Introduction
Bladder cancer (BC) is the fifth most common type of
cancer in Western countries, accounting for >130,000 mortalities
annually, worldwide (1). At any point
in time, 2.7 million people are diagnosed with or have a history of
BC (1).
Cigarette smoke is the major risk factor for BC
(2). Recently, a meta-analysis of 43
published case-control and cohort studies reported that, at
present, cigarette smokers exhibit a ~3-fold higher risk of
developing BC compared with non-smokers (3). The risk of BC has been found to
significantly correlate with the intensity and duration of
cigarette smoke exposure, whereas quitting smoking reduces this
risk (4).
Exposure to CSE or its constituents is known to
trigger a cascade of events in the multistage process of
carcinogenesis (5). A previous study
revealed that the mitogen-activated protein kinase (MAPK)/activator
protein-1 (AP-1) pathway is associated with the effects induced by
CSE (6). MAPKs are involved in the
phosphorylation and activation of the Jun and Fos proteins. Three
well-characterized subfamilies of MAPK have been identified, which
include the extracellular signal-regulated kinase (ERK)1/2, Jun
N-terminal kinase (JNK)/stress-activated protein kinase and p38
subfamilies (7). The MAPK signaling
pathway, which is upregulated in cancer cells, promotes
proliferation, differentiation and cell survival, and mediates
oncogenesis (8).
Malignant tumors exhibit aberrant cell growth, which
results from uncontrolled cell division and proliferation.
Urothelial hyperplasia has been identified as a precancerous lesion
in BC (9). Exposure to nicotine
induces the expression of cyclin D1 and proliferating cell nuclear
antigen (PCNA), which are two AP-1 target genes known to be
involved in promotion of cell cycle progression (10). Cyclin D1 monitors the cell cycle by
forming complexes with cyclin-dependent kinase 4 and 6 in the
cytoplasm. These complexes then enter the nucleus and inactivate
the cell-cycle suppressive retinoblastoma protein, thereby
promoting progression from G1 to S phase (10). By contrast, the tumor suppressor, p21,
is a key negative regulator of the cell cycle and cell
proliferation, and is often downregulated in cancer. Notably, p21
is rarely mutated or deleted (11).
However, few studies have investigated the effect of CSE on the
cell cycle in normal human urothelial cells.
Thus, the aim of the present study was to
investigate whether exposure to CSE induced proliferation in
SV-HUC-1 cells. Furthermore, the role of the MAPK/AP-1 pathway in
CSE-induced proliferation of SV-HUC-1 cells was also analyzed.
Materials and methods
Reagents
The SV-40 immortalized human urothelial SV-HUC-1
cell line was purchased from the Chinese Academy of Sciences Cell
Bank (Shanghai, China). Gibco F12K medium was purchased from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). Fetal bovine serum
(FBS) was obtained from PAA Laboratories GmbH (Pasching, Austria).
3-(4,5)-dimethylthiahiazo(−z-y1)-3,5-di-phenytetrazoliumromide
(MTT) was purchased from Sigma-Aldrich (Merck Millipore, Darmstadt,
Germany). Monoclonal rabbit phosphorylated (p-)JNK (cat. no.
AF3318; 1:1,000), p38 (cat. no. 9212; 1:1,000), ERK1/2 (cat. no.
RS-2637S; 1:1,000), p-c-Jun (cat. no. AF-3095; 1:1,000), p-c-Fos
(cat. no. 5348; 1:1,000), Jun B (cat. no. 10486-1-AP; 1:1,000), p21
(cat. no. 10355-1-AP; 1:1,000), cyclin D1 (cat. no. 2978; 1:1,000)
and PCNA (cat. no. 10205-2-AP; 1:1,000) primary antibodies were all
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Monoclonal rabbit glyceraldehyde 3-phosphate dehydrogenase (GAPDH;
cat. no. 60004-1-Ig; 1:1,000) was purchased from Proteintech
(Rosemont, IL, USA). Monoclonal rabbit Jun D antibody (cat. no.
sc-2048; 1:1,000) was purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Mouse anti-rabbit IgG secondary antibodies
(cat. no. bs-0295M; 1:10,0000) were purchased from Saihongrui
Biotechnology Co., Ltd., (Nanjing, China). Primers were synthesized
according to published sequences by Invitrogen (Thermo Fisher
Scientific, Inc.). Inhibitors of the MAPK pathway (SB203580 for
p38, SP600125 for JNK and U0126 for ERK) were purchased from
Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture and treatment
SV-HUC-1 cells were cultured in F12K medium
(Sigma-Aldrich; Merck Millipore) containing antibiotics (100
units/ml penicillin and 100 µg/ml streptomycin) in an atmosphere of
5% CO2 at 37°C. Cells were seeded in 10 cm2
flasks at a density of 1×105 cells per well. The medium
was changed every other day until cells reached 80–90% confluence,
then treated with 0, 0.10, 0.25 or 0.50% cigarette smoke extract
(CSE) or U0126 (5 µM), SB203580 (5 µM) or SP600125 (2 µM).
