Introduction
Colorectal cancer (CRC) is a malignant disease that
originates from colorectal epithelial cells (1), and is one of the most commonly diagnosed
malignancies in the world (2).
Genetic events such as rare or high-penetrance variants in the CRC
susceptibility genes and DNA mismatch repair genes have been
demonstrated to be important in the etiology of both sporadic and
familial CRC (3). However, the
carcinogenic mechanisms can only be explained in <6% of all CRC
cases (3). Therefore, there are still
numerous genetic events associated with dysregulation or mutations
in CRC patients that remain to be determined.
Long non-coding RNAs (lncRNAs) are a class of
transcripts longer than 200 nucleotides without coding potential to
be translated into proteins (4).
Numerous studies have revealed that lncRNAs were frequently
dysregulated in various diseases and had multiple functions in a
wide range of pathological processes, including apoptosis,
proliferation and invasion-metastasis of malignant tumors (5,6). For
instance, colorectal cancer associated transcript 2 was identified
by Ling et al as a lncRNA mapping to 8q24 that promoted
metastatic progression in CRC (7).
Another lncRNA, homeobox transcript antisense intergenic RNA
(HOTAIR) has been determined to exhibit higher levels in the plasma
of CRC patients than in healthy controls, and its overexpression
predicted unfavorable prognosis (8).
The association between prognosis of CRC patients and expression of
prostate cancer associated transcript 1 and metastasis associated
lung adenocarcinoma transcript 1 has also been explored (9,10). The
above studies indicated that lncRNAs are important in the
regulation of carcinogenesis in CRC, and that lncRNAs could be used
as biomarkers of diagnosis and prognosis, and could be potential
therapy targets for novel antitumor drugs. However, the function
and dysregulation of lncRNAs in CRC still remain to be explored.
Thus, the identification of differential lncRNA profiles in CRC is
required.
Array-based expression profiles regarding CRC have
been established (11). However,
these previous array-based profiles only compared protein coding
RNAs and somatic genomic alteration profiles, such as somatic copy
number alteration (11). In addition,
those array-based data contained extensive information about lncRNA
profiles, which, however, were not explored, since lncRNAs were not
the intended targets of study of the original array design.
Microarray probes thus can be re-annotated for interrogating lncRNA
expression (12), and it is possible
to build CRC lncRNA profiles based on those published array-based
datasets. The present study aimed to build CRC lncRNA profiles from
published Affymetrix Human Exon 1.0 ST arrays (Affymetrix, Inc.,
Santa Clara, CA, USA). The differential lncRNA expression profiles
from three CRC-related datasets were explored, including 44 tumor
samples, and the results were validated in another CRC array-based
dataset that comprised 166 CRC patients. The expression of those
lncRNAs that were significantly associated with prognosis was
further determined in CRC cells and cancer tissues.
Materials and methods
Microarray data
E-GEOD-31737 consisted of 20 paired CRC and adjacent
normal tissues; E-MATB-829 contained 14 paired tissues; and
E-GEOD-24550 included 166 samples from CRC patients with detailed
information about overall survival (OS) time. Data were downloaded
from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/). The Affymetrix
colon cancer dataset was downloaded from http://www.affymetrix.com/support/technical/sample_data/exon_array_data.affx
and comprised 10 paired CRC tissues. All raw CEL files of the above
datasets were obtained for exploring underlying lncRNAs.
E-GEOD-31737, E-MATB-829 and the Affymetrix datasets were used as
experimental sets to identify differentially expressed lncRNAs in
CRC, while E-GEOD-24550 was used as a validation set to screen
lncRNAs associated with OS rates.
Re-annotation of Affymetrix Exon 1.0
ST Array lncRNA probes
The microarray data were preprocessed with a
preprocessing program and re-annotated with Affymetrix CEL file
(Affymetrix, Inc.) from noncoder (http://noncoder.mpi-bn.mpg.de/#) (13). The data were normalized by MAS5.0
(included in the tools of noncoder) prior to lncRNAs annotation.
The alignment transcript cluster was filtered by the following
steps: i) Genes with >3 probes were retained; and ii) probes
were mapped to known lncRNAs in NONCODE v3.0 (which was included in
the tools of noncoder) (14). Paired
t-test analysis was used to obtain probe sets whose magnitude of
change in expression of lncRNAs between CRC tissue and adjacent
normal tissues was significant. Genes with adjusted P<0.05 and
fold-change either greater or lower than 2-fold were considered to
be differentially expressed. The differentially expressed genes of
each dataset were plotted in R version 3.1.1 (https://www.r-project.org/) using the ‘pheatmap’
package, which can be accessed via the above link.
Identifying differential lncRNAs
associated with OS in CRC
E-GEOD-24550 was used to screen differential lncRNAs
associated with the OS rate. The transcript clusters that matched
>3 probes were retained, and the differential lncRNAs that were
determined in the E-GEOD-31737, E-MATB-829 and Affymetrix datasets
were further selected.
E-GEOD-24550 provided prognostic
information for each patient
Statistical analyses were also performed using R
package version 3.1.1. Time-dependent receiver operating curve
(ROC) was applied to calculate the best cutoff values for survival
analysis (15). The lncRNAs were
further examined with the Kaplan-Meier log-rank test using the R
survival package. The survival curves were plotted with R
software.
