Introduction
Rhabdomyosarcoma (RMS) is the most common soft
tissue sarcoma in children, and represents 4.5% of all pediatric
malignancies (1–3), with an incidence of 4.5/1,000,000 cases
(4). Although the majority of RMS
cases arise sporadically, RMS may develop as a result of
Li-Fraumeni syndrome (tumor protein p53 mutations),
Beckwith-Wiedemann syndrome (11p15 defects), von Recklinghausen
disease (neurofibromatosis type 1 mutations), cardiofaciocutaneous
syndrome (B-Raf mutations) and Noonan syndrome [rat sarcoma
(RAS)/mitogen-activated protein kinase signaling pathway mutations)
(4). It has been reported that
>1/2 of patients are <10 years old when diagnosed (5,6). RMS has a
bimodal age distribution, with a first peak of incidence between
the ages of 2 and 6 years, and a second peak of incidence between
the ages of 10 and 18 years (1,7). RMS
occurs slightly more often in males than in females, but there is
no such difference in RMS that originates from the head and neck
(H&N) region (5). Nearly 35% of
all RMS tumors develop in the H&N region (5,6). Within
the H&N type, it is clinically useful to divide RMS into three
distinct categories: i) Orbital, ii) superficial and iii)
parameningeal (8). Of all H&N RMS
cases, ~44.0% are localized in the parameningeal region, which
includes the nasal cavity, nasopharynx, paranasal sinuses, middle
ear, infratemporal and pterygopalatine fossa (5), while 25.6% of H&N RMS cases occur in
the orbital region (5).
RMS develops from embryonic mesenchyme with the
potential to differentiate into skeletal muscles (9). RMS may be divided into three major
subtypes: i) Embryonal, ii) alveolar and iii) pleomorphic (10). Embryonal RMS develops as a result of
oncogenic mutations involving the anaplastic lymphoma kinase, RAS,
fibroblast growth factor receptor 4,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha or catenin-cadherin-associated protein beta 1 genes (4). By contrast, alveolar RMS arises as a
consequence of the translocation between the forkhead box protein
O1 transcription factor gene (which is located on chromosome 13)
and either the paired box (PAX)3 transcription factor gene on
chromosome 2 or the PAX7 gene on chromosome 1, and mutations in the
v-Myc avian myelocytomatosis viral oncogene neuroblastoma derived
homolog and the c-Met genes (4). Both
RMS prognosis and treatment depend on the histopathological subtype
(11). Of all patients with H&N
RMS, >1/2 have embryonal RMS, which is a favorable prognostic
factor, whereas the alveolar subtype carries a poorer prognosis
(12). Other RMS negative prognostic
factors include parameningeal localization, presence of distant
metastases, non-radical primary surgical procedure, tumor size
>5 cm, age at diagnosis >10 years old and time to tumor
relapse (1).
Oncological advancements during the last decades
have improved RMS treatment outcomes substantially (13). During the early 20th century, surgery
was the only available therapy for RMS (13,14).
Radical excision was the standard of care, and survival rates were
poor, with survival being 7–70% at that time (9).
Survival of children with RMS has improved
significantly during the past 30 years (11). This improvement may be attributed to
the development of multimodal therapy combining surgery, radio- and
chemotherapy (11). According to the
Intergroup Rhabdomyosarcoma Study (IRS) IV, the 5-year RMS survival
increased from 25% in 1970 to 73% in 2001 (15,16), but
at the same time, almost 15% of children with RMS present with
metastatic disease (group IV), and their prognosis has not improved
significantly over the last 20 years (9). Radiation therapy is generally reserved
for those patients with high-grade, unresectable tumors, and as
adjuvant therapy following resection with cancer-positive surgical
margins (4). Recent advances in
radiation therapy include intensity modulated radiation therapy,
specific computer-modulated radiotherapy and up-front radiotherapy
prior to chemotherapy treatment (9).
In conjuction, chemotherapy is the backbone of
therapy for RMS patients (4). The
three-drug combination of vincristine, dactinomycin and
cyclophosphamide (termed VAC) is considered the standard treatment
for these patients. The D9803 and CWS-91 trials, and the MMT-98 and
IRS IV studies, have all demonstrated that the addition of other
agents, including ifosfamide, etoposide or topotecan, does not
improve clinical outcomes in terms of overall response rate,
overall survival (OS) and progression-free survival, compared with
standard VAC (4,9). Recently, combined therapy based on
multidrug chemotherapy, advanced radiotherapy and more advanced
surgical procedures (including skull base procedures, microsurgery
and endoscopic and reconstructive surgery) have all contributed to
an increase in survival (17). The
formation of study groups such as the IRS (now known as the
Children's Oncology Group), the International Society of Pediatric
Oncology, the German Cooperative Soft Tissue Sarcoma Group and the
Italian Cooperative Group, has improved treatment protocols,
analyzed large-scale cohort outcomes, unified classifications and
defined RMS risk factors (17).
However, despite such advances in H&N RMS
treatment, unfavorable prognostic factors such as parameningeal
location or alveolar histopathological type reduce patient survival
rates significantly (survival rates, 49 and 44%, respectively)
(5). Surgical procedures have had a
notable influence on prognosis, with radical macro- and microscopic
resection correlating strongly with higher survival rates; however,
the treatment of patients with recurrent RMS remains challenging
(4). Local control in the form of
aggressive surgical resection and radiation therapy to sites not
irradiated previously is generally recommended, particularly in
patients with localized recurrence (4). Furthermore, there is no expert consensus
on second- or third-line salvage chemotherapy to be used (4). The choice of chemotherapy is guided by
the history of previous treatment received, and includes irinotecan
with vincristine and temozolomide; topotecan with vincristine and
doxorubicin; vinorelbine with oral cyclophosphamide; and
gemcitabine with docetaxel (4,18–21). For heavily pretreated patients,
monotherapy of temsirolimus, bevacizumab, cediranib or cixutumumab
has been proposed (4).
Materials and methods
Treatment setting
Retrospective analysis was performed on a cohort of
36 pediatric patients diagnosed with H&N RMS who were treated
at the Department of Otorhinolaryngology in the Faculty of Medicine
and Dentistry of the Medical University of Warsaw (Warsaw, Poland)
from January 2000 to December 2013. All patients underwent
treatment with current therapy protocols for RMS, according to the
guidelines of the Polish Pediatric Solid Tumors Group (15,16,22).
Therapy was conducted in collaboration with pediatric oncologists
who supervised the treatment regime. Diagnostic biopsies and
primary tumor resections were performed, as well as secondary
resections after tumor relapse or after not achieving radical
resection during previous surgery performed at other institutions.
The primary surgical objective was tumor resection with negative
margins, and obtaining acceptable cosmetic and functional outcomes.
The histopathological specimens acquired during surgery were
examined as indicated by the Polish Pediatric Solid Tumors Group
(22). After surgery, patients
returned to their referring oncology centers to continue therapy
according to treatment protocol (chemo-/radiotherapy). Patients'
treatment and outcomes were followed thereafter.
The patients' parameters included in the present
analysis were age, gender, localization of the primary tumor,
histopathological subtype, staging of the disease according to the
tumor-node-metastasis (TNM) classification for RMS (23), treatment modalities and outcomes
(Table I).
 | Table I.Patients' characteristics and
outcome. |
Table I.