Preparation of CSE
CSE was freshly prepared for each experiment by
combusting one commercial cigarette, according to a previously
reported method (12). Commercial
cigarettes (Hongtashan filter-tipped cigarettes; YuXi Cigarette
Factory, Yunnan, China), each containing 12 mg tar and 1.1 mg
nicotine, were combusted. Using a vacuum, mainstream (first-hand)
smoke was drawn through 10 ml pre-warmed (37°C) FBS-free F12K
medium supplemented with penicillin and streptomycin at a rate of 5
min/cigarette (13). The obtained CSE
stock solution was considered to contain a concentration of 100%.
The CSE stock solution was then filtered through a 0.22-µm-pore
size filter and diluted to the desired concentrations using
treatment medium. The resulting CSE solutions were applied to
epithelial cell cultures within 30 min of preparation. A control
solution was prepared using the same protocol, using unlit
cigarettes.
Cell proliferation assay
SV-HUC-1 cells (2×103 cells/well) were
seeded in 96-well plates in 200 µl F12K medium. Cells were exposed
to 0, 0.10, 0.25 or 0.50% CSE for 7 days and cell viability was
then determined by MTT assay. The media were changed every day.
Subsequent to 7 days of culture, MTT stock solution (5 mg/ml) was
added to each well to solubilize formazan crystals, and plates were
incubated for an additional 4 h at 37°C. Next, MTT solution was
removed and the crystals were solubilized in dimethyl sulfoxide.
Absorbance was measured at 490 nm using a microplate reader
(Titertek Instruments Inc., Huntsville, AL, USA). All experiments
were performed in triplicate.
Cell cycle analysis
The cell cycle distribution of SV-HUC-1 cells was
determined by flow cytometry. SV-HUC-1 cells
(5×105cells/well) were incubated in F12K medium
supplemented with 5% FBS at 37°C overnight. Cells were then
cultured in F12K medium without FBS for 6 h. Next, cells were
treated with 0, 0.1, 0.25 and 0.5% CSE for 7 days. Cells were
trypsinized, washed twice with cold phosphate-buffered saline (PBS)
and fixed overnight with 70% ethanol at 4°C followed by
resuspension in 500 µl PBS. Following the addition of 10 µl RNAse
(10 mg/ml), cells were incubated for 30 min in the dark at 37°C
then stained with 10 µl propidium iodide (1 mg/ml). Cell cycle
analysis was then performed by flow cytometry (FACSCalibur
instrument; BD Biosciences, San Jose, CA, USA). The percentage of
cells in each cell cycle phase (G0/G1, S or G2/M) was calculated
using MultiCycle 3.0 software (BD Biosciences). All experiments
were performed in triplicate.
Western blot analysis
SV-HUC-1 cells were harvested at 70–80% confluence,
washed with ice-cold PBS and lysed in RIPA buffer (Thermo Fisher
Scientific, Inc.). The concentration of precipitated proteins in
the cell lysates was measured using the BCA Protein Assay kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). Next, proteins
were diluted to equal concentrations, boiled for 5 min, separated
by 7.5–10.0% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA). After blocking with 5%
milk, the membranes were incubated with the p-JNK, p38, ERK1/2,
p-c-Jun, p-c-Fos, Jun B, Jun D, p21, cyclin D1 and PCNA primary
antibodies at 4°C overnight. The membranes were washed with PBS 3
times for 5 min each time, prior to incubation with secondary
antibodies for 1 h at room temperature. The blots were subsequently
developed using an enhanced chemiluminescence detection kit
(Amersham Biosciences, Uppsala, Sweden) and exposed to film (Kodak,
Rochester, NY, USA). GAPDH acted as the loading control.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total cellular RNA was isolated using Trizol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Total RNA was reverse-transcribed
using the Easy RT-PCR kit (Takara Bio, Inc., Otsu, Japan). qRT-PCR
was performed using the Power SYBR Green Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and an ABI 7300
Real-time PCR Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). A 20 µl sample system was used, including 1 µl
cDNA, 0.6 µl forward primer, 0.6 µl reverse primer, 7.8 µl
diethylpyrocarbonate water and 10 µl Rox-mix. PCR was performed
under the following conditions: Denaturation at 95°C for 15 sec,
followed by 40 cycles of annealing at 65°C for 30 sec and extension
at 72°C for 15 sec. The sequences of the primers (Invitrogen;
Thermo Fisher Scientific, Inc.) used for RT-PCR were as follows:
Forward, 5′-CGTGGCCTCTAAGATGAAGG-3′ and reverse,
5′-TGCGGATGATCTGTTTGTTC-3′ for cyclin D1; forward,
5′-GACACCACTGGAGGGTGACT-3′ and reverse, 5′-CAGGTCCACATGGTCTTCCT-3′
for p21; and forward, 5-′GCTGCCCAACGCACCGAATA-3′ and reverse,
5′-GAGTCAACGGATTTGGTCGT-3′ for GAPDH. Each sample was run in
triplicate. Fold changes in the expression of each gene were
calculated using the comparative threshold cycle (Ct) method using
the formula 2−ΔΔCt (14).
