Introduction
Endometrial cancer (EC) is one of the most common
gynecological malignancies worldwide (1) and is classified into estrogen-dependent
type I and estrogen-independent type II forms. The type I form is
the most common, accounting for 75–85% of all cases of EC (2). Unopposed estrogen has been shown to
increase the risk of EC development, as estrogens stimulate
endometrial cell proliferation and inhibit apoptosis (3). Additionally, diseases associated with
insulin resistance, such as obesity, type II diabetes mellitus and
polycystic ovary syndrome (PCOS), are considered significant risk
factors for the development and progression of type I EC (4–6). Insulin
resistance is a condition in which target tissues have decreased
sensitivity to insulin, and blood insulin levels consequently
increase to maintain normal glucose levels. The chronic
hyperinsulinemic state has been found to exert direct and indirect
effects that contribute to EC development (7). A large body of evidence has suggested
that women with diabetes possess a stronger association with
neoplastic processes (5), and
furthermore, diabetic patients with cancer experience increased
mortality compared with normoglycemic individuals (8). Finally, a meta-analysis study has shown
that women with diabetes have a two-fold risk for EC (9). Insulin resistance appears to play a
central role in endometrial carcinogenesis. Therefore, treatment
with insulin-sensitizing agents that act through reducing insulin
levels could offer a general approach to prevent the development of
cancer and reduce metastasis (10,11).
Metformin, an anti-hyperglycemic and insulin
sensitizing agent, is the most commonly used drug for treating type
II diabetes mellitus, as well as off-label insulin resistance (as
observed in women with PCOS) (12).
In recent years, numerous studies have indicated that metformin
could be effective as a cancer therapy, along with its traditional
role in treating diabetes (13–15).
Metformin use can prevent malignant transformation indirectly via
systemic changes (improving hyperglycemia, hyperlipidemia and
hyperinsulinemia) and via direct effects, such as suppression of
mammalian target of rapamycin via 5′ adenosine
monophosphate-activated protein kinase activation, leading to
reduced protein synthesis and cell proliferation (16,17). A
meta-analysis of 18 observational studies showed that metformin
therapy is associated with an overall 27% reduction in the risk of
developing any type of cancer in patients with type II diabetes
(18). A retrospective study showed
that metformin use is additionally associated with less recurrence
and improved overall survival in EC patients; although, when
recurrence took occurred, it was not delayed compared with that in
the control (19). However, the
majority of these previous studies possessed methodological
weaknesses and/or insufficient data. Additionally, experimental
studies revealed that metformin is an effective antiestrogenic
agent, inhibiting cell proliferation and leading to growth arrest,
as well as inducing apoptosis in EC (17,20–22).
Furthermore, recent findings have suggested that metformin is
important in suppressing the migration and invasion of cancer
cells, which could prevent metastasis (23–25).
However, in these studies metformin action was observed at high
supra-pharmacological concentrations and without estradiol
addition, an important factor in endometrial proliferative
disorders.
The present study investigated the capability of
low-dose metformin (0.1 mM) to inhibit the development of cancer
and reduce the metastatic potential of endometrial adenocarcinoma
type I in vitro using the Ishikawa cell line in the high and
normal glucose environments. Finally, the study evaluated the
efficiency of low-dose metformin to prevent EC using normal uterine
endometrial epithelial cells (eEPs) exposed to high glucose
concentrations.
Materials and methods
Isolation and culture of primary
eEPs
The primary eEPs were isolated from endometrial
biopsies (between March and August 2015) taken from healthy,
regularly cycling women (34.1±3.4 years old) undergoing
laparoscopic surgery for benign reasons. Informed consent was
obtained and the study protocol was approved by the the Clinical
Hospital of Porto Alegre (Porto Alegre, Rio Grande do Sul, Brazil)
and the University of Heidelberg (Heidelberg, Germany). Exclusion
criteria were hormonal stimulation within the preceding 3 months,
endocrinopathies, cancerous lesions and irregular menstrual
bleeding. Human endometrial tissues were digested with collagenase
type Ia (Gibco, Karlsruhe, Germany) for 2 h. Thereafter, the cells
were washed, centrifuged (800 × g, room temperature) and separated
using a 40-µm filter. The stromal cells passed through the filter
and the epithelial cells were retained. Epithelial cells were
recovered from the filter by backwashing with Dulbecco's modified
Eagle's medium (DMEM)/F12 (Gibco; Thermo Fisher Scientific Inc.,
Waltham, MA, USA).
Cell culture
The Ishikawa human endometrial adenocarcinoma cell
line was purchased from Sigma-Aldrich (Merck Millipore, Darmstadt,
Germany; European Collection of Cell Culture authenticated).
