (DTT; Luanhuabio, Shanghai, China)
and 1.8 ml of 10% Sarkosyl solution (Yantuobio, Shanghai, China) in
STE buffer was added, and the suspension was sonicated (for 3 min
on ice; Misonix Sonicator 4000; QSonica LLC, Newtown, CT, USA) to
solubilize the proteins. The lysate was centrifuged (12,000 × g; 10
min; 4°C) to separate the insolubilized fraction. The clear
supernatant was transferred to a fresh tube, to which 3 ml of 10%
Triton X-100 in STE buffer and 200 µl of glutathione-Sepharose
beads (Weijiabio, Guangzhou, China) were added. The tube was
rotated overnight at 4°C, following which the purified protein
bound to glutathione was collected by centrifugation (2 min; 600 ×
g; 4°C) and washed five times with NETN buffer supplemented with
protease inhibitors. The level of purification was determined by
SDS-PAGE, and purified proteins were stored at 4°C.
GST pull-down assays
In vitro translation of HA-Aurora A was
performed using the T7-TNT Quick Coupled Transcription-Translation
system (Promega Corporation, Madison, WI, USA) according to the
manufacturer's protocol. For in vitro binding experiments,
GST fusion proteins were incubated with 35S-labeled
in vitro-translated Aurora A protein in binding buffer
(Yubobio, Shanghai, China) consisting of 1X PBS, 0.1% NP-40, 0.5 mM
DTT and 10% glycerol, supplemented with protease inhibitors. The
interating proteins were eluted in %X Laemmli loading buffer,
boiled and separated on 12% SDS-PAGE. An autoradiography
phosphorimager screen (Molecular Dynamics, San Diego, CA, USA) was
used and scanning was performed by the Typhoon 9140 imaging system
(Molecular Dynamics).
Reporter assay
In total, 1.2×107 cells were
co-transfected with the pGL3-basic or pGL3-Aurora A reporter
construct with combinations of various plasmids using a Bio-Rad
Gene Pulser II electroporator. At 24 h post-transfection, the cells
were harvested, washed in PBS and lysed in cell lysis buffer
(BioVision, Milpitas, CA, USA). In total, 50 µl of cell lysate was
used for the reporter assay, which was performed using an LMaxII384
luminometer (Molecular Devices, Sunnyvale, CA, USA). In total, 20%
of the cell lysate was used for western blotting as aforementioned.
The transferred proteins were detected with Odyssey infrared
scanning technology (Li-Cor Biosciences, Inc.), using Alexa Fluor
680 and Alexa Fluor 800 (Molecular Probes; Thermo Fisher
Scientific, Inc. All transfections were performed three times, and
the results shown indicate the means of the data from three
independent experiments.
Lentiviral small hairpin (sh)RNA
vector constructs
For the lentivirus-mediated stable knockdown of
HPV16 E6, the E6 shRNA sequence (sequence,
5′-GGACAGAGCCCATTACAATAT-3′; LC-Bio, Hangzhou, China) was inserted
into the pGIPZ vector according to the manufacture's protocol (Open
Biosystems; GE Dharmacon, Lafayette, CO, USA), resulting in the
HPV16 E6 shRNA-expressing vector (sh-E6). In addition, a 21-mer
oligonucleotide (sequence, 5′-TCTCGCTTGGGCGAGAGTAAG-3′) that had no
significant homology to any known human messenger (m)RNA in the
databases was cloned in the same vector and used as control shRNA
(sh-C).
Virus production and transduction of
CaSki cells
Lentivirus was produced by transient transfection of
the aforementioned plasmids into HEK293T cells. A total of
2×106 HEK293T cells were seeded in 10-cm dishes
containing DMEM supplemented with 10% FBS and 1%
antibiotic-antimycotic and cultured in a 5% CO2
incubator for 24 h prior to transfection. A total of 20 µg plasmid
DNA was added to each dish for transfection, including 1.5 µg
envelope plasmid pCMV-VSV-G (catalog no. 8454; Addgene, Inc.,
Cambridge, MA, USA), 3 µg packaging plasmid pRSV-REV (catalog no.