Statistical analysis
All statistical analysis was performed using SPSS
17.0 (SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean
± standard deviation. One-way analysis of variance or the
Kruskal-Wallis test were used to analyze differences among groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CSE induces proliferation in SV-HUC-1
cells
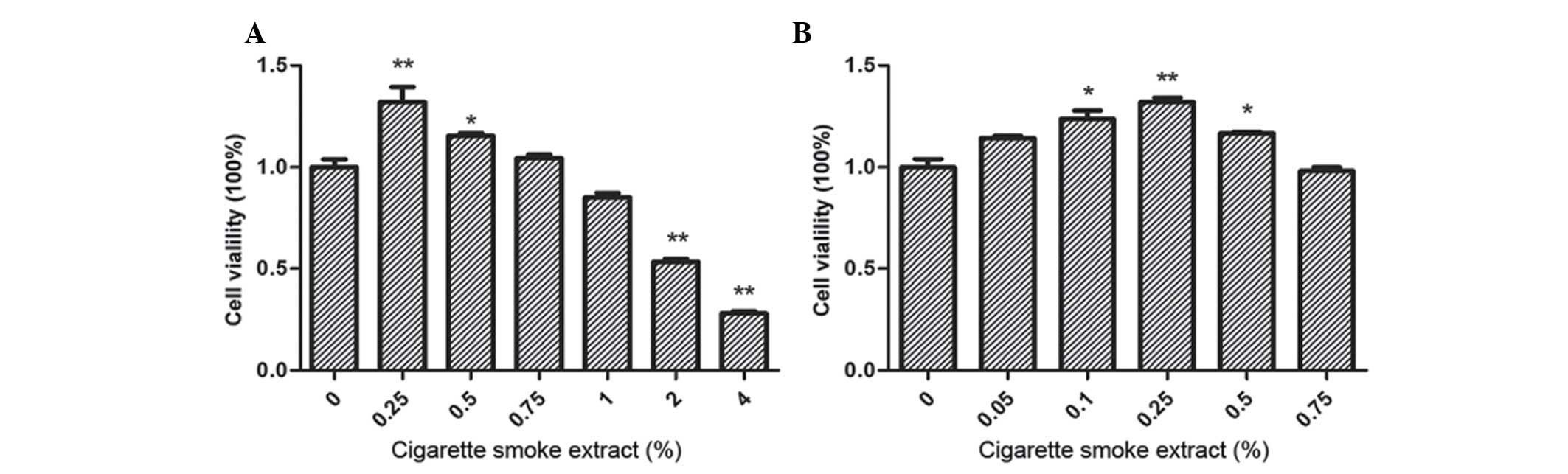
To study the cell viability of SV-HUC-1 cells
following treatment with CSE, cells were incubated with CSE (0,
0.25, 0.50, 0.75, 1, 2 and 4%) for 7 days and examined by MTT
assay. A significant increase in the cell viability of SV-HUC-1
cells following treatment with 0.25 (P<0.001) and 0.50%
(P=0.042) CSE was identified, while cell viability decreased to
<80% following treatment with CSE concentrations of ≥2%
(P<0.001), which were demonstrated to be toxic to SV-HUC-1 cells
(Fig. 1A). To further investigate the
effect of CSE treatment on the proliferation of SV-HUC-1 cells,
lower concentrations of CSE (0.25 and 0.50%) were selected to study
cell viability in further experiments. Although a CSE concentration
of 0.05% increased SV-HUC-1 cell proliferation, concentrations of
0.10, 0.25 and 0.50% CSE were selected as the optimal
concentrations for future investigation, as they resulted in the
most significant differences in proliferation (Fig. 1B).