Tissue samples collection, cell
culture and total RNA extraction
Cancerous and paired non-cancerous tissues were
consecutively collected from 20 patients with CRC who underwent
curative resection of the tumor at Beijing Chaoyang Hospital
(Beijing, China) from January 1, 2013 to March 2, 2013. These
patients were not subjected to any preoperative chemotherapy or
radiotherapy. The freshly dissected tissues were immediately frozen
in liquid nitrogen and stored at −80°C. RNA was extracted from the
CRC samples using the mirVana™ PARIS™ kit for protein and RNA
isolation (Ambion; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's protocol. The study protocol
was approved by the Medical Ethics Committee of Chaoyang Hospital
of Capital Medical University (Beijing, China), and informed
consent was obtained from each subject.
The RKO, HCT116 and HT29 cell lines were purchased
from the American Type Culture Collection (Manassas, VA, USA),
while the SW620 and SW480 cell lines were obtained from the Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
Cells were cultured with complete Dulbeccos modified Eagle medium
(Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA). Human umbilical vein endothelial cells (HUVECs)
were purchased from ScienCell Research Laboratories, Inc.
(Carlsbad, CA, USA). Total RNA from the cell lines was extracted
with TRIzol (Thermo Fisher Scientific, Inc.). Briefly, cell pellets
were resuspended in 500 µl TRIzol, and subsequently, chloroform and
isopropanol were added for RNA extraction. Finally, total RNA was
dissolved with RNase-free water. The concentration of RNA was
determined using the NanoDrop 2000 spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc., Wilmington, DE,
USA).
Reverse transcription (RT)-polymerase
chain reaction (PCR) and RT-quantitative (q) PCR
A total of 1 µg RNA from each of the samples or cell
lines was individually reversed transcribed to synthesize
complementary DNA using TranScript II Two-Step RT-PCR SuperMix
(Beijing Transgen Biotech Co., Ltd., Beijing, China), and DNase I
(Beijing Transgen Biotech Co., Ltd.) was used to degrade the