Patients' characteristics and
outcome.
| Pt no. | Age at diagnosis
(years) | Pre-treatment TNM
staging | Localization | Histological
subtype | Clinical
course | Survival |
|---|
| 1 | 10 | 3 | Parameningeal | ARMS | Local
recurrence | Died 33 months
post-ID |
| 2 | 3 | 4 | Parameningeal | ERMS/ARMS |
| Alive 11 years
post-ID |
| 3 | 4 | 3 | Other H&N | ERMS | Local
recurrence | Died 20 months
post-ID |
| 4 | 2 | 3 | Parameningeal | ERMS |
| Alive 6 years
post-ID |
| 5 | 3 | 4 | Parameningeal | ERMS | No clinical
remission | Died 24 months
post-ID |
| 6 | 4 | 3 | Parameningeal | ERMS |
| Alive 8 years
post-ID |
| 7 | 8 | 3 | Parameningeal | ERMS/ARMS |
| Alive 11 years
post-ID |
| 8 | 13 | 3 | Parameningeal | ERMS | Local
recurrence | Alive 6 years
post-ID |
| 9 | 9 | 3 | Parameningeal | ERMS |
| Alive 9 years
post-ID |
| 10 | 7 | 3 | Parameningeal | ERMS | Local
recurrence | Died 33 months
post-ID |
| 11 | 6 | 3 | Parameningeal | ERMS | Local
recurrence | Died 31 months
post-ID |
| 12 | 4 | 3 | Parameningeal | ERMS |
| Alive 13 years
post-ID |
| 13 | 22 | 3 | Parameningeal | ERMS | Local
recurrence | Died 31 months
post-ID |
| 14 | 1 | 3 | Parameningeal | ERMS | Local
recurrence | Died 37 months
post-ID |
| 15 | 10 | 3 | Parameningeal | ERMS |
| Alive 7 years
post-ID |
| 16 | 2 | 1 | Other H&N | ARMS | Local
recurrence | Alive 8 years
post-ID |
| 17 | 7 | 3 | Parameningeal | ERMS |
| Alive 12 years
post-ID |
| 18 | 4 | 3 | Parameningeal | ERMS | Local
recurrence | Died 27 months
post-ID |
| 19 | 5 | 4 | Parameningeal | ARMS | Local
recurrence | Died 42 months
post-ID |
| 20 | 4 | 3 | Parameningeal | ERMS/ARMS | No clinical
remission | Died 11 months
post-ID |
Ethics statement
All patients or their families provided written
consent to the proposed treatment. The medical data used in the
present study are anonymous and are presented in the form of
cumulative statistical analysis, without photos or personal
information that could allow the identification of each
individual.
Results
Patient inclusion
From January 2000 to December 2013, 36 pediatric
patients with H&N RMS underwent surgical treatment in the
Department of Otorhinolaryngology at the Medical University of
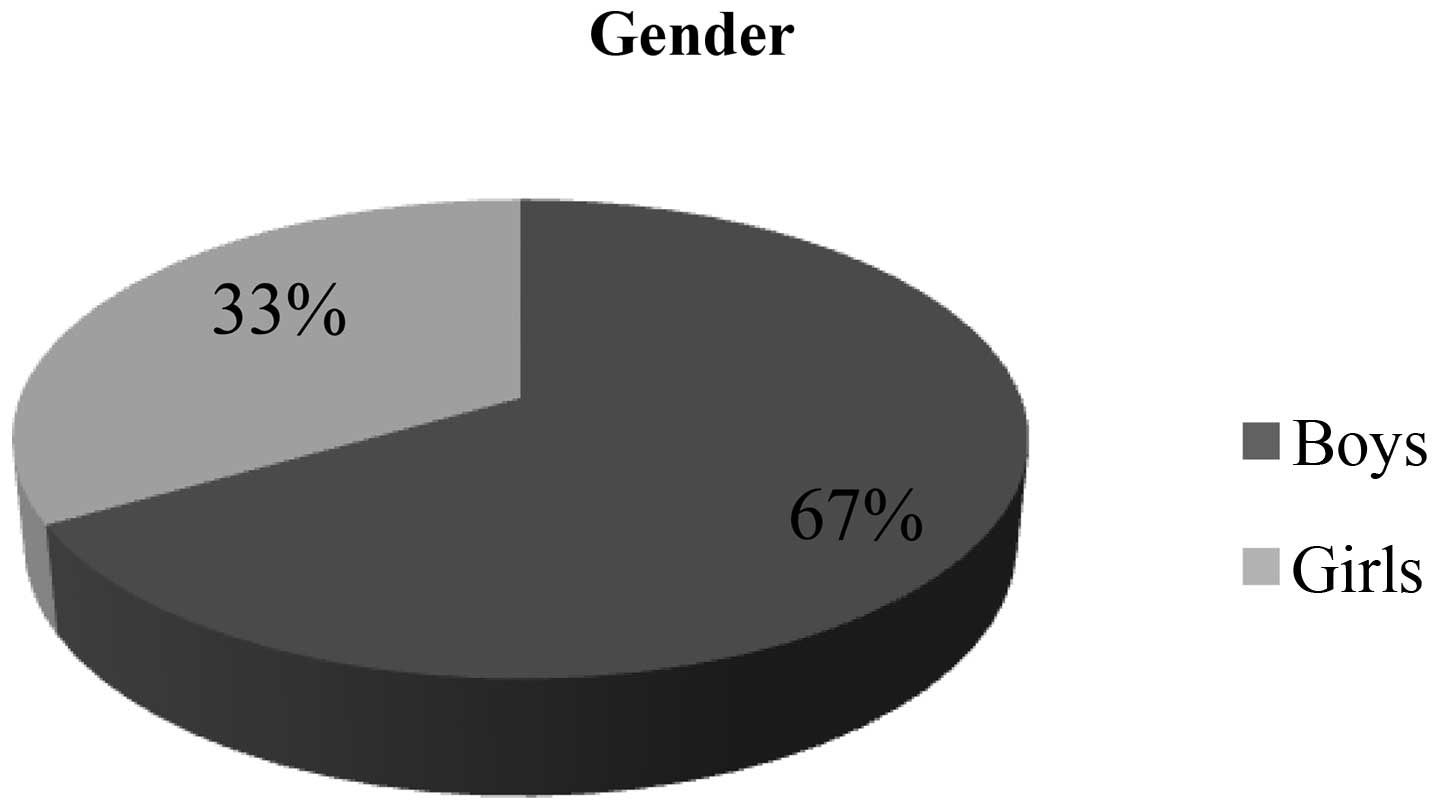
Warsaw. The cohort consisted mostly of males (67%) (Fig. 1). The mean age at the time of
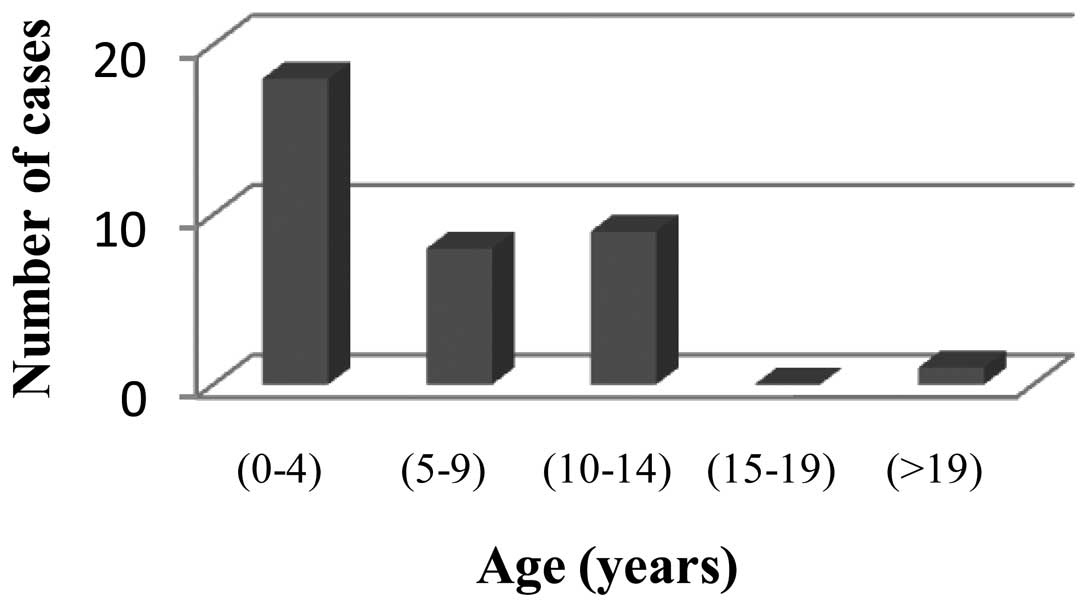
diagnosis was 7 years, and the patient age ranged from 21 months to
22 years (Fig. 2).