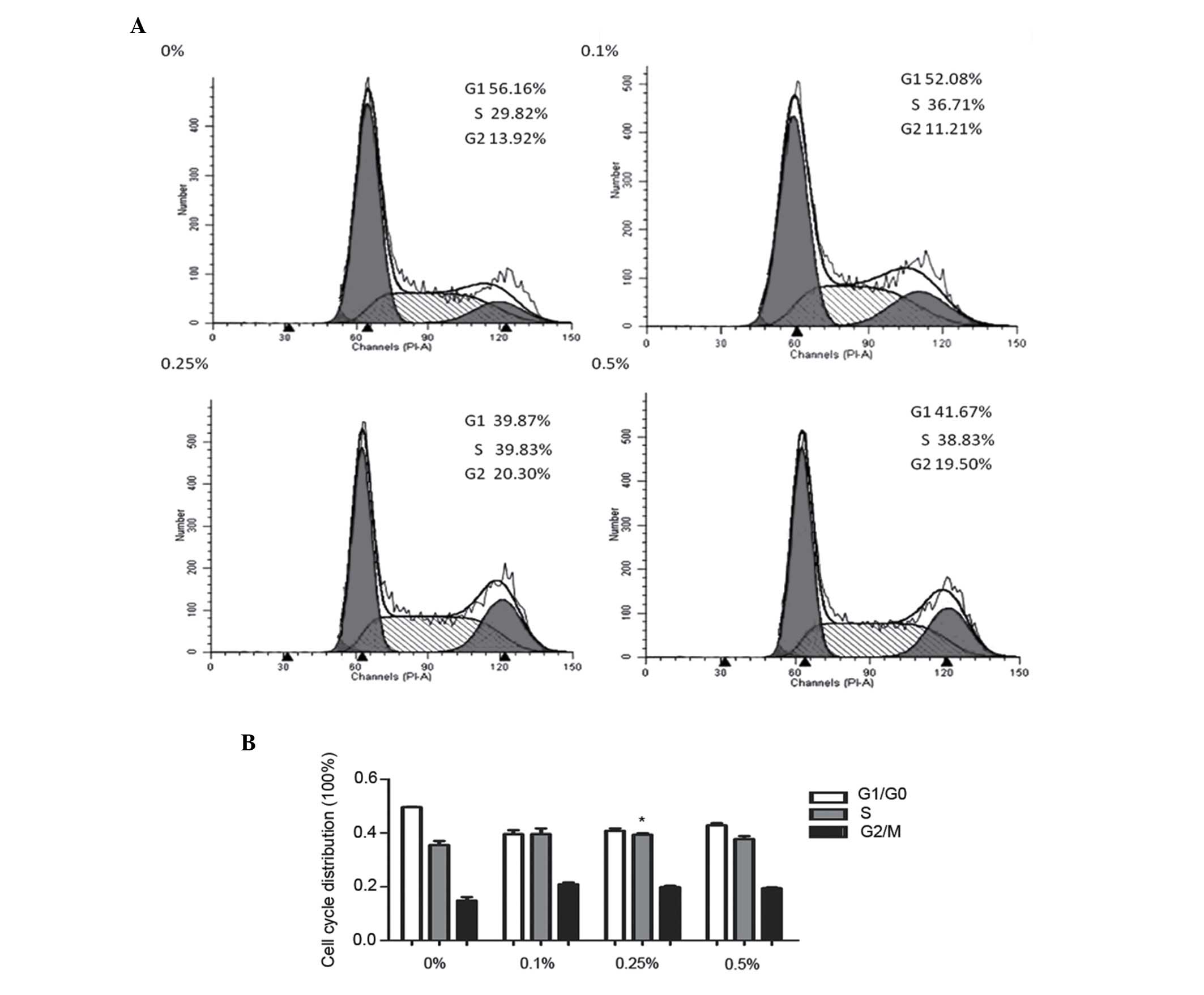
CSE promotes SV-HUC-1 cell
transformation from G1 to S phase
To examine whether the proliferative effects of CSE
in SV-HUC-1 cells were mediated via cell cycle modulation, the cell
cycle distribution in SV-HUC-1 cells was investigated by flow
cytometry. Low concentrations of CSE (0.10, 0.25 and 0.50%)
promoted SV-HUC-1 cell transformation from G1 to S phase (Fig. 2A and B). Notably, a total of 39.83% of
SV-HUC-1 cells treated with 0.25% CSE entered S phase, which was
significantly increased compared with the control group (29.82%;
P=0.032). Next, the expression of proteins associated with the cell
cycle were determined; the results revealed that the expression of
cyclin D1 and PCNA were markedly increased following 0.10, 0.25 and
0.50% CSE treatment. Notably, the expression of p21 was attenuated
by CSE treatment and the expression of p21 almost disappeared
following treatment with 0.5% CSE (Fig.