genomic DNA. The primers for VPS9 domain containing 1
(VPS9D1)-antisense (AS) 1 were as follows: Forward primer,
5′-GCTTCAGGCGTGTTTTCCC-3′ and reverse primer,
5′-CCCAGAGGCCTTTTCCGTT-3′. The primers for family with sequence
similarity 83 member H (FAM83H)-AS1 were as follows: Forward
primer, 5′-CCGAGCGAGGATATTGAG-3′ and reverse primer,
5′-AACACCAACATCAGAGACC-3′. The primers for glyceraldehyde
3-phosphate dehydrogenase (GAPDH) were as follows: Forward primer,
5′-AATCCCATCACCATCTTCCA-3′ and reverse primer,
5′-TGGACTCCACGACGTACTCA-3′. RT-qPCR was performed using a SYBR
Premix Ex Taq™ II kit (Takara Biotechnology Co., Ltd.,
Dalian, China) with the corresponding sense and antisense primers.
The RT-qPCR protocol consisted of an initial denaturation step at
95°C for 10 min, followed by 40 cycles of 95°C for 10 sec, 60°C for
34 sec and 72°C for 10 sec. The quantification cycle (Cq) was
determined, and the relative lncRNA expression was calculated using
the 2−∆Cq method (16)described by the manufacturer, using
GAPDH as the calibrator gene. The diagram of 2−∆Cq
lncRNA relative expression was plot, and the difference between
2−∆Cq lncRNA relative expression in normal and cancer
tissues was calculated with a two-tailed non-parametric
Mann-Whitney U test using GraphPad Prism 6.0 software (GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Differentially expressed lncRNAs in
CRC
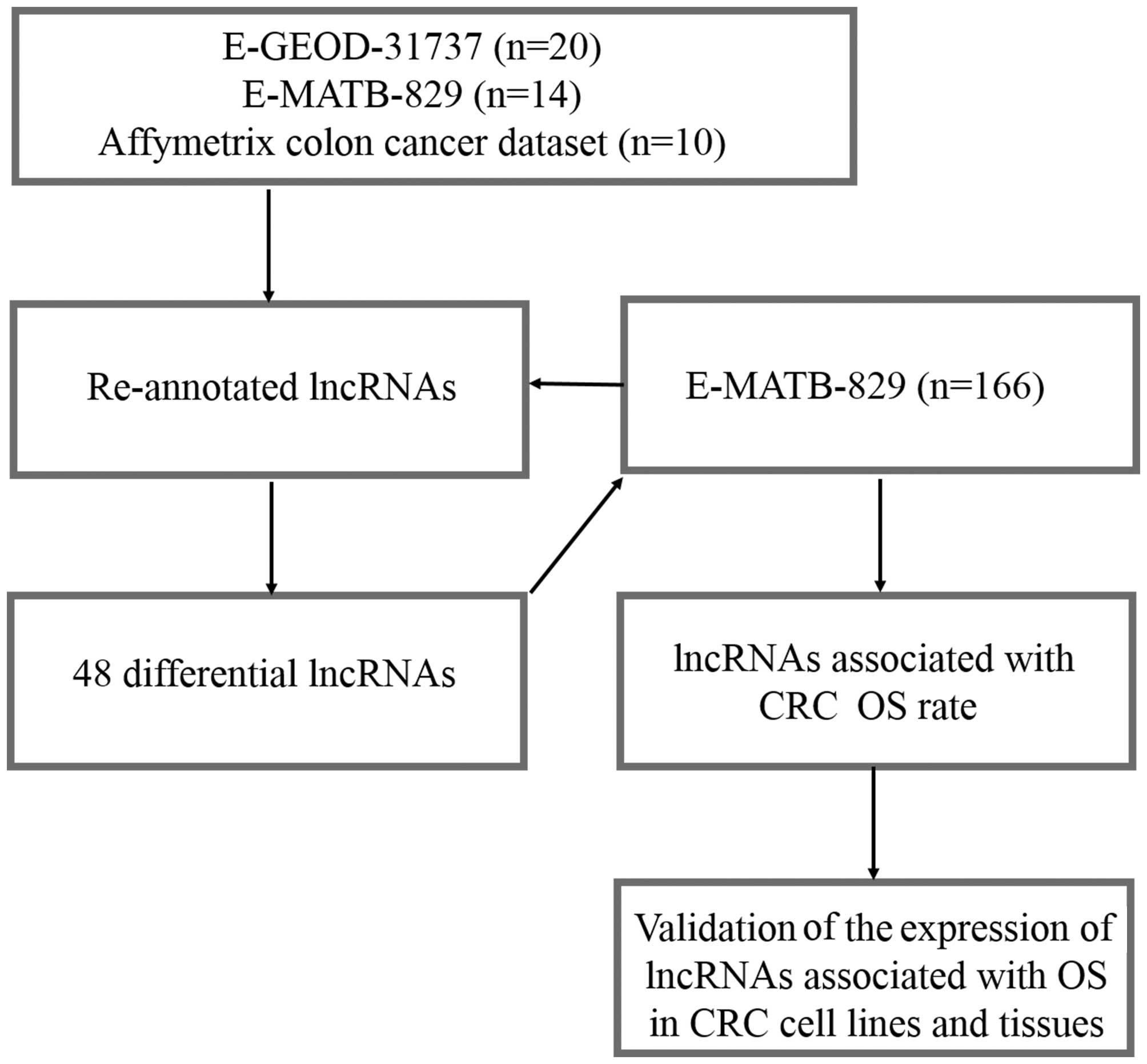
A pipeline was designed to re-annotate and validate
the lncRNAs from four different Affymetrix arrays associated with
CRC (Fig. 1). A total of 462
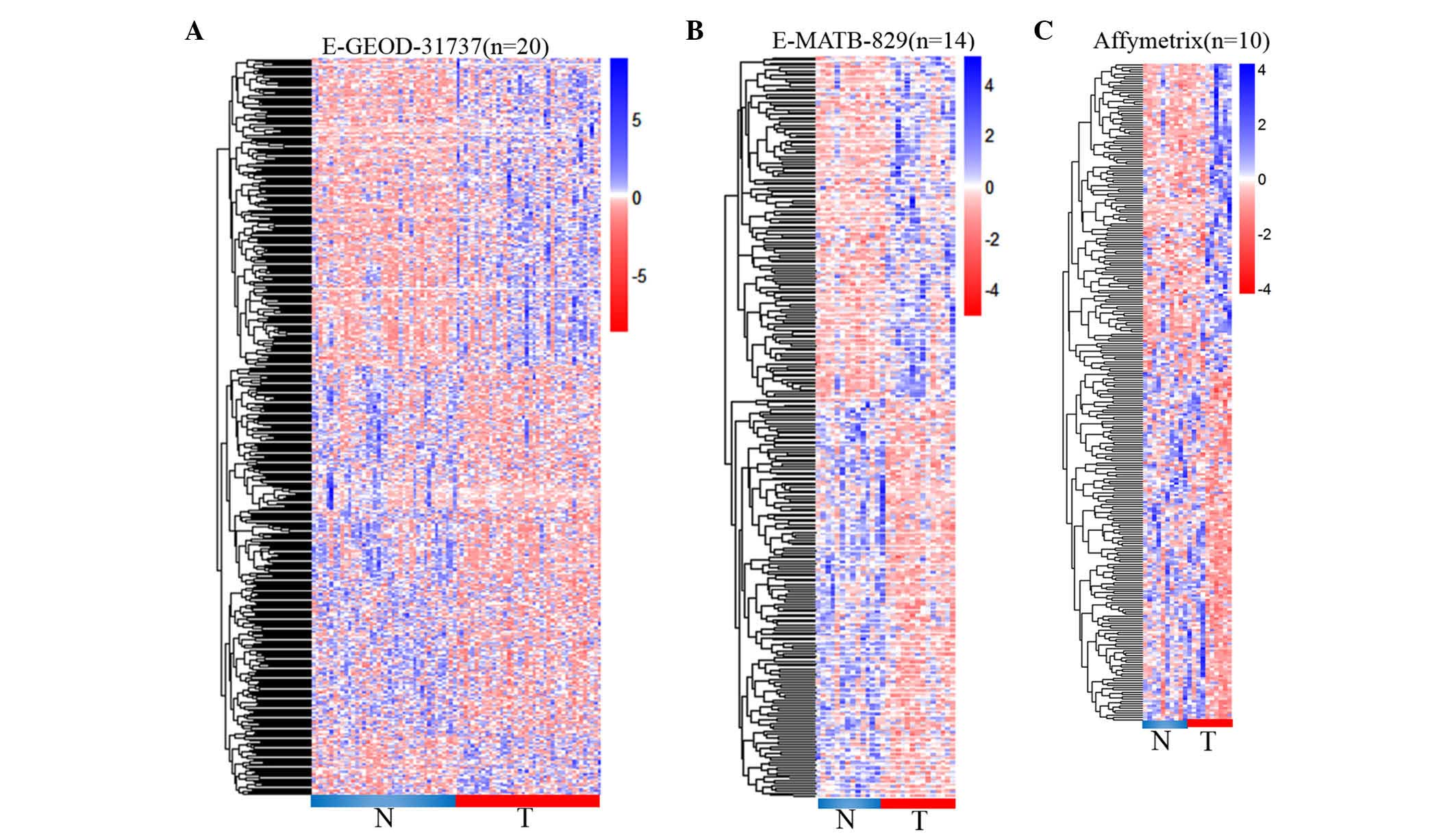
differential lncRNAs were determined from E-GEOD-31737. Among them,
229 lncRNAs were downregulated by >2-fold in cancerous tissues
(P<0.05) compared with paired normal tissues. Meanwhile, 233
lncRNAs were noticed to be upregulated in cancerous tissues with
>2-fold-change in expression and P<0.05 (Fig. 2A). Furthermore, 286 differential
lncRNAs were detected in the E-MATB-829 dataset, including 153
downregulated and 133 upregulated lncRNAs, with >2-fold-change
in expression and P<0.05 (Fig.