Cancer localization
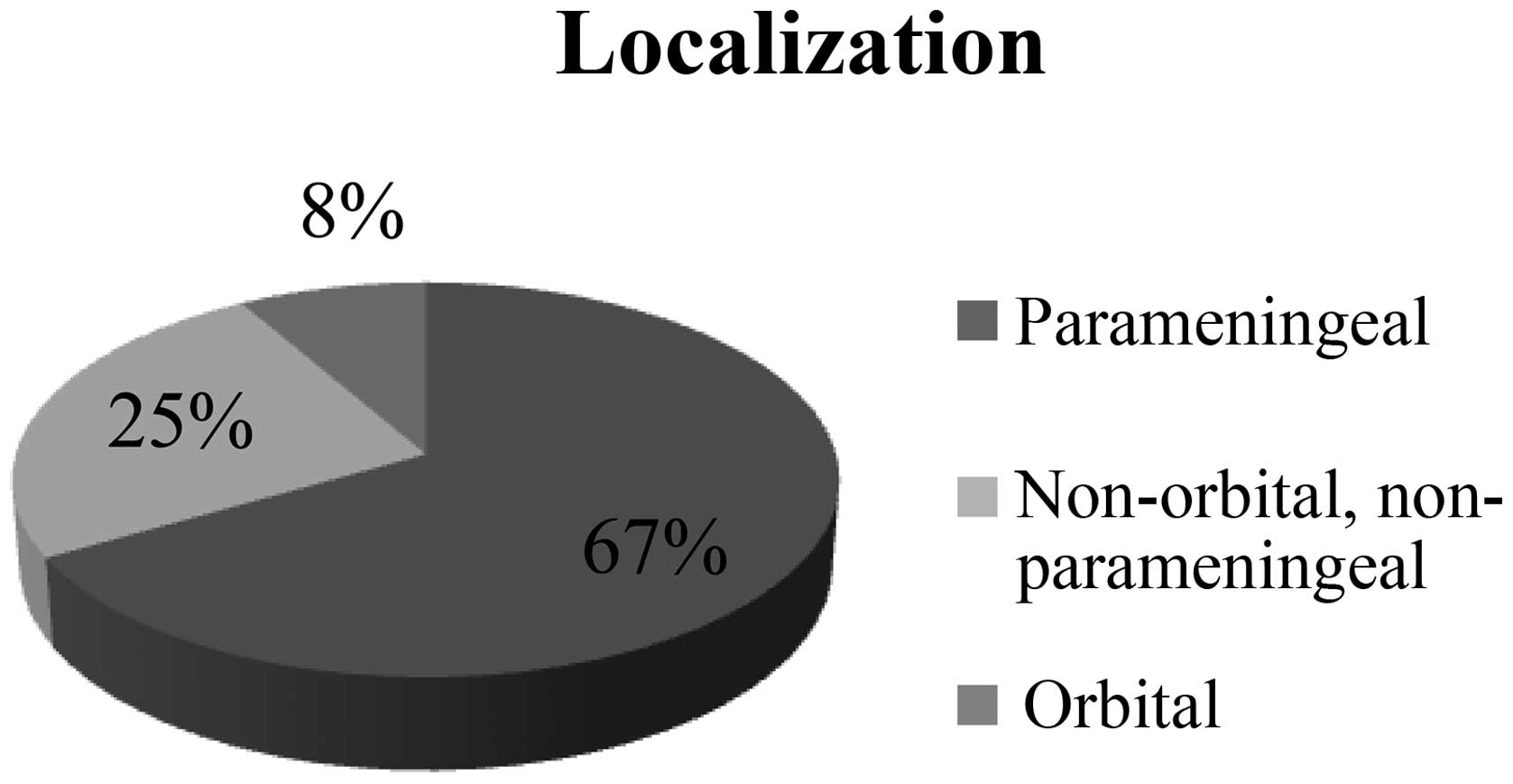
A total of 24 patients (67%) developed RMS in the
parameningeal region. RMS primarily infiltrated the orbital region
in 3 patients (8%). In 9 other cases (25%), the cancer was
localized in different regions of the H&N (Fig. 3).
Cancer histopathology
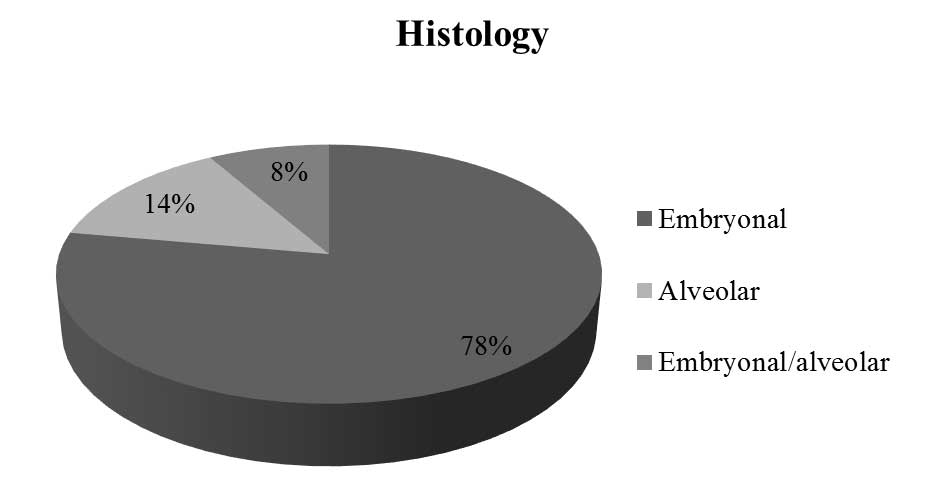
The most commonly observed RMS histopathology was
embryonal subtype (28 cases, 78%).