2C-D).
MAPK/AP-1 pathway is involved in the
proliferative effects induced by CSE in SV-HUC-1 cells
The MAPK/AP-1 pathway is hypothesized to be involved
in the effects induced by CSE in SV-HUC-1 cells. Therefore,
SV-HUC-1 cells were treated with 0, 0.10, 0.25 or 0.50% CSE for 7
days followed by western blot analysis to investigate whether CSE
activates the MAPK/AP-1 pathway. The results revealed that CSE
activated p-ERK1/2, P38 and JNK (Fig.
3A). However, the AP-1 proteins exhibited varying sensitivity
to CSE (Fig. 3B). For example,
p-c-Jun was more sensitive to 0.25% CSE, while Jun B was more
sensitive to 0.1% CSE; whereas p-c-Fos was continuously activated
by CSE, which indicates that at higher concentrations of CSE, the
effects of Jun D attenuation are more evident.
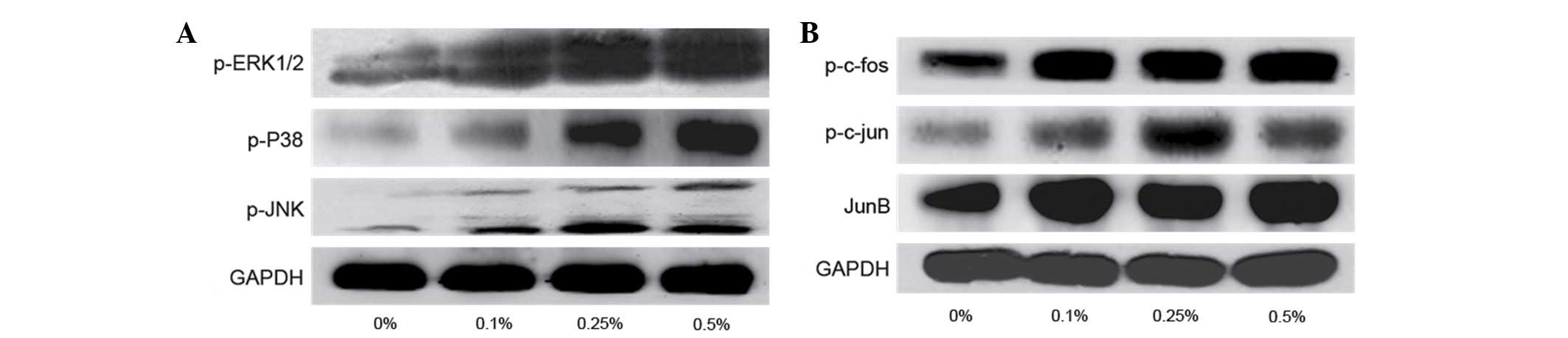 | Figure 3.(A) Expression of proteins involved in
the mitogen-activated protein kinase pathway, including p-ERK1/2,
p-p38 and p-JNK, in SV-HUC-1 cells following treatment with 0.10,
0.25 and 0.50% CSE for 7 days. p-ERK1/2, p-P38 and p-JNK protein
expression was increased following CSE treatment. (B) Expression of
proteins involved in activator protein 1 pathway, including
p-c-Jun, p-c-Fos, Jun B and Jun D in SV-HUC-1 cells following
treatment with CSE for 7 days. The expression of p-c-Jun, p-c-Fos
and Jun B were markedly increased following CSE treatment. p-,
phosphorylated; ERK, extracellular signal regulated protein kinase;
JNK, Jun N-terminal kinase; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
MAPK pathway inhibitors reverse
CSE-induced proliferation in SV-HUC-1 cells
To investigate the role of the MAPK pathway in
CSE-induced cell proliferation of SV-HUC-1 cells, cells were
incubated with DMSO (control group), 0.10 or 0.25% CSE in
combination with U0126 (5 µM), SB203580 (5 µM) or SP600125 (2 µM)
for 7 days. An MTT assay was conducted to analyze cell viability
and the inhibitory effects induced, following treatment with MAPK
inhibitors (Fig. 4A). The results
revealed that U0126 and SP600125 markedly reversed CSE-induced
proliferation in SV-HUC-1 cells (P<0.001; 0.25%CSE vs.
0.25%CSE+U0126/SP600125). SB203580 marginally attenuated the effect
of CSE; however, the effect was not reversed, which indicates that
p38 is not involved in the proliferative effects induced by
CSE.