2B). Another CRC dataset downloaded from the Affymetrix website
was observed to contain 166 differential lncRNAs with
>2-fold-change in expression and P<0.05, including 78
downregulated and 88 upregulated lncRNAs in cancerous tissues
(Fig. 2C).
Comparison of differential lncRNA
expression in three datasets
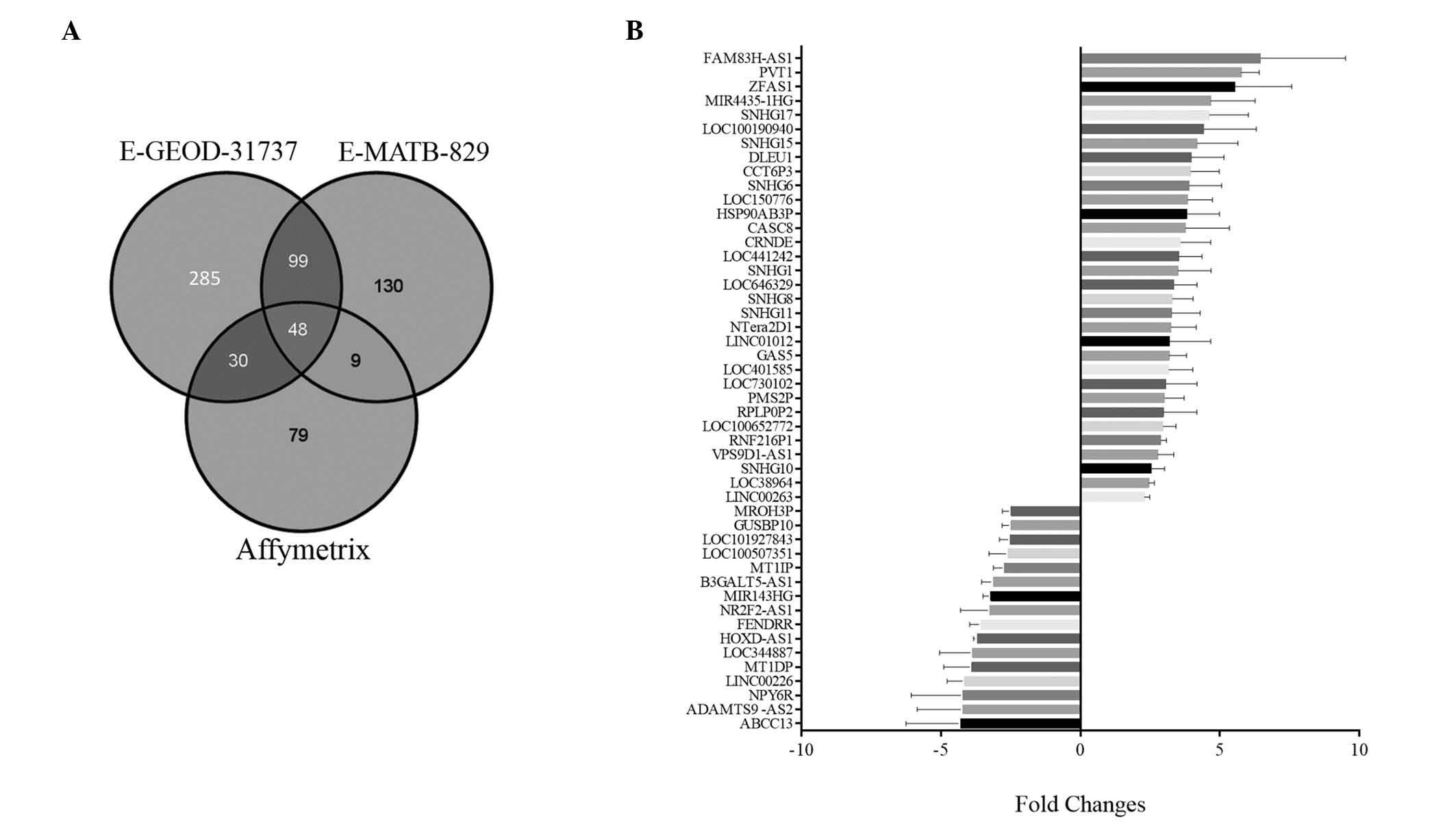
Venn diagrams were applied for comparative analysis
of the differential lncRNAs identified in the above three datasets,
as shown in Fig. 3A. A total of 48
differential lncRNAs were commonly detected in the three datasets,
including 16 downregulated lncRNAs (whose mean values ranged from
−4.31 to −2.49, based on calculating their fold-changes in the
three datasets) and 32 upregulated lncRNAs, with mean values
ranging from 2.29 to 6.46 (Fig.