In 5 patients (14%) alveolar subtype of RMS was
diagnosed, and in other 3 patients (8%), tumor tissue consisted of
both alveolar and embryonal (mixed alveolar/embryonal RMS)
(Fig. 4).
Cancer diagnosis
Only 2 patients had their primary diagnostic
biopsies performed in the Czerniakowski Hospital (Warsaw, Poland),
while 16 patients were referred to the Department of
Otorhinolaryngology at the Czerniakowski Hospital due to cancer
relapse or previous non-radical surgical treatment. Of them, 75%
had previously undergone surgery for RMS. A total of 18 patients
were referred to the Czerniakowski Hospital for primary surgical
treatment following induction chemotherapy, with radiotherapy if
required.
Cancer stage
In the analyzed cohort, the majority of patients
presented with advanced disease. Among the patients referred to the
Department of Otorhinolaryngology at the Czerniakowski Hospital for
primary surgical treatment, 16 had stage III RMS disease according
to TNM classification, 3 patients presented with lung metastasis,
and 1 patient had bone marrow involvement.
Surgical treatment
Lateral, anterior, basal-through and combined
surgical approaches were used depending on the localization and
extent of disease progression. The most common localization was the
skull base, and craniotomy was the most common surgical approach
(20 cases). One patient had a concurrent nasal cavity surgical
approach, while 2 others had a concurrent pharyngotomy. In 9 cases,
access was achieved through rhinotomy with partial maxillectomy, if
required; 2 cases involved a sublabial approach; 1 patient
underwent petrosectomy; and 3 patients required lymphadenectomy due
to disease stage. In other cases, different surgical procedures,
including parotidectomy and resection of tumor of the
parapharyngeal space or submandibular region, were performed if
necessary.
Early complications manifested as 1 case of hematoma
in the postoperative site, which required surgical intervention,
and 1 case of cutaneous flap necrosis. Among the late
complications, there were 3 cases of trismus after orbitozygomatic
craniotomy; rehabilitation relieved symptoms in 2 of these
patients, while 1 patient required surgical treatment. Hypernasal
speech developed in 2 other cases, caused by losses of soft palate
tissue. Salivary fistula ending in the external acoustic meatus
formed in 1 patient. The majority of children suffered from various
grade cosmetic defects, which depended on the extent of surgical
resection. In total, 4 patients had to undergo orbital exenteration
due to tumor infiltration of the orbit and eyeball. In 2 cases, the
orbital content, including the bony structures, was removed, and
microvascular flap reconstruction was performed to close the
postoperative site. One patient underwent reconstructive surgery to
enable the usage of oculi prosthesis. In 1 case, reconstructive
surgery in the orbital region was postponed until achieving
complete clinical remission.
Treatment outcomes
Out of 20 patients treated with primary surgical
resection, 10 patients succumbed to disease, 8 of them due to a
disease relapse, while 2 others did not achieve clinical remission.
Of the remaining patients, 2 were lost on follow-up, and 7 patients
are currently free of disease with relapse-free survival at 6–13
years post-diagnosis. At present, 1 patient is treated due to a
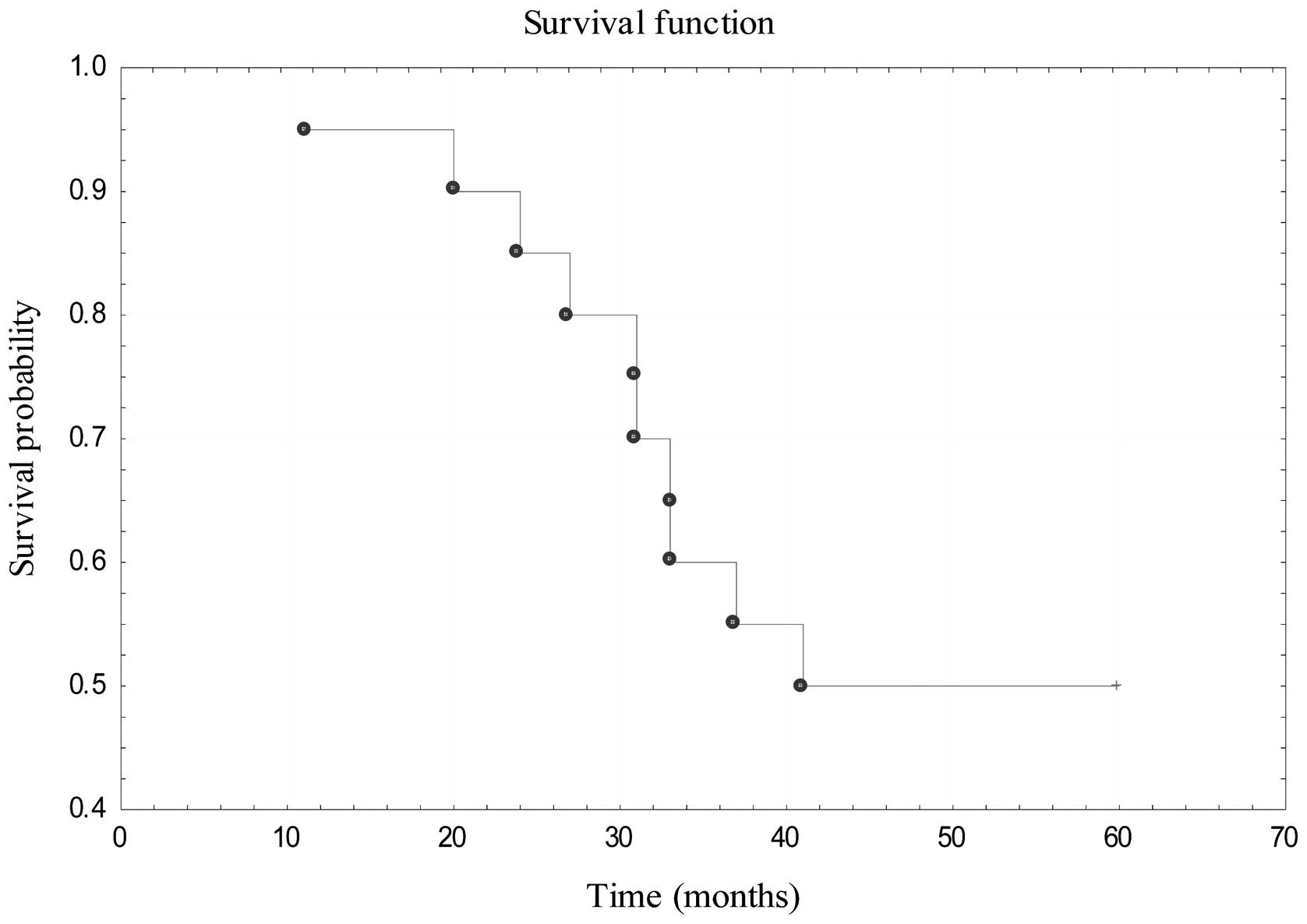
disease relapse. The 5-year OS for patients treated with primary
surgical resection was estimated to be 50% (Fig. 5). In the cohort of 16 patients
referred to the Czerniakowski Hospital to treat RMS relapse, 12