12251 Addgene, Inc.), 5 µg packaging plasmid pMDLg/Prre (catalog
no. 12251; Addgene, Inc.) and 10.5 µg lentiviral vector plasmid.
The precipitation was formed by adding the plasmids to a final
volume of 438 µl H2O and 62 µl 2 M CaCl2, and
mixing well. A total of 500 µl of 2X
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered
saline (Yeasenbio, Shanghai, China) was then added and the solution
was incubated at room temperature for 30 min. Chloroquine (Haoran
Bio) was added to the 10-ml plated media 5 min prior to
transfection at a final concentration of 25 µM. Subsequent to 12 h
of chloroquine treatment, the medium was replaced with DMEM
supplemented with 10% FBS, 10 mM HEPES and 10 mM sodium butyrate
(Sangonbio, Shanghai, China). The medium was replaced again 10 h
later by DMEM supplemented with 10% FBS and 10 mM HEPES. The
conditioned medium was collected four times at 12 h intervals,
filtered through 0.45 µm pore-size cellulose acetate filters, and
stored on ice. The virus was concentrated by centrifugation at
70,000 × g for 2.5 h. The concentrated virus was resuspended in
RPMI-1640 (Qcbio, Shanghai, China) then used to infect
106 cells in the presence of 20 µm/ml Polybrene (Haoran
Bio). Following 72 h, puromycin was added to a final concentration
of 2 µg/ml for the selection of transfected cells. GFP
immunofluorescence was assessed using an Olympus IX71 microscope
(Olympus, Tokyo, Japan) filtered with 560 nm excitation and 645 nm
emission filters. The cells were grown to 80% confluency in the
presence of 2 µg/ml puromycin prior to western blot analysis as
aforementioned.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from cells was extracted using TRIzol
reagent and cDNA was generated using a Superscript II Reverse
Transcription kit (Invitrogen; Thermo Fisher Scientific, Inc.). The
primers for RT-qPCR were as follows: Aurora A sense,
5′-GGAGAGCTTAAAATTGCAGATTTTG-3′ and antisense,
5′-GGCAAACACATACCAAGAGACCT-3′; HPV E6 sense,
5′-GACCCAGAAAGTTACCACAG-3′ and antisense,
5′-CACAACGGTTTGTTGTATTG-3′; and GAPDH sense,
5′-CTCCTCTGACTTCAACAGCG-3′ and antisense,
5′-GCCAAATTCGTTGTCATACCAG-3′. cDNA was amplified using 10 µl Master
Mix from the DyNAmo SYBR green RT-qPCR kit (MJ Research; Bio-Rad
Laboratories, Inc.), 1 mM of each primer, and 2 µl of the cDNA
product in a 20 µl total volume. Thirty cycles of 1 min at 94°C, 30
sec at 55°C, and 40 sec at 72°C were followed by 10 min at 72°C in
an MJ Research Opticon II thermocycler (Bio-Rad Laboratories,
Inc.). A melting curve analysis was performed to verify the
specificity of the amplified products. The values for the relative
levels of change were calculated using the 2−ΔΔCq method
(24), and each sample was tested in
triplicates.
Results
HPV16 E6 combines with Aurora A
It has been shown that Aurora A accumulates and
functions as an inhibitor of p53 in the majority of cancer cells.
In order to determine the association between HPV16 E6 and Aurora
A, these two molecules were first confirmed to form a complex by
co-immunoprecipitation assays. HEK293 cells were co-transfected
with expression constructs for Flag-tagged HPV16 E6 and HA-tagged
Aurora A, with empty vectors acting as negative controls.