3B).
Screening lncRNAs associated with CRC
prognosis in E-GEOD-24550
The 48 differential transcript clusters identified
in the above three datasets were further validated in E-GEOD-24550.
Time-dependent ROC curve applied for setting the best cutoff
values, in addition to Kaplan-Meier survival rate, were used to
analyze the effects of their overexpression on CRC survival rate.
CRC patients were divided into two groups according to best cutoff
values of their normalized values. Kaplan-Meier analysis was used
to explore the association of lncRNAs with survival rate. Among
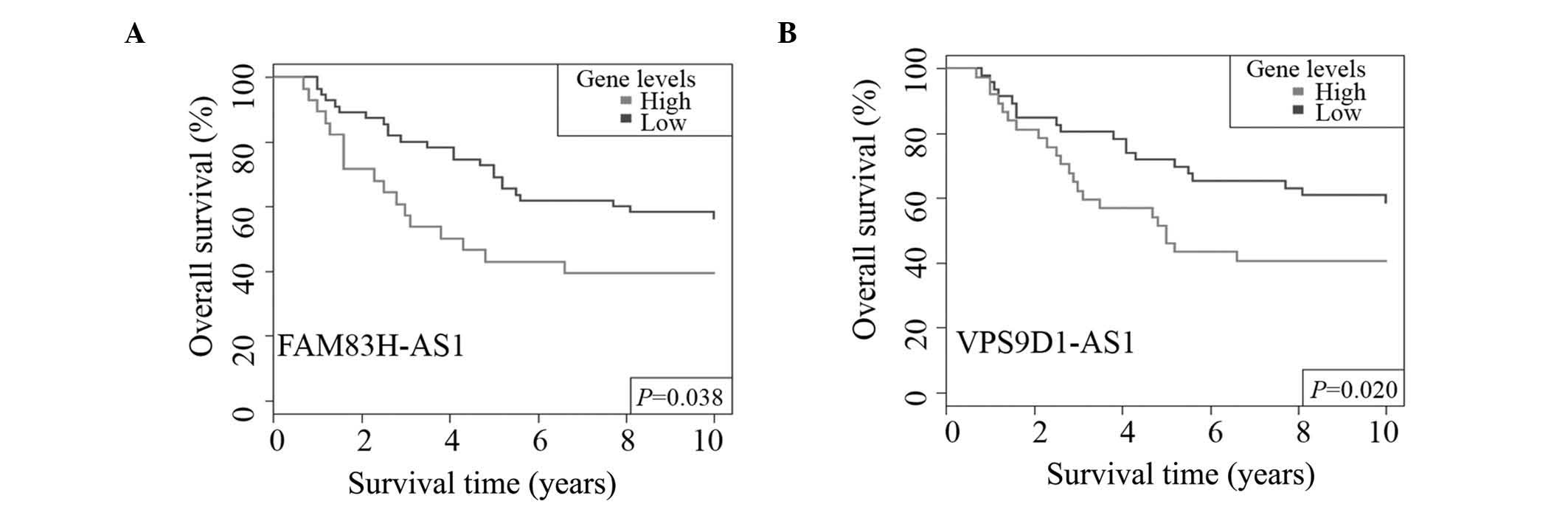
them, there were two lncRNAs that exhibited significant association
with CRC OS rate. FAM83H-AS1 was determined to have a significant
association with CRC OS rate (P=0.038, Fig. 4A). The overexpression of VPS9D1-AS1
was associated with CRC shorter OS time (P=0.020, Fig. 4B), and indicated a poor prognosis.
Thus, both lncRNAs had the potential to be CRC prognostic
biomarkers.
Validation of lncRNAs in human CRC
cell lines and tissues
The present study next validated the lncRNAs that
appeared to be associated with OS in human CRC cell lines and
tissues. Firstly, specific primers were designed to clone their
sequence from six CRC cell lines, including HUVEC, SW480, HCT116,
RKO, HT29 and SW620. Agarose gel electrophoresis analysis revealed
that CRC cell lines had relative higher levels of both FAM83H-AS1
and VPS9D1-AS1 lncRNAs in comparison with HUVECs (Fig. 5A). With the exception of RKO cells,
the cell lines HCT116, SW480, HT29 and SW620 expressed FAM83H-AS1,
while the expression of FAM83H-AS1 could not be detected in HUVECs.