Ishikawa cells were grown in minimal essential medium (MEM;
Sigma-Aldrich; Merck Millipore) supplemented with 5% fetal bovine
serum (FBS; HyClone; Thermo Fisher Scientific Inc.), 1%
penicilin-streptomycin (Gibco; Thermo Fisher Scientific Inc.), 1%
non-essential amino acids (Sigma-Aldrich; Merck Millipore) and 5
µg/ml bovine insulin (Sigma-Aldrich; Merck Millipore), while the
primary eEPs were maintained in DMEM/F12 supplemented with 5% FBS
and 1% penicillin-streptomycin at 37°C in a humidified atmosphere
with 5% CO2.
For the experiments in a high glucose environment,
the cells were cultured in various conditions with DMEM/F12 medium
containing 17 mM glucose, and for the experiments in a normal
glucose environment, the cells were cultured with MEM containing
5.5 mM glucose. The media for all conditions were supplemented with
10−8 M estradiol. The different conditions included: a)
Control group, medium only; b) insulin group, medium plus 100 ng/ml
insulin; c) metformin group, medium plus 0.1 mM metformin; and d)
insulin plus metformin group, medium plus 100 ng/ml insulin and 0.1
mM metformin. All chemicals were purchased from Sigma-Aldrich
(Merck Millipore).
Migration and invasion analysis
To assess the ability of cellular motility in the
different conditions, Transwell filters (6.5 mm in diameter; 5-µm
pore size; Corning Inc., Corning, NY, USA) were used. The cells
were harvested as aforementioned and suspended in serum-free
medium. The Transwell filter was coated with 100 ml medium for the
migration assay or with 100 µl Matrigel (1:1 dilution; Corning
Matrigel Growth Factor Reduced; Corning Inc.) for the invasion
assay. Thereafter, the primary eEPs (1×104 in 200 µl) or
Ishikawa cells (2.5×105 in 200 µl) were added with the
individual treatment conditions into the Transwell chamber. Medium
containing 5% FBS was added to the lower well, then the Transwells
were incubated for 16 h. Non-migrated/invading cells were removed
with a cotton swab, and migrated/invading cells were fixed in 3.7%
paraformaldehyde in phosphate-buffered saline, permeabilized with
100% methanol and stained with Giemsa. Cell migration/invasion,
repeated five times, was quantified by blinded counting of the
number of migrated/invaded cells in each insert in four different
areas. Data are expressed as the percentage compared with the
control group.
Cell proliferation assays
The CellTiter Glo Luminescent assay (Promega
Corporation, Madison, WI, USA) was used to evaluate the relative
cell proliferation of Ishikawa cells exposed to high and normal
glucose conditions. The cells were seeded into 96-well plates at a
density of 5,000 cells/well in 100 µl drug-free medium overnight.
The cells were then treated with the indicated treatment (control,
insulin, metformin or insulin plus metformin) for 72 h. After this
period, the plates were equilibrated at room temperature for 30
min, and 100 µl of CellTiter Glo reagent was added in each well,
mixed for 2 min and incubated at room temperature for 10 min. The
luminescence was detected by a Centro LB 960 Microplate Luminometer
(Berthold Technologies GmbH and Co. KG, Bad Wildbad, Germany).
In order to evaluate the sensitivity of the
epithelial carcinoma cells to varying doses of metformin in high
and normal glucose conditions, the Ishikawa cells were seeded into
96-well plates at a density of 5,000 cells/well in 100 µl drug-free
medium overnight. The cells were then treated with increasing doses
of metformin (0, 0.1, 1 and 5 mM) for 72 h. A CellTiter Glo
Luminescent assay was performed as aforementioned. The effect of
the different treatments was calculated as a fold-change compared
with the control group. Each experiment was performed in
sextuplicate and repeated three times.
Statistical analysis
Quantitative data are represented as the mean ±
standard error of the mean of at least three independent
experiments. SPSS version 23 (IBM SPPS, Armonk, NY, USA) was used
to perform a one-way analysis of variance and Turkey's post-hoc
test, a generalized estimating equation test or Student's t-test,
as appropriate. P<0.05 was considered to indicate a
statistically significant difference.