Whole-cell extracts of the transfected HEK293 cells were
precipitated with anti-HA antibody, and the precipitates were
analyzed by western blot analysis with anti-Flag antibody. The
transfected Flag-HPV16 E6 and HA-Aurora A were found to associate
with each other, but not empty vectors, indicating a specific
interaction between ectopically expressed HPV16 E6 and Aurora A
(Fig. 1A). Whether HPV16 E6 combines
with Aurora A in cervical carcinoma cell lines was then
investigated. Endogenously expressed HPV16 E6 was
immunoprecipitated from HPV16 positive cervical carcinoma CaSki and
SiHa cells. The co-immunoprecipitation of Aurora A was monitored by
the polyclonal antibody reactive to Aurora A. The HPV-negative
cervical carcinoma C33A cell line was used as a negative control.
The results revealed that HPV16 E6 formed a stable complex with
Aurora A (Fig. 1B). An in
vitro binding assay was also performed to determine whether
HPV16 E6 directly interacts with Aurora A. The GST-E6 and GST
expression constructs were bacterially expressed and incubated with
in vitro translated 35S-labeled Aurora A, and the
GST pull-down assay result showed that GST-E6 beads, but not GST
alone, precipitated a significant amount of Aurora A protein with
radioactivity. The result showed there is a direct association
between HPV16 E6 and Aurora A (Fig.
1C). This interaction between HPV16 E6 and Aurora A was also
shown by an immunostaining assay. Endogenous HPV16 E6 accumulated