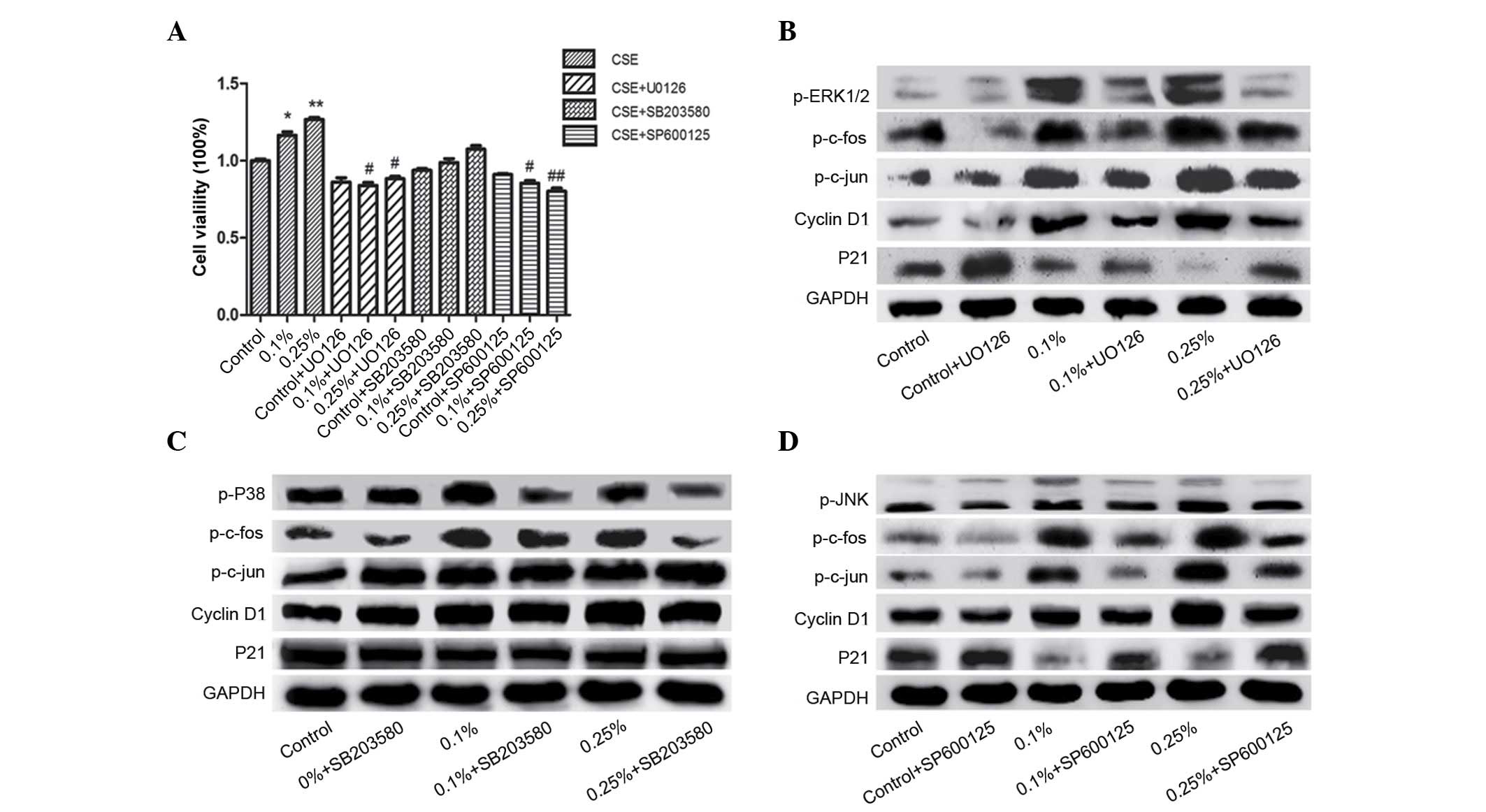 | Figure 4.(A) Fold change in cell viability of
SV-HUC-1 cells treated with 0, 0.10 and 0.25% CSE for 7 days in
combination with 5 µM U0126, 5 µM SB203580 or 2 µM SP600125. Data
are representative of three independent experiments and are
expressed as the mean ± standard deviation. *P<0.05 vs. control
group (0% CSE + dimethyl sulfoxide); #P<0.05
represents CSE ± inhibitor group vs. respective CSE group. (B)
Protein expression of AP-1 pathway and cell cycle markers in
SV-HUC-1 cells following combined treatment with CSE and 5 µM U0126
for 7 days. The upregulated expression of p-c-Fos, p-c-Jun, cyclin
D1 following CSE treatment was decreased by U0126, while U0126
inhibited the CSE-induced downregulation of p21. (C) Protein
expression of AP-1 pathway and cell cycle markers in SV-HUC-1 cells
following combined treatment with CSE and 5 µM SB203580 for 7 days.