Results
Low-dose metformin inhibits the
insulin effect on the migration and invasion in the EC cell
line
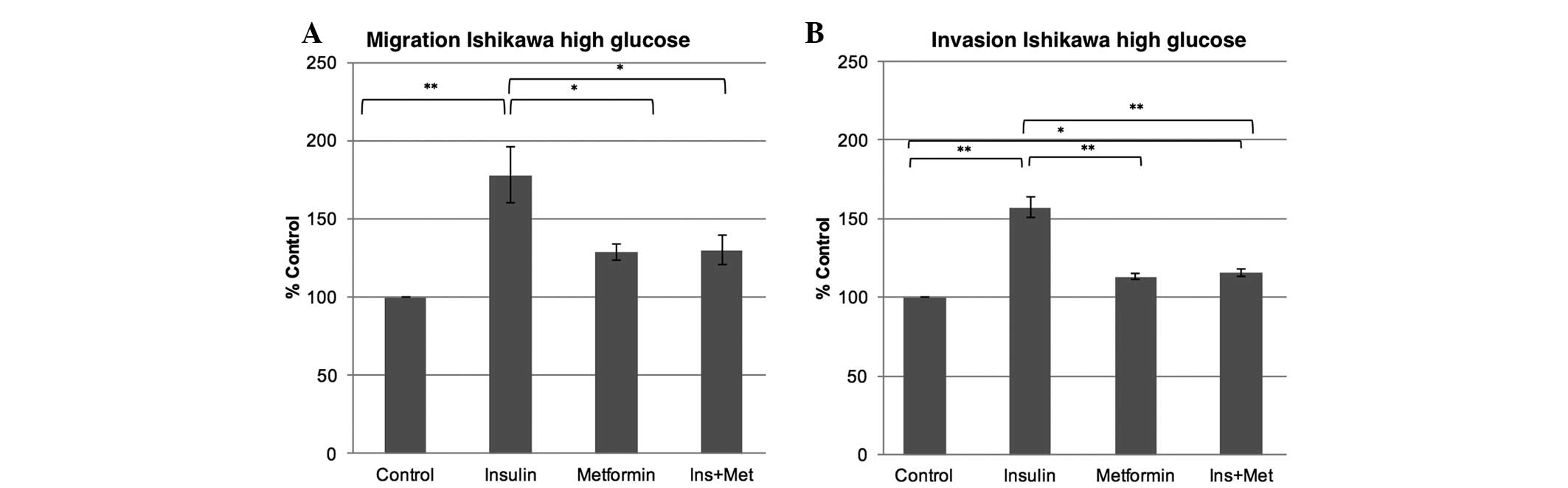
The present study examined the effect of a
hyperinsulinemic environment with or without metformin treatment on
the migration and invasion ability of Ishikawa cells when exposed
to a high or normal glucose environment. In a high glucose
environment, insulin exposure increased the migration by 77%
(P<0.001) and the invasion capability by 57% (P<0.001) in the
Ishikawa cells compared with the control. While metformin alone did
not confer any change in the migration and invasion capabilities of
the Ishikawa cells in a high glucose environment when compared with
the control, the metformin co-treatment (insulin plus 0.1 mM
metformin) markedly attenuated the insulin effect, leading to a
decrease in the migration ability by 47% (P=0.023) and in the
invasion ability by 42% (P<0.001), therefore reducing the
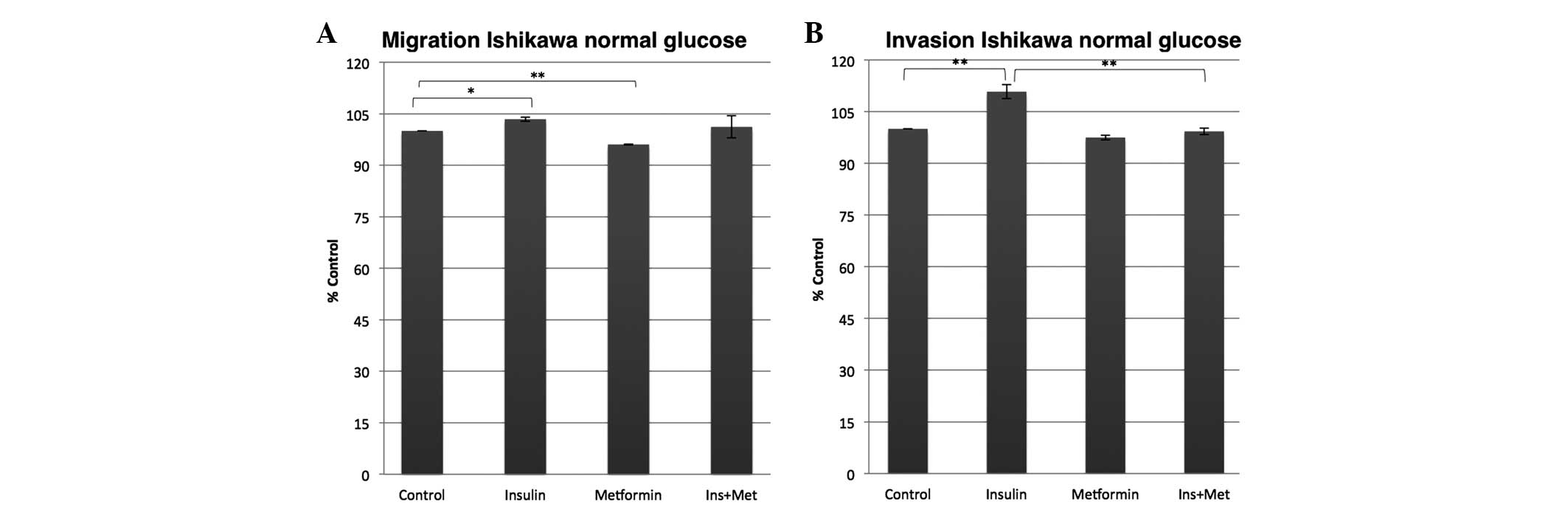
insulin effect (Fig. 1). In the
normal glucose environment, however, the insulin and the metformin
effects were less dominant compared with those in the high glucose
environment. While the insulin exposure of the Ishikawa cells only
slightly increased the migration and invasion ability by 3%
(P=0.032) and 10% (P=0.006), respectively, compared with the
control, the metformin co-treatment almost completely abolished the
insulin effect on the invasion potential, as the metformin
co-treatment inhibited the insulin effect on the invasion ability
to a control rate (Fig. 2). Metformin
alone on the other hand significantly decreased the capability of
the Ishikawa cells to migrate by 4% (P=0.001) and non-significantly
decreased their ability to invade by 3% (P=0.069) compared with the
control.