in the nucleus of CaSki cells co-localized with Aurora A (Fig. 1D).
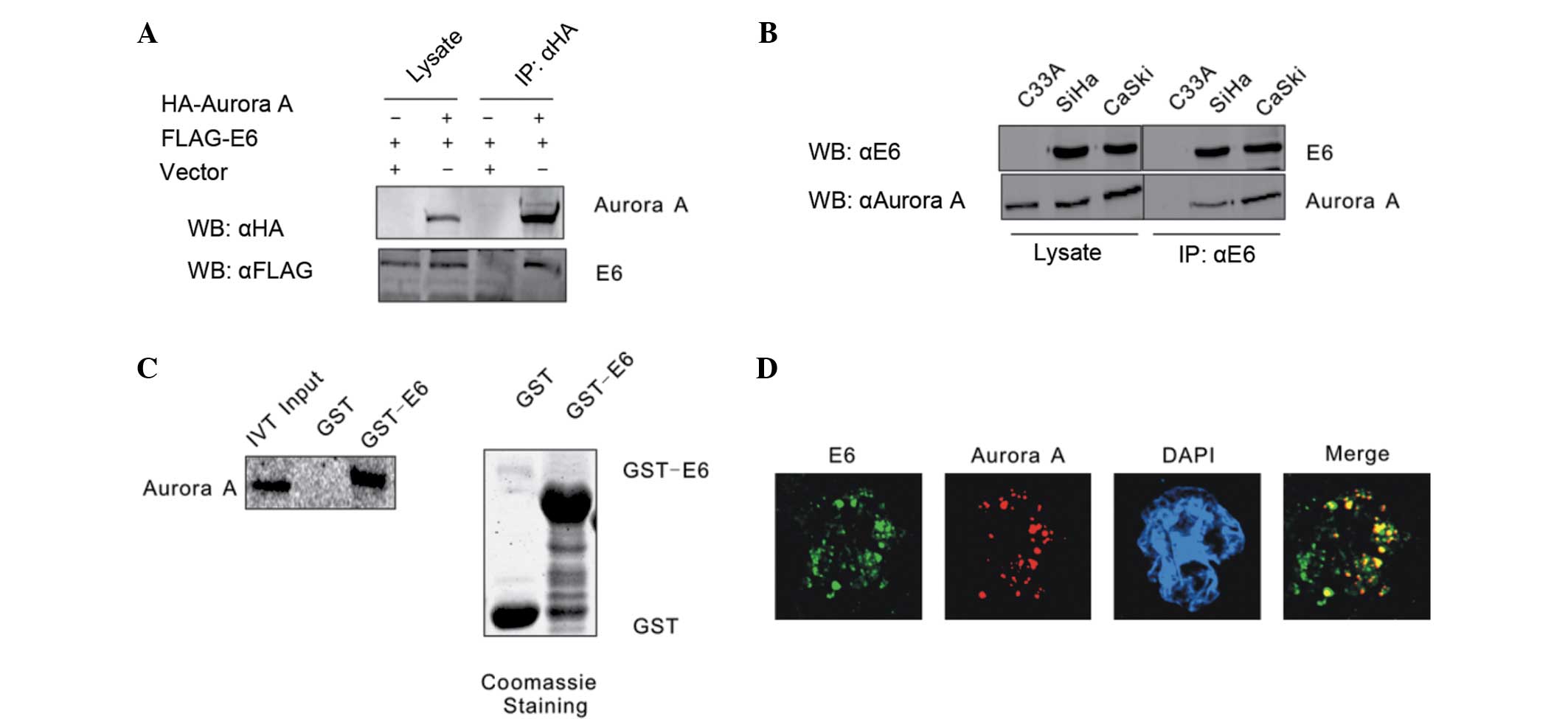 | Figure 1.HPV16 E6 combines with Aurora A. (A)
HEK293 cells were co-transfected with Flag-E6 and HA-Aurora A,
balanced with an empty vector. The cell lysates were subjected to
IP with a HA-specific antibody and protein expression was detected
by WB. (B) Lysates from the HPV-negative cervical carcinoma C33A
cell line and the two HPV16-positive SiHa and CaSki cell lines were
subjected to IP with a HPV16 E6 specific antibody C1P5. The
expression of the indicated proteins was detected by WB. (C) Either
GST control or GST-E6 beads were incubated with Aurora A in
vitro-translated 35S-radiolabeled protein. In
addition, 5% of in vitro translated protein input was used
as a comparison. Precipitated proteins were resolved by SDS-PAGE,
exposed to phosphorimager screen and scanned by the Typhoon 9410
imaging system. Coomassie blue staining of SDS-PAGE-resolved
purified GST and GST-E6 proteins was shown in the right panel. (D)
Colocalization of endogenous HPV16 E6 and Aurora A in CaSki cells.
HPV, human papillomavirus; IP, immunoprecipitation; HA,
hemagglutinin; WB, western blotting; GST, glutathione
S-transferase; SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel
electrophoresis; DAPI, 4′,6-diamidino-2-phenylindole; IVT, in
vitro translation. |
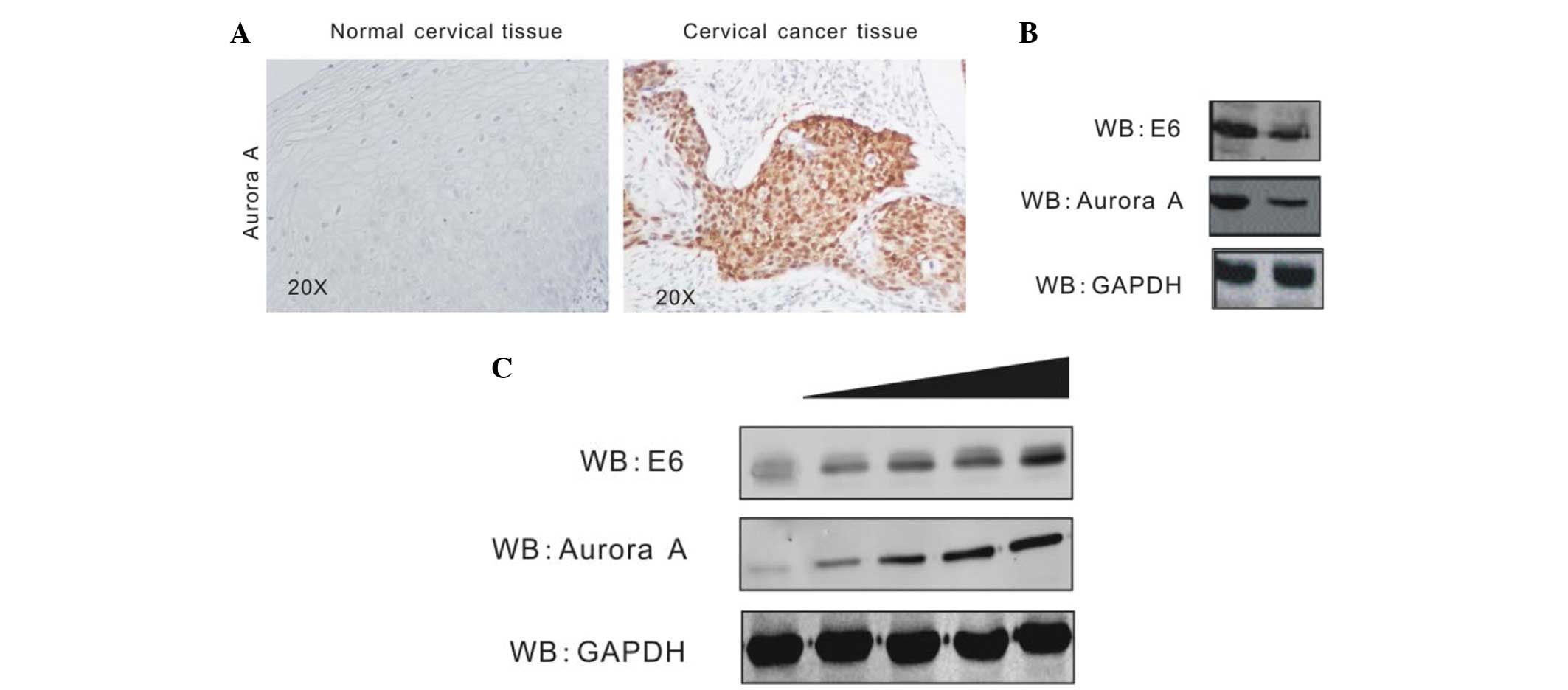
HPV16 E6 enhances Aurora A