The expression of p-c-Jun was decreased by SB203580. The expression
of p-c-Fos and cell cycle regulators, cyclin D1 and p21, was not
decreased following SB203580 treatment. (D) Protein expression of
AP-1 pathway and cell cycle markers in SV-HUC-1 cells following
combined treatment with CSE and 2 µM SP600125 for 7 days. p-c-Fos,
p-c-Jun, cyclin D1 and p21 were decreased following treatment with
SP600125. CSE, cigarette smoke extract; p-, phosphorylated; ERK,
extracellular signal regulated protein kinase; JNK, Jun N-terminal
kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; AP-1,
activator protein-1. |
Western blot analysis revealed similar results.
p-ERK1/2 was significantly downregulated and the expression of
p-c-Jun and p-c-Fos was attenuated by U0126 treatment after CSE
treatment (Fig. 4B). Furthermore,
U0126 reversed the CSE-induced upregulation of cyclin D1 and
downregulation of p21 expression. However, SB203580 exhibited no
effect on cyclin D1 or p21 expression following CSE treatment
(Fig. 4C). Additionally, SP600125
treatment markedly inhibited the effects of CSE, and the expression
of downstream p-c-Fos and p-c-Jun were strongly inhibited.
Furthermore, the expression of cyclin D1 and p21 were decreased
following SP600125 treatment (Fig.
4D). Notably, following treatment with SP600125, the observed
protein levels of cyclin D1 and p21 were similar to the levels
exhibited in the control group, which indicates that SP600125 may
be present a potential chemopreventative agent. U0126, SB203580 and
SP600125 treatment exhibited little effect on the control group and
mainly diminished the effect induced by CSE.
Discussion
The present study demonstrated that exposure to CSE
activated the MAPK/AP-1 pathway and induced cell proliferation in
SV-HUC-1 cells. Furthermore, inhibitors of the ERK1/2 and JNK
pathways reversed the proliferation induced by exposure to CSE.
However, the inhibition of p38 did not reverse the proliferation
induced by CSE.
BC is one of the most common tumors of the urinary
tract. It is a disease that peaks in older patients, and ~90% of
cases are pathologically diagnosed as urothelial carcinoma
(15). Cigarette smoke is considered
to be the major risk factor for BC (3). Thus, elucidating the processes involved
in the transformation of normal urothelial cells to malignant cells
induced by exposure to cigarette smoke may aid the development of
early targeted interventions for BC. To date, >60 carcinogens,
which are present in mainstream smoke, sidestream smoke and the
particulate phase of CSE, including polycyclic aromatic
hydrocarbons, benzo[a]pyrene, nitrosamines and aromatic amines,
have been identified (16). As the
risk of BC is significantly associated with the intensity and
duration of exposure to cigarette smoke, CSE was selected as a
stimulus in the present study, as it has been widely used in
previous BC studies (17,18).
Previous studies have demonstrated that the
MAPK/AP-1 pathway is involved in the effects induced after
treatment with CSE (6,19). The Ras-MAPK signaling pathway is a
well-established target for anticancer therapies, due to its
central role in regulating the growth and survival of cells
(19). BC is a disease caused by
alterations in several cellular processes, characterized by
constitutive activation of the Ras-MAPK pathway (20). In the present study, CSE was found to
activate the ERK1/2, P38 and JNK pathways. The activation of the
ERK signaling pathway, also known as the p42/p44 MAP kinase
pathway, is a major determinant in the control of cell growth,
differentiation and survival (21).
Furthermore, ERK1/2 inhibitors have been administered as
anti-cancer drugs (22). The present
study results revealed that ERK1/2 inhibition reversed the
proliferative effects induced by CSE, which may aid with the
development of novel therapies for bladder cancer. Notably, a
previous study demonstrated that JNK and p38 are involved in
arsenic-induced activating transcription factor 2 activation in
SV-HUC-1 cells (23). However, few
studies have investigated CSE-induced AP-1 activation in SV-HUC-1
cells, as demonstrated in the present study. The results of the
present study revealed that ERK1/2 and JNK regulated the expression
of AP-1 proteins involved in the proliferative effects induced by
CSE (Fig. 4B and D).
The AP-1 transcription factor is a hetero or
homodimeric complex that comprises members of the proto-oncogene
Jun (c-Jun, Jun B and Jun D) and Fos (c-Fos, Fos B, Fra-1 and
Fra-2) protein families. c-Fos and c-Jun are a pair of
transcription factors that, in combination with other associated
proteins, form the AP-1 transcription factor complex (24). AP-1 exhibits a crucial role in the
carcinogenesis of tumors via the promotion of cell proliferation,
invasion and metastasis. c-Jun expression has been found to
positively correlate with an increasing tumor stage in BC (25).