Low-dose metformin is effective in
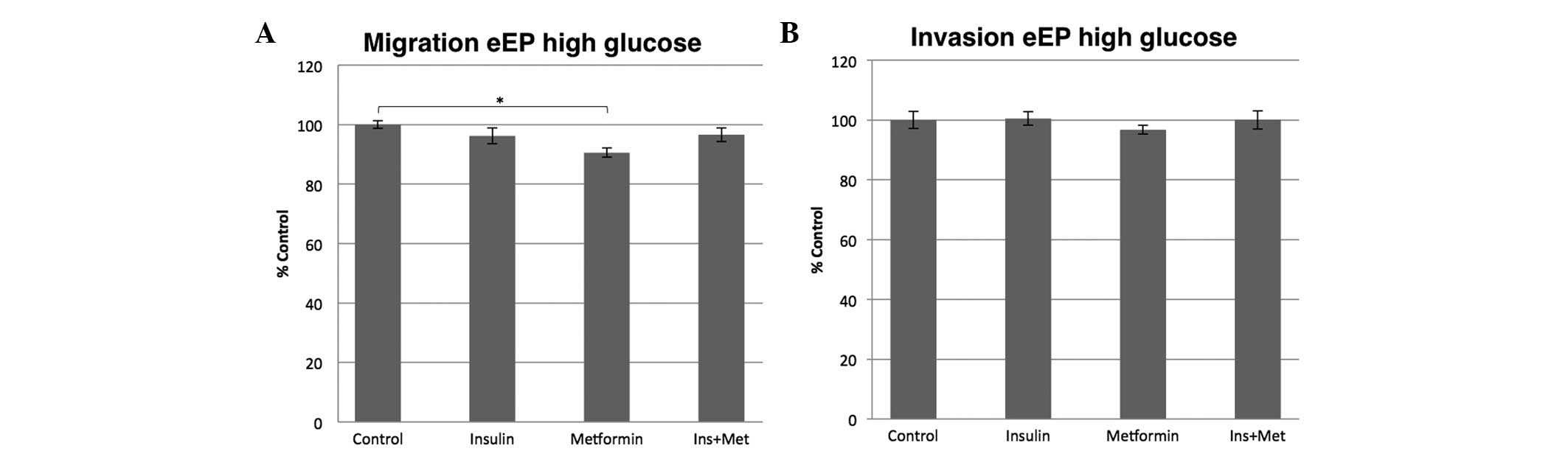
primary eEPs
To investigate the efficiency of metformin
treatment, the effect of metformin on normal eEPs was analyzed.
When the primary eEPs were cultured in a high glucose environment,
insulin alone or in combination with metformin did not change the
migration or invasion potential after 16 h. However, metformin
alone inhibited the migration potential significantly by 10%
(P<0.001) and the invasion capacity non-significantly by 4%
(P=1.000) compared with the control (Fig.
3).