expression
Aurora A has been shown to aberrantly accumulate in
numerous types of cancer cells (25,26). In
order to investigate Aurora A expression and its association with
HPV in cervical cancer, the present study assessed the protein
level of Aurora A in HPV-positive cervical cancer tissue and
HPV-negative normal cervical tissue by immunohistochemistry assays.
The result revealed that Aurora A is highly expressed in cervical
cancer tissue, but barely expressed in normal cervical tissue
(Fig. 2A). To determine that the
elevated Aurora A level is due to HPV16 E6, stable CaSki cell lines
carrying HPV16 E6 knockdown (Sh-E6) or control (Sh-Cr) were created
by transduction of the cells with shRNA-containing lentivirus
followed by selection of lentivirus-expressing cells using
puromycin (23). The Aurora A level
was determined by western blot analysis. The protein level of
Aurora A in E6-knockdown cells was evidently decreased compared
with the control cells (Fig. 2B). In
addition, the present study confirmed the effect of HPV16 E6 on
Aurora A expression by transient transfection. In total,
1.5×107 HEK293 cells were transiently transfected either
with the empty vector or increasing amounts of Flag-E6. At 36 h
post-transfection, the cells were harvested, lysed and individually
subjected to western blot analysis with antibodies against Aurora
A, HPV16 E6 and GAPDH. HPV16 E6 induces Aurora A expression in a
dose-dependent manner (Fig. 2C).