patients succumbed to disease, 1 patient was lost during follow-up
and 3 patients remain in remission 8 years and 3 months on average
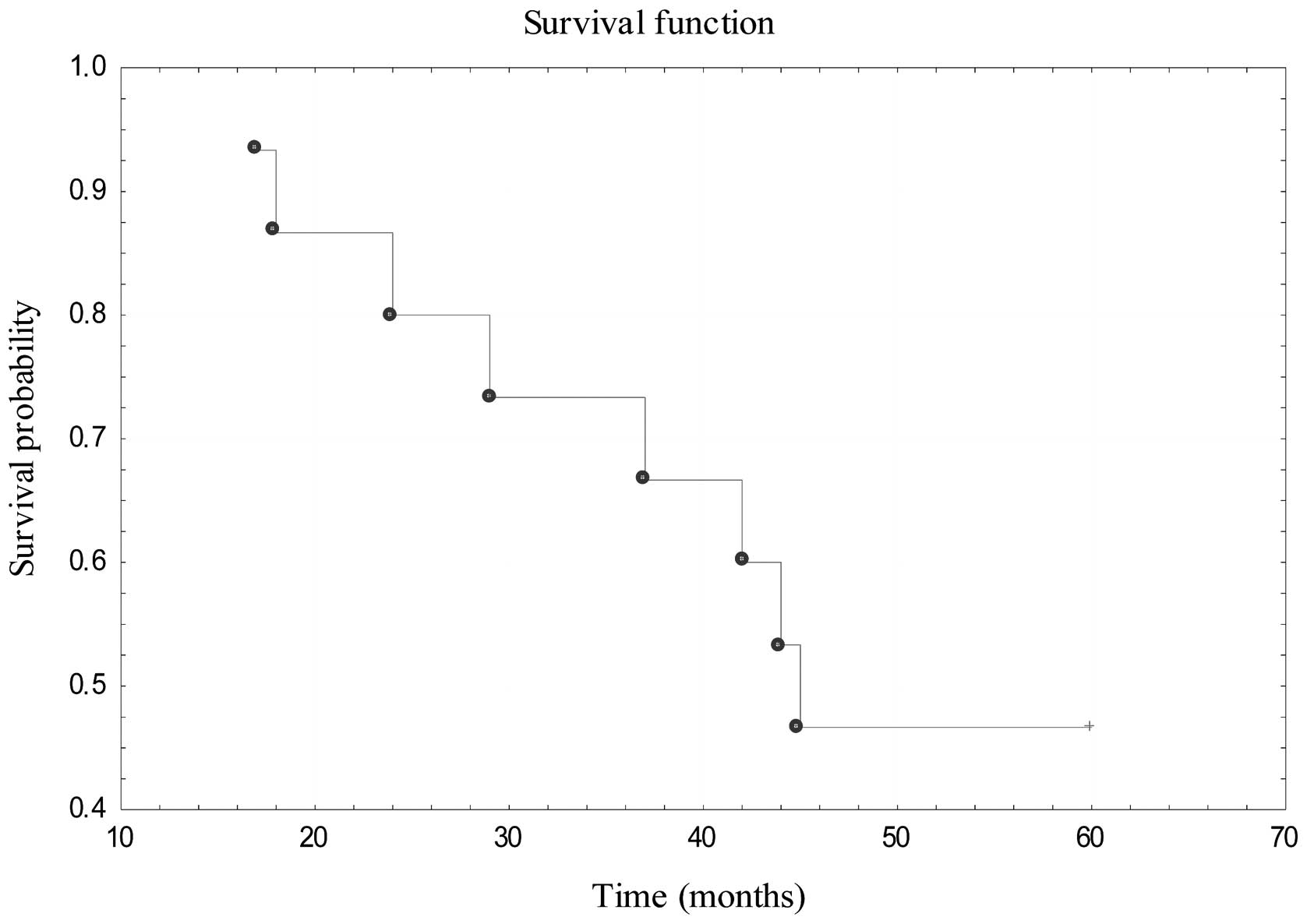
after the procedure. The 5-year OS for those patients was estimated
to be 46.67% (Fig. 6).
Discussion
The treatment strategies for children with H&N
RMS have changed drastically over the last 30 years (11). Until the early 1960s, the gold
standard of treatment was primary surgical resection with possible
irradiation of the postoperative site in cases of non-radical
procedure or infiltration of surgical margins (24). This approach resulted in disturbingly
low survival rates (5–9%) (25,26). The
introduction of the first chemotherapeutics revolutionized soft
tissue sarcoma treatment (25).
Long-standing comparative analysis of numerous subsequent treatment
protocols resulted in defining combination therapy (multidrug
chemotherapy with radiotherapy and surgery) as the gold standard of
treatment (9,13). RMS requires multidisciplinary care,
and all patients with RMS should be placed on protocol-driven
therapy (9). The protocol must define
the timing of chemotherapy, radiation therapy and surgery (9). Whether complete surgical excision is
feasible, it may be performed prior to the initiation of
chemotherapy or upon induction treatment (9). Surgery should be determined by an
experienced pediatric H&N surgeon (9). The necessity for radiotherapy is based
on the site of the primary tumor and the completeness of surgical
excision (9). At present, the surgeon
is a vital member of a multidisciplinary team that plans individual
treatment for every RMS patient (14). Surgical duties include assessing
preoperative classification, performing proper biopsies, planning
and conducting surgical resections with consideration of chemo- and
radiotherapy, and evaluating postoperative classification (14). The essential aim of surgical treatment
is the complete and radical resection of the primary RMS tumor with
an acceptable functional and cosmetic result (27). Radical resection with
histopathologically confirmed cancer-free surgical margins warrants
a favorable prognosis (16,28). Surgical procedures play a major role
in controlling cancer locally, particularly in patients who, due to
their age (<2 years), are disqualified from irradiation
(11,27). Recommendations from different study
groups regarding the range of resection and optimal resection time
varies substantially, and are under current analysis and discussion
(27). It is also clear that when
considering surgical treatment, the patient benefits from treatment
in a large center where there is full access to a broad expertise,
including a full preoperative assessment with diagnostic imaging to
determine if there is bone erosion, intracranial spread or
metastatic disease, which would be contraindications for surgery
(29).
The signs and symptoms of pediatric H&N RMS vary
widely with these sites of involvement (8). Orbital and eyelid RMSs are readily
identified, thereby facilitating early diagnosis and treatment
(8). Children may demonstrate mild
facial asymmetry, nasal congestion and serous otitis media. If RMS
is localized in the paranasal sinuses, extensive growth prior to
identification is possible, thus initiation of treatment may be
delayed, and infiltration of adjacent vital structures may further
complicate the management of these patients (8). Localization of H&N RMS
parameningeally or in the skull base also remains a challenge for
surgeons (11). Sarcomas located
parameningeally grow relatively large without any signs or
symptoms, which delays treatment introduction (8,10).