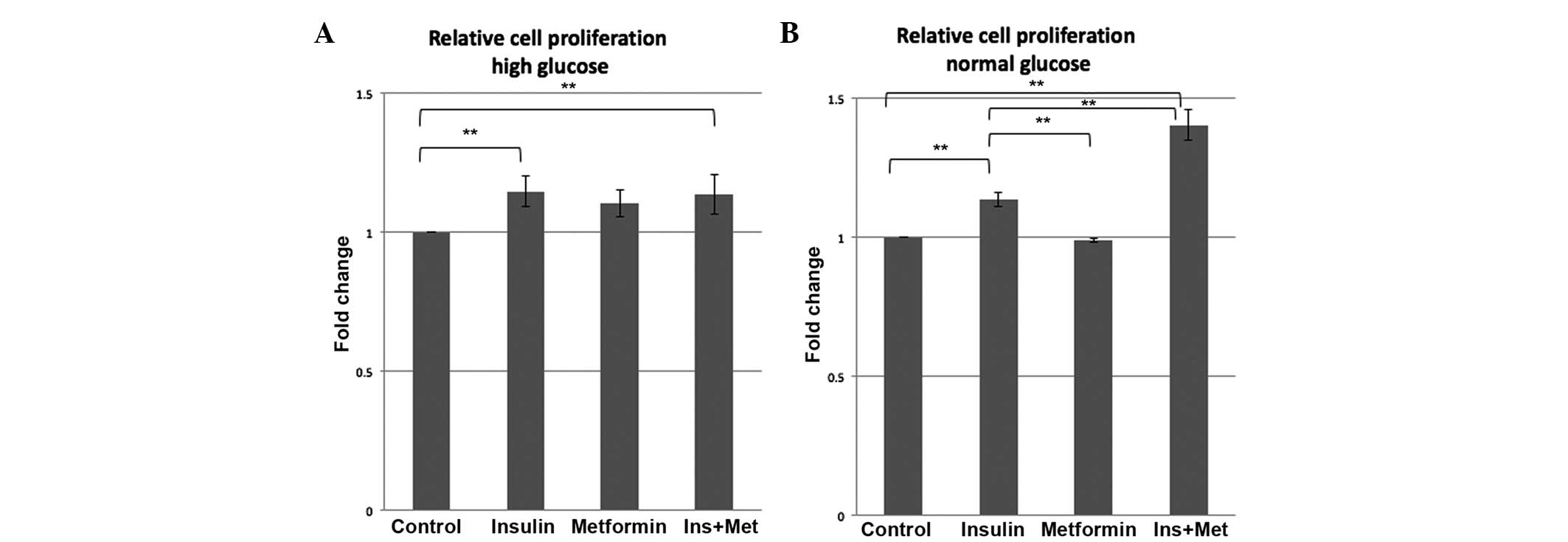
Low-dose metformin does not decrease
the proliferation of the EC cell line
To determine if the glucose environment affects the
proliferation potential of epithelial cancer cells, Ishikawa cells
were treated using the four treatment conditions (control, insulin,
metformin and insulin plus metformin) for 72 h in media containing
high or normal glucose. As shown in Fig.
4, insulin treatment increased the proliferation of the
Ishikawa cells by 1.15-fold in the high glucose condition (P=0.029)
and by 1.13-fold in the normal glucose condition (P=0.003) compared
to the control group. However, treatment with 0.1 mM metformin did
not have a significant effect on Ishikawa cell proliferation
compared with the control group in the 72 h, either in a high or
normal glucose environment. Furthermore, in this treatment period,
0.1 mM metformin in combination with insulin was not able to
inhibit the stimulatory insulin effect on the proliferation
potential in high glucose and even lead to an enhanced
proliferation potential in the normal glucose condition compared
with the control (1.40-fold; P<0.001).
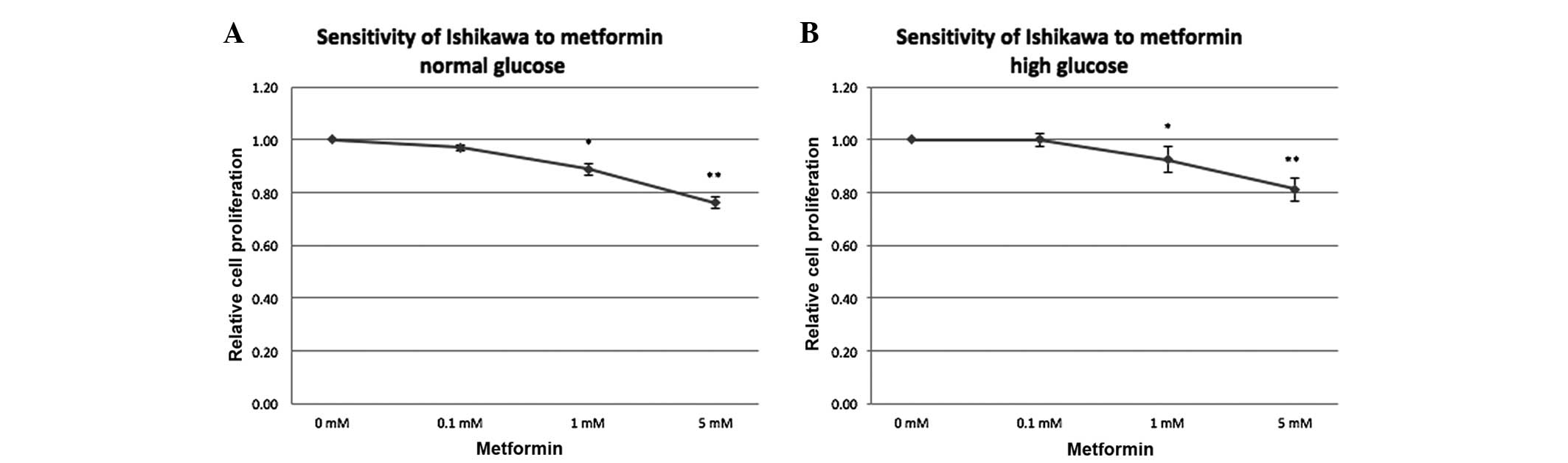
Metformin decreases EC cell line
proliferation in a dose-dependent manner
In order to test the sensitivity of the EC cell line
to metformin at different concentrations in a high and normal
glucose environment, the proliferation of Ishikawa cells treated
with metformin (0, 0.1, 1 and 5 mM) in medium containing high and
normal levels of glucose for 72 h was measured (Fig. 5). Independent of the glucose
concentration in the medium, the proliferation potential was
unchanged at 0.1 mM during 72 h (1.0-fold with high glucose and
0.97-fold with normal glucose relative to the control), while
reduced proliferation was noted with 1 mM metformin (0.92-fold with
high glucose; P=0.037), followed by a further reduction with
increasing metformin concentration (0.81-fold at 5 mM with high
glucose; P=0.002). In a normal glucose condition, a slightly more
pronounced effect was noted compared with that in the high glucose
environment (0.89-fold for 1 mM, P=0.027; and 0.79-fold for 5 mM,
P=0.002).