VPS9D1-AS1 was observed to be expressed in all CRC cell lines at
higher levels than in HUVECs. In addition, cancerous tissues and
paired non-cancerous tissues were collected from 20 patients with
CRC to determine the levels of FAM83H-AS1 and VPS9D1-AS1 by
RT-qPCR. The relative expression of FAM83H-AS1 in cancerous tissues
was demonstrated to be 1.5-fold higher in 16 (80%) CRC patients
compared with paired non-cancerous tissues. The mean fold-change of
FAM83H-AS1 was 62.00. In addition, the results of a paired t-test
confirmed that the 2−∆∆Cq values of FAM83H-AS1 (relative
to GAPDH) were significantly higher in CRC tissues (P=0.0325) than
in normal tissues (Fig. 5B).
Furthermore, there were 13 (65%) CRC patients with >1.5
fold-change in VPS9D1-AS1 expression, with a mean value of 41.34,
ranging from 0.14 to 584.07 in cancerous tissues. The
2−∆∆Cq values of VPS9D1-AS1 also were observed to be
significantly higher in cancerous tissues compared with normal
tissues (P=0.0110, Fig. 5C).
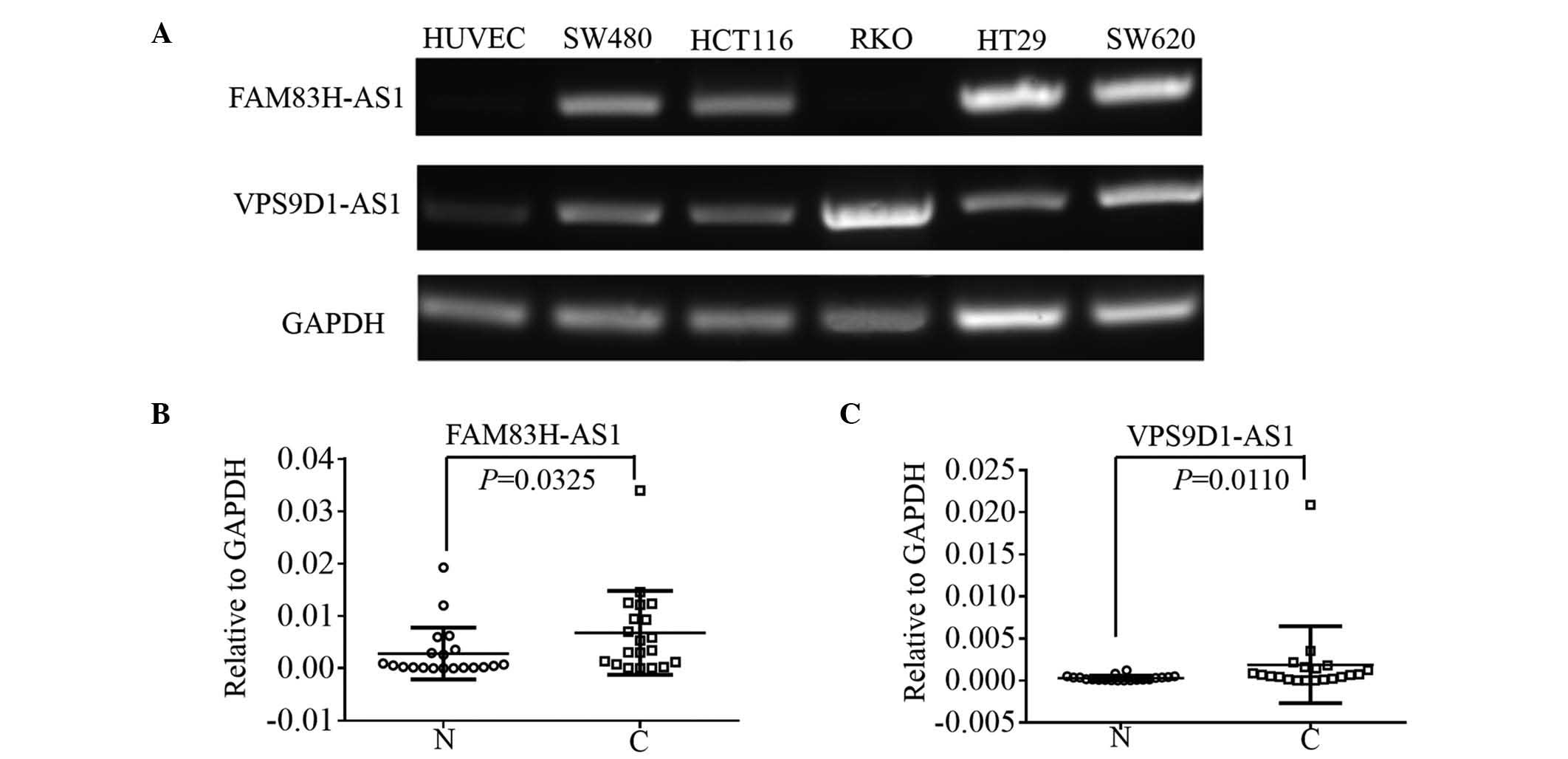 | Figure 5.(A) RT-PCR was used to detect the
expression of FAM83H-AS1 and VPS9D1-AS1 in HUVEC, SW480, HCT116,
RKO, HT29 and SW620 cells. GAPDH was used as the internal reference
gene. RT-quantitative PCR was used to validate the expression of
(B) FAM83H-AS1 and (C) VPS9D1-AS1 in cancerous and paired
non-cancerous tissues from 20 colorectal cancer patients. The
paired t–test was used to compare the statistical difference
of 2−∆Cq between tumor and non-cancerous tissues. GAPDH
was used as the internal reference gene. N, normal; C, cancer;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HUVEC, human
umbilical vein endothelial cell; FAM83H, family with sequence
similarity 83 member H; AS, antisense; VPS9D1, VPS9 domain
containing 1; RT-PCR, reverse transcription-polymerase chain
reaction. |
Discussion
Considering that protein-coding genes only comprise
~2% of the human genome, the majority of transcripts of human or
mammalian genomes have lost the potential to be translated into
proteins, and a large proportion of them are ncRNAs (17). For the past few years, RNA sequencing
or array-based strategies have been used to search lncRNAs
transcribed from the human genome (18,19).