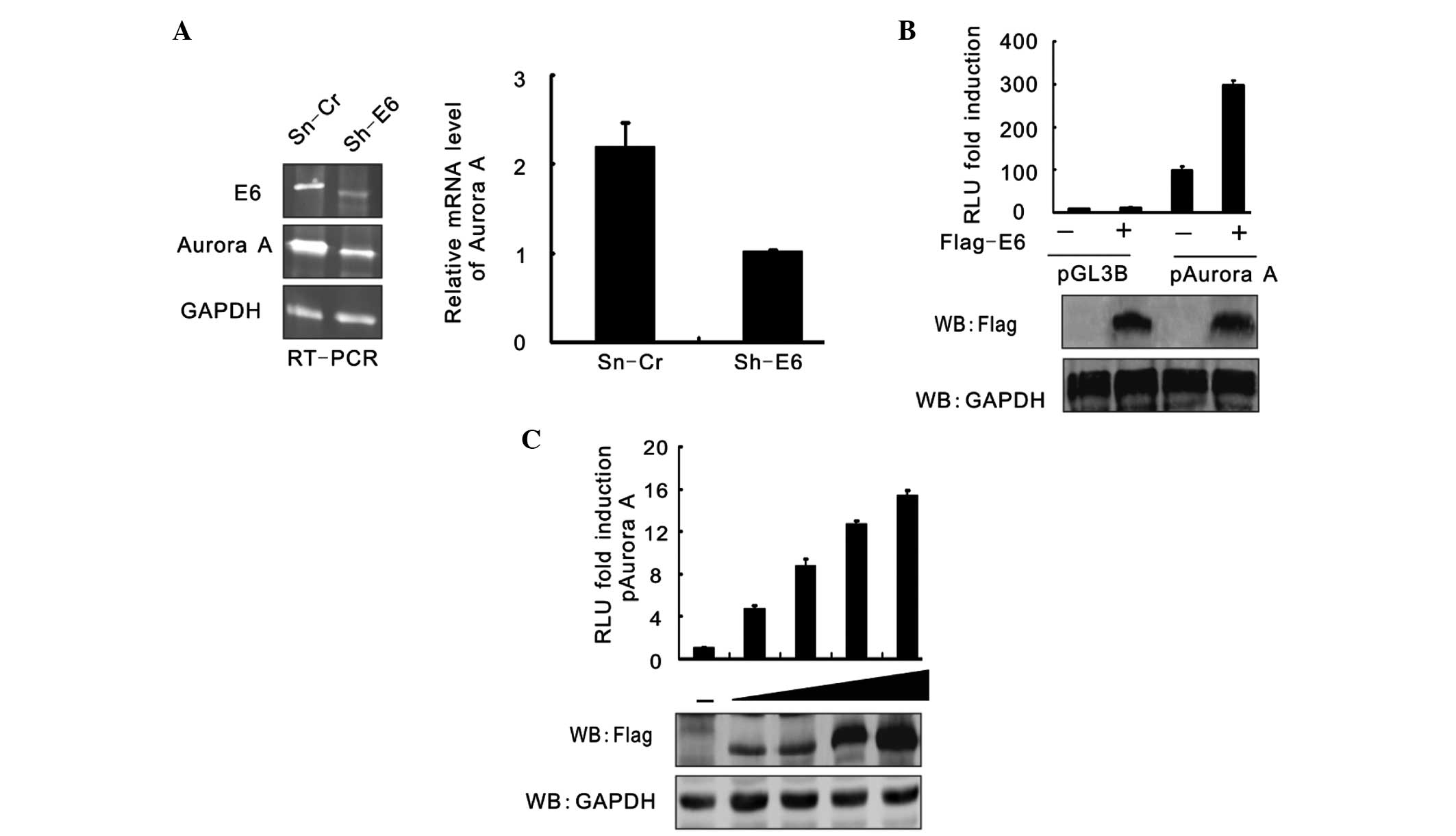
HPV16 E6 affects Aurora A expression
at transcription level
RT-PCR analysis was used to assess the effect of
HPV16 E6 on Aurora A at the transcription level. The results showed
that the Aurora A mRNA level was also significantly reduced when
HPV16 E6 expression was inhibited (Fig.
3A). To confirm this result, the luciferase reporter gene
driven by the Aurora A promoter was generated and assessed using a
reporter assay. HEK293 cells were cotransfected with pGL3-basic or
pGL3-Aurora A and either Flag-E6 or the empty vector, and the cells
were harvested at 24 h post-transfection. The cell lysates were
subjected to a luciferase reporter assay. The results were
presented as the relative luciferase unit (RLU) compared with
pGL3-basic and vector alone-cotransfected cells. Data is expressed
as the means ± standard deviation of three independent experiments.
The immunoblotting results of Flag-tagged E6 and GAPDH are shown in
Fig. 3B. A reporter assay was
performed of pGL3-Aurora A promoter cotransfection with an
increasing amount (0, 5, 10, 15, 20 mg) of Flag-E6 (Fig. 3C). At 24 h post-transfection, the
cells were harvested and subjected to a luciferase reporter assay.