Discussion
Considering the increasing level of metformin
research in cancer therapy, as well as in patients with type II
diabetes mellitus, thus extending its use to the non-diabetic
population, the present study investigated the effectiveness of
low-dose metformin in EC development and prevention. In order to
analyze metformin as a cancer prevention agent and/or cancer
progression treatment in endometrial adenocarcinoma type I eEPs and
endometrial epithelial cancer cells were exposed to metformin in
either a high glucose or normal glucose medium with or without
insulin substitution.
Glucose is an essential nutrient that supports
cellular energy homeostasis. The normal serum glucose level is
usually maintained at ~5.5 mM. However, increased blood glucose
levels and hyperglycemia contribute to growth and carcinogenesis in
EC (26), and act as a critical link
between the observed increased cancer risk in patients with type II
diabetes (5). Furthermore, in
conditions such as PCOS, insulin resistance can be
obesity-independent. Despite a normoglycemic state, the associated
hyperinsulinemia is believed to be a promoting factor not only for
cancer initiation, but also for cancer progression (7,27).
Several studies have suggested that high glucose
levels create an optimal environment for cancer cells to exhibit a
resistance to metformin (28–30). The current study evaluated the insulin
action and the metformin efficiency on the metastatic potential of
EC cells when exposed to a high and normal glucose environment. The
findings showed that insulin treatment in each glucose condition
increased the migration and invasion ability of the endometrial
epithelial cancer cells, however, this effect was more pronounced
in the high glucose environment. In addition, low-dose metformin
effectively attenuated the effects of insulin action in high and
normal glucose conditions, leading to a less profound effect.
Notably, the results in the normal glucose environment showed a
decrease in the ability of the cells to migrate when treated with
metformin compared with the control group. However, a significant
difference could not be found between the metformin and control
groups in a high glucose environment, but compared with the normal
glucose environmen, a tendency of cancer cell resistance was
present when exposed to high glucose levels. By contrast, insulin
alone or in combination with metformin showed no effect on primary
eEPs exposed to high glucose conditions with regard to their
migration or invasion potential after 16 h. We assume that primary
eEPs do not have the metastatic potential observed in cancer cells.
Furthermore the short incubation of 16 h may not be long enough for
the insulin to activate the invasion potential of these cells.
However, metformin alone was able to reduce the inherent migration
capability of the eEPs. These results support the evidence that a
high glucose environment interferes in the metformin effect on
cancer cells.
To the best of our knowledge, the present study
shows for the first time the ability of metformin to inhibit the
metastatic potential of endometrial epithelial cancer cells, while
the metformin efficiency in the inhibition of migration and
invasion has already been reported in a dose-dependent manner in
other types of cancer cells, including ovarian (23) and prostate (31) cancer cells, and osteosarcoma (32). Recently, Han et al reported an
increased ability of adhesion and invasion in two EC cell lines,
suggesting that targeting glucose metabolism may be a promising
therapeutic strategy for the treatment of EC (33).
Besides the potential anti-metastatic effect of
metformin, several studies have indicated that metformin may be a
useful anti-proliferation agent for EC cells. Cantrell et al
showed for the first time that metformin potently inhibits growth
in two EC cell lines [half maximal inhibitory concentration
(IC50) of 1 mM metformin] after 72 h of treatment in a
normal glucose environment (17).