However, those strategies have an expensive cost. Thus, the
re-purposing of microarray probes for constructing lncRNA
expression profiles in patients with cancer is a cost-effective
approach that has been employed by numerous researchers (20). The Affymetrix Exon 1.0 ST Array
contains almost 5.4 million probes and is designed to determine the
expression of each of the exons of a gene individually (13). In that array, all isoforms of a gene
are combined to be a ‘transcript cluster’, and each exon of the
transcript cluster is defined as a ‘probe set’ (13). In a previous study, each of the four
probes designed to match a gene represented the expression of the
gene (13). Various other
microarray-based platforms could also be re-annotated for lncRNAs,
such as Affymetrix U133A (21,22).
GATExplorer (http://bioinfow.dep.usal.es/xgate/principal.php) and
ncFANs (http://english.ict.cas.cn/)
re-annotated several microarrays from Affymetrix for protein and
lncRNAs, and revealed that the Affymetrix Exon 1.0 ST Array
contains considerably more probes matching lncRNAs than other
microarray platforms (23,24). That was the reason why these datasets
were collected from previous studies based on the Affymetrix Exon
1.0 ST Array.
In the present study, 462, 286 and 166 differential
lncRNAs were identified, respectively, in CRC tissues based on
three Exon 1.0 ST databases. A number of the identified lncRNAs had
been demonstrated by previous studies to exhibit dysregulated
levels in several types of cancer (13). Among them, HoxA transcript at the
distal tip RNA antisense RNA (HOTTIP), has been reported to display
dysregulated levels in cancerous tissues and to be involved in
hepatocarcinogenesis (25). In the
present study, HOTTIP was detected to exhibit dysregulation in CRC
cancer tissues (E-GEOD-31737). Additionally, the variance
transcripts of HOTAIR, including HOTAIRM1, were determined to
exhibit differential levels in CRC cancer tissues (E-GEOD-31737 and
E-MATB-829) (8). However, variances
associated with measurement error were not comparable across
different studies (26). In the
present study, it was observed that large samples created a large
number of differential lncRNAs, and 48 differentially expressed
lncRNAs were detected in all three datasets. It may be necessary to
further explore more lncRNAs with differential expression in CRC
tissues; however those 48 lncRNAs identified in the present study
have the most credible probability to be tumor suppressor or
oncogenic genes during the process of tumorigenesis.
To more correctly screen CRC-related lncRNAs, 48
differential lncRNAs were validated in other datasets with larger
samples. Future studies may be based in those lncRNAs. Furthermore,
the present study has identified for the first time two lncRNAs
that have the potential to be prognostic biomarkers for CRC
patients. These lncRNAs were initially demonstrated to be
overexpressed in tumor tissues by three datasets, and the results
revealed that they could promote tumor progression. The validation
in CRC cell lines or tissues also confirmed that those lncRNAs had
higher levels in cancer cell lines and tissues than in normal cell
lines or tissues. Further studies should be focused on these
lncRNAs for exploring their detailed mechanism of promotion of CRC
development.
In conclusion, the current study employed
bioinformatics methods and screened 462 differential lncRNAs in
CRC. A total of 48 lncRNAs were confirmed by three datasets based
on Exon 1.0 ST arrays. Another array-based dataset allowed us to
screen two lncRNAs possessing the potential to be prognostic
biomarkers of CRC patients. In addition, these two lncRNAs were
validated to exhibit higher levels in cancerous tissues in
comparison with paired non-cancerous tissues from CRC patients.
Those primary findings supported the idea that these two lncRNAs
(FAM83H-AS1 and VPS9D1-AS1) may be the key regulators for
controlling CRC carcinogenesis and development, and it may be
urgent to validate their levels in a large cohort for exploring
their clinical significance and identifying their mechanisms of
action in CRC.
Acknowledgements
The present study was supported by Beijing Natural
Science Foundation (Beijing, China; grant no. 7154199) and National
High Technology Research and Development Program (Beijing, China;
grant no. 2012AA02A506).
References
|
1
|
Ogino S, Chan AT, Fuchs CS and Giovannucci
E: Molecular pathological epidemiology of colorectal neoplasia: An
emerging transdisciplinary and interdisciplinary field. Gut.