Therefore, the primary tumor size often makes radical resection
impossible (8). Parameningeal RMS
tends to infiltrate the skull base and expand intracranially, which
worsens prognosis (1). Complicated
skull base anatomy as well as plurality of important vital and
functional anatomical structures limits surgical procedures,
including impeded choice of optimal surgical approach,
impossibility to resect the tumor en block, risk of losing
important functional performance, inability to achieve acceptable
cosmetic effect and difficulties at maintaining cancer-free
surgical margins (30). All these
properties may cause poor prognosis, and explain the necessity for
more aggressive systemic treatment in patients with parameningeal
RMS (1). Surgical treatment of
malignant neoplasia originating in the skull base requires great
experience and collaboration of different specialists, including
H&N surgeons, neurosurgeons, oral and maxillofacial surgeons,
and plastic surgeons (14).
Therefore, only selected medical centers offer such treatment.
Primary RMS site in the orbital region and other more accessible
regions of the H&N provides favorable prognosis (with exclusion
of parameningeal location, as aforementioned), since it involves
earlier symptoms occurrence, facilitates decision-making regarding
implementation of further diagnostic imaging [such as computed
tomography (CT) and magnetic resonance imaging (MRI)] and decreases
time to surgery (30).
Children with localized H&N RMS are able to
undergo complete surgical resection with low long-term surgical
morbidity (31). It was reported
that, by undergoing complete surgical resection, these patients
avoid radiotherapy and its long-term complications, with no
compromise in survival (31).
Patients in whom radical resection with healthy tissues margin is
impossible should receive neoadjuvant chemotherapy (27,32). A
postponed procedure must be performed upon achieving satisfactory
response to chemotherapy (32).
Second-look surgery must be undertaken to resect viable tumors
after the administration of definitive local therapy (1). In the IRS III, patients underwent
induction chemotherapy and radiotherapy prior to second-look
surgery (33,34). Surgery was delayed as much as 20
weeks. In that trial, 52% of patients undergoing secondary
operations, upon achieving only a minor response, were converted to
a complete remission status by surgery (33,34).
According to the IRS III, >1/2 of patients with
localized disease had previously undergone only subtotal resection
or biopsy as a surgical treatment (33). Patients classified after surgical
intervention in this group were at higher risk of cancer relapse
than patients with completely resected localized tumor or tumor
grossly resected with the evidence of microscopic residual disease
only (33). Since the majority of
treatment failures apply to patients with relapse in the primary
site, radiotherapy plays a substantial role in local RMS treatment.
In addition, involvement of lymph nodes at primary diagnosis
predicts a higher risk of local and distant treatment failure
compared with children with negative lymph nodes (35). Smith et al (28) suggested the use of radiation therapy
for every patient with confirmed microscopically cancer remnants,
affection of regional lymph nodes and RMS histopathological subtype
linked with unfavorable prognosis. Furthermore, according to a
report by the Polish Pediatric Solid Tumors Group, radiotherapy
should be used in every patient with parameningeal RMS (15).
Since survival rates in patients with RMS have
improved with more effective treatment regimens, more long-term
treatment-associated complications have been described (8). The most frequent difficulties are
associated with radiation exposure, and occasional problems result
from the cytotoxic effects of chemotherapy (8). The most frequently identified
difficulties involve speech and memory skills (8). Other common complications include facial
and statural growth retardation, neuroendocrine dysfunction, visual
or orbital difficulties, hearing loss, intellectual and academic
delays, and development of secondary malignancies (8). In RMS survivors, secondary leukemias
were linked to etoposide therapy, and bone sarcomas in the sites of
radiation treatment have been reported (35). In terms of otorhinolaryngology and
H&N surgery long-term care, H&N RMS treatment may result in
dentofacial abnormalities that affect the patient's quality of life
(29). In patients with H&N RMS
who were followed at the Dental Service of the Memorial Sloan
Kettering Cancer Center (New York City, NY, USA) and were alive and
free of disease with ≥5-year follow-up, multiple clinical and
radiographic dentofacial abnormalities were observed, including
facial asymmetry, enamel defects, bony hypoplasia, trismus,
velopharyngeal insufficiency, tooth and root agenesis, malformed or
missing teeth, microdontia, maxillary and mandibular hypoplasia,
disturbance in root development, poor tooth development, root
stunting and xerostomia (8,29). The care of the long-term survivor
requires a multidisciplinary approach, including early involvement
of the dental specialists (29).
Furthermore, behavioral problems, including hyperactivity,
depression, immaturity and suicidal behaviors, have also been
observed in certain RMS patients (8).
The majority of RMS complications are encountered within the first
10 years after radiotherapy and chemotherapy (8).
A limited number of studies in the literature
comment on the surgical treatment of pediatric H&N RMS. The
majority of publications concern population studies,
epidemiological analyses or multinational collaborations
summarizing the efficacy of combined therapy treatment according to
differing treatment protocols (13).
Moretti et al (36), from the Otorhinolaryngological Unit at
the Clinical Hospital of The University of São Paulo Medical School
(São Paulo, Brazil), described a group of 24 patients suffering
from RMS of the H&N. Similar to the present study, the
majority of cases were parameningeal RMS, but only 4 patients
received surgical treatment as a part of combined therapy, and in 2
cases, resection was not radical. Fyrmpas et al (37) reported 14 patients with RMS of the
nasal area and paranasal sinuses. Among them, 6 underwent primary
surgical resections with subsequent chemotherapy and, if required,
radiotherapy. Surgical approaches included combined approach
(endoscopic and external) in 3 cases, sublabial approach
(mid-facial degloving) in 2 cases and intranasal approach in 1
case. Resection was not radical in 2 cases. Only 1 patient suffered
from progressive disease and consequently succumbed to disease
(37).
In the current study, a heterogenous patient cohort
that was treated at Department of Otorhinolaryngology at the
Czerniakowski Hospital is presented. Among these patients, ~1/2 of
them were referred due to previous treatment failure for salvage
surgery. A significant number of tumors were parameningeal, which
is an anatomical space difficult for surgical access and correlated
with an unfavorable long-term prognosis. Out of 12 patients with a
favorable prognosis due to tumor localization, 10 were operated in
the Department of Otorhinolaryngology at the Czerniakowski Hospital
due to cancer relapse, 9 of whom had previously undergone surgical
treatment in different medical centers. The majority of patients
(18 in total) had locally advanced cancer after induction
chemotherapy and, possibly, radiotherapy. In the majority of cases,
residual tumor was localized in the surrounding area of the
infratemporal and pterygopalatine fossa; thus, the preferable
surgical approach orbitozygomatic craniotomy. Skin incision was
performed in line with the coronal suture, and subsequently
lengthened longitudinally down along the anterior border of the
auricle. This minimized postoperative scarring. Revelation of skin,
fascial layer, temporal muscle and zygomatic arch excision provided
excellent access to the infratemporal fossa, which allowed the
identification and occasional preservation of the trigeminal nerve
and maxillary artery. Furthermore, it eased complete resection of
the pterygoid muscles with their bone insertion (pterygoid
processes, temporomandibular joint and part of the mandibular
ramus) in cases of muscle infiltration. The unilateral resection of
the temporomandibular joint caused minor face asymmetry, and led to
trismus in 3 cases, 1 of who required surgical correction while 2
others only underwent rehabilitation.
In 9 patients with RMS localized in the limits of
the nasopharynx and/or maxillo-ethmoidal complex, the preferred
surgical approach was lateral rhinotomy with partial maxillectomy.
This approach allowed access into the nasal cavity structures, and
following excision of the medial part of the maxilla with frontal
process, it enabled the revision of the entire maxillary sinus.