Takahashi et al demonstrated that after 48 h of treatment
with increasing concentrations of metformin in normal glucose
conditions, ≥5 mM metformin significantly reduce the number of
viable cells (IC50 of 6.78 mM metformin) (21). Furthermore, the metformin inhibitory
proliferation effect has been reported for certain other types of
cancer. Wu et al showed that metformin inhibits the
proliferation of ovarian cancer cells in a dose- and time-dependent
manner in vitro (IC50 of 16.67 mM metformin) and
in vivo in a nude mouse model (23). Kato et al examined the effects
of metformin in prostate cancer cell proliferation using the MTS
assay and cell counting. The number of viable cells of three
prostate cancer cell lines decreased significantly after incubation
with metformin (IC50 of 5 mM metformin) for 48 h
(31). However, in the present study,
in the Ishikawa endometrial epithelial cancer cell line, it was
observed that the insulin treatment increased cell proliferation,
without any reduction in proliferation by the addition 0.1 mM
metformin regardless of the glucose concentration present. This
apparent discrepancy may result from the high micromolar metformin
concentrations used in the previous studies, which are much greater
than the steady-state levels in the patients' plasma. The present
study observed the relative cell proliferation sensitivity to
metformin in the EC cell line in the range between 1 and 5 mM in
each glucose environment, although this was more pronounced in the
normal glucose condition. Moreover, these findings were in the
presence of estradiol, an important factor in endometrial
proliferative disorders, but not used in the published endometrial
studies.
In addition, the epidemiological and laboratory
studies remain controversial. Numerous details of the action of
metformin remain to be identified, and the risk of harm must be
considered when designing novel metformin-based therapies. The
knowledge gained from the combination of tumor genetics, patient
metabolic profiles and the cellular environment will assist in
determining the tumor preventative or treatment efficiency of
metformin. We expect to improve the understanding of the mechanisms
linking glucose metabolism, hyperinsulinemia and metformin
treatment with EC development and progression. According to the
present findings, it is indicated that metformin at 0.1 mM is not
efficient enough to decrease the proliferation in 72 h of treatment
in an EC cell line. However, in this concentration, metformin can
inhibit the insulin effect by decreasing the high cellular invasion
and migration potential in high and normal glucose environments,
demonstrating an anti-metastatic effect of metformin in endometrial
epithelial cancer cells. Nevertheless, further investigations are
necessary to improve the knowledge of metformin as an effective
targeted therapy in cancer, as well as a long term treatment to be
efficient in the inhibition of target genes and proteins connected
to the development and progression of cancer.
Acknowledgements
This study was generously supported by National
Council for Scientific and Technological Development (CNPq; grant
no. 470636/2012-2), Incentive Fund for Research and Events of
Clinics Hospital of Porto Alegre (FIPE-HCPA; grant no. 14-0267),
Coordination for the Improvement of Higher Education Personnel
(CAPES) and the Excellence Initiative from the University of
Heidelberg (grant no. 7.013.14).
References
|
1
|
No authors listed, . Cancer statistics.
JAMA. 310:9822013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garg K and Soslow RA: Endometrial
carcinoma in women aged 40 years and younger. Arch Pathol Lab Med.
138:335–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clarke CL and Sutherland RL: Progestin
regulation of cellular proliferation. Endocr Rev. 11:266–301. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nead KT, Sharp SJ, Thompson DJ, Painter
JN, Savage DB, Semple RK, Barker A; Australian National Endometrial
Cancer Study Group (ANECS); Perry JR, Attia J, et al: Evidence of a
causal association between insulinemia and endometrial cancer: A
mendelian randomization analysis. J Natl Cancer Inst. 107:pii:
djv178. 2015.PubMed/NCBI
|
|
5
|
Szablewski L: Diabetes mellitus:
Influences on cancer risk. Diabetes Metab Res Rev. 30:543–553.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
SGO Clinical Practice Endometrial Cancer
Working Group; Burke WM, Orr J, Leitao M, Salom E, Gehrig P,
Olawaiye AB, Brewer M, Boruta D, Villella J, et al: Endometrial
cancer: A review and current management strategies: Part I. Gynecol
Oncol. 134:385–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mu N, Zhu Y, Wang Y, Zhang H and Xue F:
Insulin resistance: A significant risk factor of endometrial
cancer. Gynecol Oncol. 125:751–757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barone BB, Yeh HC, Snyder CF, Peairs KS,
Stein KB, Derr RL, Wolff AC and Brancati FL: Long-term all-cause
mortality in cancer patients with preexisting diabetes mellitus: A
systematic review and meta-analysis. JAMA. 300:2754–2764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friberg E, Orsini N, Mantzoros CS and Wolk
A: Diabetes mellitus and risk of endometrial cancer: A
meta-analysis. Diabetologia. 50:1365–1374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bowers LW, Rossi EL, O'Flanagan CH,
deGraffenried LA and Hursting SD: The role of the insulin/IGF
system in cancer: Lessons learned from clinical trials and the
energy balance-cancer link. Front Endocrinol (Lausanne).
6:772015.PubMed/NCBI
|
|
11
|
Gallagher EJ and LeRoith D: Obesity and
diabetes: The increased risk of cancer and cancer-related
mortality. Physiol Rev. 95:727–748. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shao R, Li X, Feng Y, Lin JF and Billig H:
Direct effects of metformin in the endometrium: A hypothetical
mechanism for the treatment of women with PCOS and endometrial
carcinoma. J Exp Clin Cancer Res. 33:412014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Umene K, Banno K, Kisu I, Yanokura M,
Nogami Y, Tsuji K, Masuda K, Ueki A, Kobayashi Y, Yamagami W, et
al: New candidate therapeutic agents for endometrial cancer:
Potential for clinical practice (review). Oncol Rep. 29:855–860.