60:397–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jia WH, Zhang B, Matsuo K, Shin A, Xiang
YB, Jee SH, Kim DH, Ren Z, Cai Q, Long J, et al: Genome-wide
association analyses in East Asians identify new susceptibility
loci for colorectal cancer. Nat Genet. 45:191–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ,
Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al: A long noncoding RNA
activated by TGF-β promotes the invasion-metastasis cascade in
hepatocellular carcinoma. Cancer Cell. 25:666–681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prensner JR, Iyer MK, Sahu A, Asangani IA,
Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, et al:
The long noncoding RNA SChLAP1 promotes aggressive prostate cancer
and antagonizes the SWI/SNF complex. Nat Genet. 45:1392–1398. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ling H, Spizzo R, Atlasi Y, Nicoloso M,
Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Guo Z, et al:
CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic
progression and chromosomal instability in colon cancer. Genome
Res. 23:1446–1461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Svoboda M, Slyskova J, Schneiderova M,
Makovicky P, Bielik L, Levy M, Lipska L, Hemmelova B, Kala Z,
Protivankova M, et al: HOTAIR long non-coding RNA is a negative
prognostic factor not only in primary tumors, but also in the blood
of colorectal cancer patients. Carcinogenesis. 35:1510–1515. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ge X, Chen Y, Liao X, Liu D, Li F, Ruan H
and Jia W: Overexpression of long noncoding RNA PCAT-1 is a novel
biomarker of poor prognosis in patients with colorectal cancer. Med
Oncol. 30:5882013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng HT, Shi DB, Wang YW, Li XX, Xu Y,
Tripathi P, Gu WL, Cai GX and Cai SJ: High expression of lncRNA
MALAT1 suggests a biomarker of poor prognosis in colorectal cancer.
Int J Clin Exp Pathol. 7:3174–3181. 2014.PubMed/NCBI
|
|
11
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long noncoding RNAs in human cancer. Nat Struct
Mol Biol. 20:908–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao Q, Liu C, Yuan X, Kang S, Miao R,
Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction
of long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gellert P, Ponomareva Y, Braun T and
Uchida S: Noncoder: A web interface for exon array-based detection
of long non-coding RNAs. Nucleic Acids Res. 41:e202013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bu D, Yu K, Sun S, Xie C, Skogerbø G, Miao
R, Xiao H, Liao Q, Luo H, Zhao G, et al: NONCODE v3.0: Integrative
annotation of long noncoding RNAs. Nucleic Acids Res. 40:(Database
Issue). D210–D215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lambert J and Chevret S: Summary measure
of discrimination in survival models based on cumulative/dynamic
time-dependent ROC curves. Stat Methods Med Res. 2014.
|
|
16
|
Yang M, Tian J, Guo X, Yang Y, Guan R, Qiu
M, Li Y, Sun X, Zheng Y, Zhang Y, et al: Long noncoding RNA are
aberrantly expressed in human papillary thyroid carcinoma. Oncol
Lett. 12:544–552. 2016.PubMed/NCBI
|
|
17
|
Batista PJ and Chang HY: Long noncoding
RNAs: Cellular address codes in development and disease. Cell.
152:1298–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ching T, Huang S and Garmire LX: Power
analysis and sample size estimation for RNA-Seq differential
expression. RNA. 20:1684–1696. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Chen Z, Tian L, Zhou C, He MY, Gao
Y, Wang S, Zhou F, Shi S, Feng X, et al: LncRNA profile study
reveals a three-lncRNA signature associated with the survival of
patients with oesophageal squamous cell carcinoma. Gut.
63:1700–1710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu X, Zhang Y, Williams J, Antoniou E,
McCombie WR, Wu S, Zhu W, Davidson NO, Denoya P and Li E: Parallel
comparison of Illumina RNA-Seq and Affymetrix microarray platforms
on transcriptomic profiles generated from 5-aza-deoxy-cytidine
treated HT-29 colon cancer cells and simulated datasets. BMC
Bioinformatics. 14(Suppl 9): S12013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michelhaugh SK, Lipovich L, Blythe J, Jia
H, Kapatos G and Bannon MJ: Mining Affymetrix microarray data for
long non-coding RNAs: Altered expression in the nucleus accumbens
of heroin abusers. J Neurochem. 116:459–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Risueno A, Fontanillo C, Dinger ME and De
Las Rivas J: GATExplorer: Genomic and transcriptomic explorer;
mapping expression probes to gene loci, transcripts, exons and
ncRNAs. BMC Bioinformatics. 11:2212010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liao Q, Xiao H, Bu D, Xie C, Miao R, Luo
H, Zhao G, Yu K, Zhao H, Skogerbø G, et al: ncFANs: A web server
for functional annotation of long non-coding RNAs. Nucleic Acids
Res. 39:(Web Server Issue). W118–W124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quagliata L, Matter MS, Piscuoglio S,
Arabi L, Ruiz C, Procino A, Kovac M, Moretti F, Makowska Z,
Boldanova T, et al: Long noncoding RNA HOTTIP/HOXA13 expression is
associated with disease progression and predicts outcome in
hepatocellular carcinoma patients. Hepatology. 59:911–923. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woo Y, Affourtit J, Daigle S, Viale A,
Johnson K, Naggert J and Churchill G: A comparison of cDNA,
oligonucleotide, and Affymetrix GeneChip gene expression microarray
platforms. J Biomol Tech. 15:276–284. 2004.PubMed/NCBI
|