Furthermore, acceptable exposition of the ethmoid bone granted
insight into possible infiltration towards the base of the anterior
crania fossa.
Independently from the surgical approach,
macroscopic assessment of the tumor infiltration margins was
difficult, and intraoperative histopathological examination of the
surgical margins previously altered by radiochemotherapy was not
always evident. Due to those limitations, the scope of operation
relied mainly on the result from imaging studies (CT and MRI).
Adequate assessment of tissue material acquired during surgery
required specific experience and often involved immunohistochemical
staining; thus, the reference center evaluated and verified every
histopathological result. After surgery was performed at the
Department of Otorhinolaryngology at the Czerniakowski Hospital,
patients continued therapy according to treatment protocols at
their respective referring oncology centers. Patients were
postoperatively monitored, due to collaboration between the
Department of Otorhinolaryngology of the Faculty of Medicine and
Dentistry of the Medical University of Warsaw and the
aforementioned oncology centers providing subsequent therapy.
However, not all patients reported for follow-up, mainly due to the
distance from their place of residence and ongoing systemic
therapy. The relatively low survival rate in the group of patients
operated at the primary treatment stage may result from the
advanced stage of the disease at the moment of diagnosis (95% of
patients exhibited stage ≥3) and unfavorable tumor localization
(86% of the group). In the group operated due to a disease relapse,
the tumor was localized mainly in a favorable site, thus resulting
in a relatively good survival rate.
It should be emphasized that management of H&N
RMS must be interdisciplinary at the time of diagnosis. The
surgical approach should be based on the individual characteristics
of each patient. It is important to explain the possible
complications of surgery, such as functional losses, cosmetic
defects and permanent limitations, to parents and to the child, if
necessary. In case of parameningeal RMS, obtaining negative
surgical margins is often impossible; therefore, subsequent
adjuvant therapy is indispensable. Experienced pathologists should
examine both tumor en block and intraoperative tissue
samples to assess the effectiveness of the surgical treatment.
Surgeons should provide radiologists evaluating postsurgical CT or
MRI scans with specific information regarding the surgical
procedure and descriptions on how the operative site was
reconstructed, as close collaboration in this field prevents
misdiagnosis of disease relapse, which could lead to needless
reoperations. 18F-fluorodeoxyglucose positron emission
tomography/CT should become a part of the standard diagnostic
algorithm for children diagnosed with RMS, although dedicated bone
imaging with 99Tc-methylene diphosphonate bone imaging
may not be necessary in these patients. To reduce the radiation
exposure of pediatric RMS patients to a minimum, as demanded by the
As Low As Reasonably Achievable principle (38,39), the
number of imaging procedures should be as low as possible, since
children are 10–15 times more sensitive to radiation than adults
(40). Furthermore, children with RMS
are often under surveillance for numerous years with serial CT
imaging; thus, the development of novel imaging approaches to
reduce radiation exposure is required (41). The role of second-look surgery after
chemotherapy and radiation remains unknown, and trials must be
designed in order to develop guidelines for such patients. It is
likely that molecular biology research exploring the basic genetic
mechanisms of RMS tumorigenesis and metastases development in the
future will lead to the development of novel treatment strategies.
Immunotherapy, targeted therapy and anti-angiogenic agents are
potential new therapy modalities in pediatric H&N RMS (35).
Acknowledgements
The present study was supported by the Medical
University of Warsaw (Warsaw, Poland) statutory funding and the
Military Institute of Medicine (Warsaw, Poland) statutory funding.
Treatment costs were covered by the Polish National Health Fund
(Warsaw, Poland). A.M.C. was supported by a SONATA grant from the
National Science Centre (NCN; Warsaw, Poland; grant no.
UMO-2012/05/D/NZ5/01844) and a WIM intramural grant from the
Military Institute of Medicine [grant no. 1/8863 (355)]. A.M.C and
C.S. were supported by an OPUS grant from the NCN (grant no.
UMO-2011/01/B/NZ5/02822).
References
|
1
|
Dasgupta R and Rodeberg DA: Update on
rhabdomyosarcoma. Semin Pediatr Surg. 21:68–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huh WW and Skapek SX: Childhood
rhabdomyosarcoma: New insight on biology and treatment. Curr Oncol
Rep. 12:402–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leaphart C and Rodeberg D: Pediatric
surgical oncology: Management of rhabdomyosarcoma. Surg Oncol.
16:173–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ray A and Huh WW: Current state-of-the-art
systemic therapy for pediatric soft tissue sarcomas. Curr Oncol
Rep. 14:311–319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turner JH and Richmon JD: Head and neck
rhabdomyosarcoma: A critical analysis of population-based incidence
and survival data. Otolaryngol Head Neck Surg. 145:967–973. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pappo AS, Shapiro DN, Crist WM and Maurer
HM: Biology and therapy of pediatric rhabdomyosarcoma. J Clin
Oncol. 13:2123–2139. 1995.PubMed/NCBI
|
|
7
|
Miller RW, Young JL Jr and Novakovic B:
Childhood cancer. Cancer. 75(Suppl 1): S395–S405. 1995. View Article : Google Scholar
|
|
8
|
Holsinger FC, Weeks BH, Hicks MJ and
Friedman EM: Contemporary concepts in the management of pediatric
rhabdomyosarcoma. Curr Opin Otolaryngol Head Neck Surg. 10:91–96.
2002. View Article : Google Scholar
|
|
9
|
Hayes-Jordan A and Andrassy R:
Rhabdomyosarcoma in children. Curr Opin Pediatr. 21:373–378. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahmed AA and Tsokos M: Sinonasal
rhabdomyosarcoma in children and young adults. Int J Surg Pathol.
15:160–165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stevens MC: Treatment for childhood
rhabdomyosarcoma: The cost of cure. Lancet Oncol. 6:77–84. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wachtel M, Dettling M, Koscielniak E,
Stegmaier S, Treuner J, Simon-Klingenstein K, Bühlmann P, Niggli FK
and Schäfer BW: Gene expression signatures identify
rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23)
translocation fusing PAX3 to NCOA1. Cancer Res. 64:5539–5545. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gradoni P, Giordano D, Oretti G, Fantoni M
and Ferri T: The role of surgery in children with head and neck
rhabdomyosarcoma and Ewing's sarcoma. Surg Oncol. 19:e103–109.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodeberg DA, Paidas CN, Lobe TL, Brown K,
Andrassy RJ, Crist WM and Wiener ES: Surgical principles for
children/adolescents with newly diagnosed rhabdomyosarcoma: A
report from the Soft Tissue Sarcoma Committee of the Children's
Oncology Group. Sarcoma. 6:111–122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zalewska-Szewczyk B, Kazanowska B,
Mlynarski W, Dłużniewska A, Drożyńska E, Kurylak A, Nurzyńska-Flak
J, Rybczyńska A, Pietniczka-Załęska M, Rychłowska-Pruszyńska M, et
al: The analysis of clinical course and outcome of soft tissue
sarcoma in parameningeal localization in children treated according
to the CWS 96 protocol - a report of the Polish Paediatric Solid
Tumours Group. Wspolczesna Onkol. 12:116–120. 2008.
|
|
16
|
Kazanowska B, Reich A, Reich M, Balcerska
A, Balwierz W, Bodalski J, Dłuzniewska A, Drozyńska E, Katski K,
Kijowski J, et al: Remaining problems and controversies in the
management of childhood head and neck soft tissue sarcomas:
Retrospective (national) Multicenter Study of the Polish Pediatric
Solid Tumors Group. Pediatr Hematol Oncol. 21:349–362. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Radzikowska J, Kukwa W, Kukwa A, Czarnecka
A and Krzeski A: Rhabdomyosarcoma of the head and neck in children.