The results were presented as the RLU fold compared with the
pGL3-Aurora A and empty vector alone-cotransfected cells. HPV16 E6
raised the RLU of pGL3-Aurora A in a dose-dependent manner.
Discussion
In the present study, it was demonstrated that HPV16
E6 combines with Aurora A and enhances its expression. The results
are consistent with other studies that have shown that Aurora A is
frequently overexpressed in a variety of human tumors and
cancer-derived cell lines (27–29).
Aurora A, B and C are highly conserved
serine/threonine kinases that play vital and distinct roles in the
process of chromosomal segregation, such as chromosome
condensation, alignment, control of spindle checkpoints, chromosome
segregation and cytokinesis, and these kinases have been identified
as oncogenes (14,30). Specifically, Aurora A localizes to
centrosomes and spindle microtubules proximal to centrosomes during
mitosis, as this kinase is required for the assembly of the mitotic
spindle, where Aurora A accumulates on centrosomes at the spindle
poles during prophase until the cell reaches metaphase. The
expression and activity of Aurora A kinase varies as the cell cycle
progresses. The levels are low in the G1/S phase, upregulated
during the G2/M phase and rapidly reduced subsequent to mitosis
(31). Aberrant expression of Aurora
A induces oncogenic transformation (32) and inhibition of Aurora kinase activity
leads to the failure of multiple events in mitosis, such as
incorrect separation of centriole pairs, misalignment of
chromosomes on the metaphase plate and incomplete cytokinesis
(33). Thus, it is reasonable to
conclude that the Aurora kinases may be good molecular therapeutic
targets for cancer.
In the present study, a novel mechanism of HPV
oncogene E6 in carcinogenesis is shown. However, further
elucidation is required. In the next step, we will further analyze
the role of Aurora A in the cell cycle and apoptosis of cervical
cancer cells.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos., 81171649
and 81572054).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausen Zur H: Papillomaviruses and cancer:
From basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scheffner M, Huibregtse JM, Vierstra RD
and Howley PM: The HPV-16 E6 and E6-AP complex functions as a
ubiquitin-protein ligase in the ubiquitination of p53. Cell.
75:495–505. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang H, Mo P, Ren S and Yan C: Activating
transcription factor 3 activates p53 by preventing E6-associated
protein from binding to E6. J Biol Chem. 285:13201–13210. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen JJ, Reid CE, Band V and Androphy EJ:
Interaction of papillomavirus E6 oncoproteins with a putative
calcium-binding protein. Science. 269:529–531. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee SS, Weiss RS and Javier RT: Binding of
human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the
Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci
USA. 94:6670–6675. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ronco LV, Karpova AY, Vidal M and Howley
PM: Human papillomavirus 16 E6 oncoprotein binds to interferon
regulatory factor-3 and inhibits its transcriptional activity.
Genes Dev. 12:2061–2072. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Underbrink MP, Howie HL, Galloway DA,
Bedard KM and Koop JI: E6 proteins from multiple human
betapapillomavirus types degrade Bak and protect keratinocytes from
apoptosis after UVB irradiation. J Virol. 82:10408–10417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao Q, Srinivasan S, Boyer SN, Wazer DE
and Band V: The E6 oncoproteins of high-risk papillomaviruses bind
to a novel putative GAP protein, E6TP1 and target it for
degradation. Mol Cell Biol. 19:733–744. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Dakic A, Zhang Y, Dai Y, Chen R and
Schlegel R: HPV E6 protein interacts physically and functionally
with the cellular telomerase complex. Proc Natl Acad Sci USA.