2013.PubMed/NCBI
|
|
14
|
Febbraro T, Lengyel E and Romero IL: Old
drug, new trick: Repurposing metformin for gynecologic cancers?
Gynecol Oncol. 135:614–621. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He H, Ke R, Lin H, Ying Y, Liu D and Luo
Z: Metformin, an old drug, brings a new era to cancer therapy.
Cancer J. 21:70–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Viollet B, Guigas B, Garcia N Sanz,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation-implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franciosi M, Lucisano G, Lapice E,
Strippoli GF, Pellegrini F and Nicolucci A: Metformin therapy and
risk of cancer in patients with type 2 diabetes: Systematic review.
PLoS One. 8:e715832013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ko EM, Walter P, Jackson A, Clark L,
Franasiak J, Bolac C, Havrilesky LJ, Secord AA, Moore DT, Gehrig PA
and Bae-Jump V: Metformin is associated with improved survival in
endometrial cancer. Gynecol Oncol. 132:438–442. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tabrizi AD, Melli MS, Foroughi M,
Ghojazadeh M and Bidadi S: Antiproliferative effect of metformin on
the endometrium-a clinical trial. Asian Pac J Cancer Prev.
15:10067–10070. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takahashi A, Kimura F, Yamanaka A,
Takebayashi A, Kita N, Takahashi K and Murakami T: Metformin
impairs growth of endometrial cancer cells via cell cycle arrest
and concomitant autophagy and apoptosis. Cancer Cell Int.
14:532014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sarfstein R, Friedman Y, Attias-Geva Z,
Fishman A, Bruchim I and Werner H: Metformin downregulates the
insulin/IGF-I signaling pathway and inhibits different uterine
serous carcinoma (USC) cells proliferation and migration in
p53-dependent or -independent manners. PLoS One. 8:e615372013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu B, Li S, Sheng L, Zhu J, Gu L, Shen H,
La D, Hambly BD, Bao S and Di W: Metformin inhibits the development
and metastasis of ovarian cancer. Oncol Rep. 28:903–908.
2012.PubMed/NCBI
|
|
24
|
Chou CC, Lee KH, Lai IL, Wang D, Mo X,
Kulp SK, Shapiro CL and Chen CS: AMPK reverses the mesenchymal
phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a
signaling axis. Cancer Res. 74:4783–4795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bao B, Azmi AS, Ali S, Zaiem F and Sarkar
FH: Metformin may function as anti-cancer agent via targeting
cancer stem cells: The potential biological significance of
tumor-associated miRNAs in breast and pancreatic cancers. Ann
Transl Med. 2:592014.PubMed/NCBI
|
|
26
|
Lambe M, Wigertz A, Garmo H, Walldius G,
Jungner I and Hammar N: Impaired glucose metabolism and diabetes
and the risk of breast, endometrial, and ovarian cancer. Cancer
Causes Control. 22:1163–1171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moran LJ, Hutchison SK, Norman RJ and
Teede HJ: Lifestyle changes in women with polycystic ovary
syndrome. Cochrane Database Syst Rev. CD0075062011.
|
|
28
|
Sinnett-Smith J, Kisfalvi K, Kui R and
Rozengurt E: Metformin inhibition of mTORC1 activation, DNA
synthesis and proliferation in pancreatic cancer cells: Dependence
on glucose concentration and role of AMPK. Biochem Biophys Res
Commun. 430:352–357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Menendez JA, Oliveras-Ferraros C, Cufí S,
Corominas-Faja B, Joven J, Martin-Castillo B and Vazquez-Martin A:
Metformin is synthetically lethal with glucose withdrawal in cancer
cells. Cell Cycle. 11:2782–2792. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhuang Y, Chan DK, Haugrud AB and
Miskimins WK: Mechanisms by which low glucose enhances the
cytotoxicity of metformin to cancer cells both in vitro and in
vivo. PLoS One. 9:e1084442014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kato H, Sekine Y, Furuya Y, Miyazawa Y,
Koike H and Suzuki K: Metformin inhibits the proliferation of human
prostate cancer PC-3 cells via the downregulation of insulin-like
growth factor 1 receptor. Biochem Biophys Res Commun. 461:115–121.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen X, Hu C, Zhang W, Shen Y, Wang J, Hu
F and Yu P: Metformin inhibits the proliferation, metastasis, and
cancer stem-like sphere formation in osteosarcoma MG63 cells in
vitro. Tumour Biol. 36:9873–9883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han J, Zhang L, Guo H, Wysham WZ, Roque
DR, Willson AK, Sheng X, Zhou C and Bae-Jump VL: Glucose promotes
cell proliferation, glucose uptake and invasion in endometrial
cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol.
138:668–675. 2015. View Article : Google Scholar : PubMed/NCBI
|