Contemp Oncol (Pozn). 19:98–107. 2015.PubMed/NCBI
|
|
18
|
McNall-Knapp RY, Williams CN, Reeves EN,
Heideman RL and Meyer WH: Extended phase I evaluation of
vincristine, irinotecan, temozolomide, and antibiotic in children
with refractory solid tumors. Pediatr Blood Cancer. 54:909–915.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wagner LM, Perentesis JP, Reid JM, Ames
MM, Safgren SL, Nelson MD Jr, Ingle AM, Blaney SM and Adamson PC:
Phase I trial of two schedules of vincristine, oral irinotecan, and
temozolomide (VOIT) for children with relapsed or refractory solid
tumors: A Children's Oncology Group phase I consortium study.
Pediatr Blood Cancer. 54:538–545. 2010.PubMed/NCBI
|
|
20
|
Casanova M, Ferrari A, Bisogno G, Merks
JHM, De Salvo GL, Meazza C, Tettoni K, Provenzi M, Mazzarino I and
Carli M: Vinorelbine and low-dose cyclophosphamide in the treatment
of pediatric sarcomas: Pilot study for the upcoming European
Rhabdomyosarcoma Protocol. Cancer. 101:1664–1671. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rapkin L, Qayed M, Brill P, Martin M,
Clark D, George BA, Olson TA, Wasilewski-Masker K, Alazraki A and
Katzenstein HM: Gemcitabine and docetaxel (GEMDOX) for the
treatment of relapsed and refractory pediatric sarcomas. Pediatr
Blood Cancer. 59:854–858. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kazanowska B and Chybicka A: Nowotwory
tkanek miękkichZalecenia Postępowania Diagnostyczno -
Terapeutycznego W Nowotworach Złośliwych. Krzakowski M: 1st. Via
Medica Gdansk; Gdańsk: pp. 868–893. 2012, (In Polish).
|
|
23
|
Lawrence W Jr, Anderson JR, Gehan EA and
Maurer H: Children's Cancer Study Group: Pretreatment TNM staging
of childhood rhabdomyosarcoma: A report of the Intergroup
Rhabdomyosarcoma Study Group Pediatric Oncology Group. Cancer.
80:1165–1170. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dito WR and Batsakis JG: Rhabdomyosarcoma
of the head and neck. An appraisal of the biologic behavior in 170
cases. Arch Surg. 84:582–588. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Healy GB, Upton J, Black PM and Ferraro N:
The Role of surgery in rhabdomyosarcoma of the head and neck in
children. Arch Otolaryngol Head Neck Surg. 117:1185–1188. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Masson JK and Soule EH: Embryonal
rhabdomyosarcoma of head and neck. Report on eighty-eight cases. Am
J Surg. 110:585–591. 1965.
|
|
27
|
Gosiengfiao Y, Reichek J and Walterhouse
D: What is new in rhabdomyosarcoma management in children? Pediatr
Drugs. 14:389–400. 2012. View Article : Google Scholar
|
|
28
|
Smith LM, Anderson JR, Qualman SJ, Crist
WM, Paidas CN, Teot LA, Pappo AS, Link MP, Grier HE, Wiener ES, et
al: Which patients with microscopic disease and rhabdomyosarcoma
experience relapse after therapy? A report from the soft tissue
sarcoma committee of the children's oncology group. J Clin Oncol.
19:4058–4064. 2001.PubMed/NCBI
|
|
29
|
Estilo CL, Huryn JM, Kraus DH, Sklar CA,
Wexler LH, Wolden SL and Zlotolow IM: Effects of therapy on
dentofacial development in long-term survivors of head and neck
rhabdomyosarcoma: The memorial sloan-kettering cancer center
experience. J Pediatr Hematol Oncol 25: 215–222, 2003.
Holsinger
FC, Weeks BH, Hicks MJ and Friedman EM: Contemporary concepts in
the management of pediatric rhabdomyosarcoma. Current Opinion in
Otolaryngology & Head and Neck Surgery. 10:91–96. 2002.
|
|
30
|
Dickson PV and Davidoff AM: Malignant
neoplasms of the head and neck. Semin Pediatr Surg. 15:92–98. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Daya H, Chan HS, Sirkin W and Forte V:
Pediatric rhabdomyosarcoma of the head and neck: Is there a place
for surgical management? Arch Otolaryngol Head Neck Surg.
126:468–472. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walterhouse D and Watson A: Optimal
management strategies for rhabdomyosarcoma in children. Paediatr
Drugs. 9:391–400. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wharam MD, Meza J, Anderson J, Breneman
JC, Donaldson SS, Fitzgerald TJ, Michalski J, Teot LA, Wiener ES
and Meyer WH: Failure pattern and factors predictive of local
failure in rhabdomyosarcoma: A report of group III patients on the
third Intergroup Rhabdomyosarcoma Study. J Clin Oncol.
22:1902–1908. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crist W, Gehan EA, Ragab AH, Dickman PS,
Donaldson SS, Fryer C, Hammond D, Hays DM, Herrmann J, Heyn R, et
al: The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol.
13:610–630. 1995.PubMed/NCBI
|
|
35
|
Dagher R and Helman L: Rhabdomyosarcoma:
An overview. Oncologist. 4:34–44. 1999.PubMed/NCBI
|
|
36
|
Moretti G, Guimarães R, Oliveira KM,
Sanjar F and Voegels RL: Rhabdomyosarcoma of the head and neck: 24
cases and literature review. Braz J Otorhinolaryngol. 76:533–537.
2010.(In English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fyrmpas G, Wurm J, Athanassiadou F,
Papageorgiou T, Beck JD, Iro H and Constantinidis J: Management of
paediatric sinonasal rhabdomyosarcoma. J Laryngol Otol.
123:990–996. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McCarville MB: Imaging techniques used in
the diagnosis of pediatric tumorsPediatric Malignancies: Pathology
and Imaging. Parham DM, Khoury JD and McCarville MB: Springer
Science and Business Media; New York, NY: pp. 7–18. 2015
|
|
39
|
Brody AS, Frush DP, Huda W and Brent RL:
American Academy of Pediatrics Section on Radiology. Radiation risk
to children from computed tomography. Pediatrics. 120:677–682.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brenner D, Elliston C, Hall E and Berdon
W: Estimated risks of radiation-induced fatal cancer from pediatric
CT. AJR Am J Roentgenol. 176:289–296. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Walter F, Czernin J, Hall T,
Allen-Auerbach M, Walter MA, Dunkelmann S and Federman N: Is there
a need for dedicated bone imaging in addition to 18F-FDG PET/CT
imaging in pediatric sarcoma patients? J Pediatr Hematol Oncol.
34:131–136. 2012. View Article : Google Scholar : PubMed/NCBI
|