106:18780–18785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hengstermann A, Linares LK, Ciechanover A,
Whitaker NJ and Scheffner M: Complete switch from Mdm2 to human
papillomavirus E6-mediated degradation of p53 in cervical cancer
cells. Proc Natl Acad Sci USA. 98:1218–1223. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bischoff JR and Plowman GD: The
Aurora/Ipl1p kinase family: Regulators of chromosome segregation
and cytokinesis. Trends Cell Biol. 9:454–459. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW,
Sahin A, Brinkley BR and Sen S: Tumour amplified kinase STK15/BTAK
induces centrosome amplification, aneuploidy and transformation.
Nat Genet. 20:189–193. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giovinazzi S, Morozov VM, Summers MK,
Reinhold WC and Ishov AM: USP7 and Daxx regulate mitosis
progression and taxane sensitivity by affecting stability of
Aurora-A kinase. Cell Death Differ. 20:721–731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang H, He L, Kruk P, Nicosia SV and Cheng
JQ: Aurora-A induces cell survival and chemoresistance by
activation of Akt through a p53-dependent manner in ovarian cancer
cells. Int J Cancer. 119:2304–2312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Q, Kaneko S, Yang L, Feldman RI,
Nicosia SV, Chen J and Cheng JQ: Aurora-A abrogation of p53 DNA
binding and transactivation activity by phosphorylation of serine
215. J Biol Chem. 279:52175–52182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chien CY, Tsai HT, Su LJ, Chuang HC, Shiu
LY, Huang CC, Fang FM, Yu CC, Su HT and Chen CH: Aurora-A signaling
is activated in advanced stage of squamous cell carcinoma of head
and neck cancer and requires osteopontin to stimulate invasive
behavior. Oncotarget. 5:2243–2262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meraldi P, Honda R and Nigg EA: Aurora-A
overexpression reveals tetraploidization as a major route to
centrosome amplification in p53−/− cells. EMBO J.
21:483–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo Y, Meng XK, Wang Q, Wang Y and Shang
H: The ING4 binding with p53 and induced p53 acetylation were
attenuated by human papillomavirus 16 E6. PLoS One. 8:e714532013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCq method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu X, Li Z, Song Y, Wang R, Han L, Wang
Q, Jiang K, Kang C and Zhang Q: AURKA induces EMT by regulating
histone modification through Wnt/β-catenin and PI3K/Akt signaling
pathway in gastric cancer. Oncotarget. 21:April 21–2016.(Epub ahead
of print).
|
|
26
|
Li Z, Sun Y, Chen X, Squires J,
Nowroozizadeh B, Liang C and Huang J: p53 mutation directs AURKA
overexpression via miR-25 and FBXW7 in prostatic small cell
neuroendocrine carcinoma. Mol Cancer Res. 13:584–591. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanaka M, Ueda A, Kanamori H, Ideguchi H,
Yang J, Kitajima S and Ishigatsubo Y: Cell-cycle-dependent
regulation of human aurora A transcription is mediated by periodic
repression of E4TF1. J Biol Chem. 277:10719–10726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marumoto T, Zhang D and Saya H: Aurora-A -
a guardian of poles. Nat Rev Cancer. 5:42–50. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Warner SL, Bearss DJ, Han H and Von Hoff
DD: Targeting Aurora-2 kinase in cancer. Mol Cancer Ther.
2:589–595. 2003.PubMed/NCBI
|
|
30
|
Carmena M and Earnshaw WC: The cellular
geography of Aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kimura M, Matsuda Y, Yoshioka T and Okano
Y: Cell cycle-dependent expression and centrosome localization of a
third human Aurora/IpI1-related protein kinase, AIK3. J Biol Chem.
274:7334–7340. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kimura M, Kotani S, Hattori T, Sumi N,
Yoshioka T, Todokoro K and Okano Y: Cell cycle-dependent expression
and spindle pole localization of a novel human protein kinase, Aik,
related to aurora of Drosophila and yeast Ipl1. J Biol Chem.
272:13766–13771. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marumoto T, Honda S, Hara T, Nitta M,
Hirota T, Kohmura E and Saya H: Aurora-a kinase maintains the
fidelity of early and late mitotic events in HeLa cells. J Biol
Chem. 278:51786–51795. 2003. View Article : Google Scholar : PubMed/NCBI
|