Bladder carcinogenesis has been reported to be
initiated by the clonal expansion of genetically altered cells that
include normal mucosa and premalignant lesions (26). In the present study, CSE treatment
resulted in the upregulation of cyclin D1 and PCNA expression and
the downregulation of p21 expression in SV-HUC-1 cells via the AP-1
pathway. Cyclin D1 and PCNA are AP-1 target genes that are involved
in cell proliferation. Cyclin D1 gene regulatory sequences contain
two AP-1 binding sites. Several AP-1 proteins, including c-Jun and
c-Fos, bind these sites to subsequently activate cyclin D1
expression (27). Cyclin D1 is a gene
that controls the cell cycle and promotes cell cycle progression
through G1-phase by forming active holoenzymes with
cyclin-dependent kinase (CDK)4 and CDK6, which leads to
phosphorylation of retinoblastoma protein (28). Cyclin D1 overexpression has been
demonstrated to be an independent adverse risk factor in
metastasizing BC (29). Similarly,
the PCNA gene contains AP-1 binding sites in the promoter region
and thus its expression is regulated by AP-1 activity (30). Consistent with the results of the
present study, a number of previous studies have demonstrated that
exposure to cigarette smoke and nicotine induces the expression of
PCNA (31,32). Additionally, p21 is a
well-characterized PCNA partner that has been identified in a
protein complex that contains PCNA, cyclin D1 and CDK. The p21
protein exhibits two various inhibitory effects on the entry of a
cell into S phase: Inhibition of the kinase activity of CDK and
inhibition of DNA replication via interactions with PCNA (33). According to the results of the present
study, ERK1/2 and JNK inhibition attenuated the proliferative
effects triggered by CSE.
In addition, the present study indicated that the
inhibition of p38 did not reverse the proliferative effect on
SV-HUC-1 cells induced by CSE. As shown in Fig. 4C, SB203580 did not suppress the
CSE-induced expression of p-c-Fos, which may indicate why the
proliferative effects induced by CSE were not reversed following
SB203580 treatment. However, numerous signaling pathways are
associated with exposure to cigarette smoke, including the nuclear
factor-κB and phosphoinositide 3-kinase/protein kinase B pathways
(31), therefore further study is
required.
The results of the present study confirmed that
exposure to CSE induces proliferation in normal human urothelial
cells. The ERK1/2 and JNK pathways are important in the regulation
of CSE-induced proliferation of SV-HUC-1 cells via the AP-1
pathway. These findings revealed the effects of CSE on the
proliferation of normal human urothelial cells and also highlighted
the importance of the MAPK/AP-1 pathway in the development of
CSE-induced pathogenesis, which may provide novel evidence with
regard to the molecular mechanisms of BC development.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (Beijing, China; grant nos.
1373005, 81072330 and 81202194) and the Priority Academic Program
Development of Jiangsu Higher Education Institutions (Nanjing,
China), the Anhui Public Welfare Research Linkage Plan (Hefei,
China; grant no. 1501ld04045) and the Anhui Medical University
Scientific Research Funds (Hefei, China; grant no. H0514).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boffetta P: Tobacco smoking and risk of
bladder cancer. Scand J Urol Nephrol Suppl. 45–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeegers MP, Kellen E, Buntinx F and van
den Brandt PA: The association between smoking, beverage
consumption, diet and bladder cancer: A systematic literature
review. World J Urol. 21:392–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neto JA Colli, Zen Júnior JH, Del Negro A,
Andreollo NA, Araujo MR and Tincani AJ: Tobacco experimental model
to induce urinary bladder neoplasms. Rev Col Bras Cir. 41:56–60.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nature Rev Cancer.
3:733–744. 2003. View
Article : Google Scholar
|
|
6
|
Zhong CY, Zhou YM, Douglas GC, Witschi H
and Pinkerton KE: MAPK/AP-1 signal pathway in tobacco smoke-induced
cell proliferation and squamous metaplasia in the lungs of rats.
Carcinogenesis. 26:2187–2195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
9
|
Montironi R, Cheng L, Scarpelli M,
Mazzucchelli R and Lopez-Beltran A: How much do you know about
benign, preneoplastic, non-invasive and invasive neoplastic lesions
of the urinary bladder classified according to the 2004 WHO scheme?
Diagn Pathol. 6:312011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chu M, Guo J and Chen CY: Long-term
exposure to nicotine, via ras pathway, induces cyclin D1 to
stimulate G1 cell cycle transition. J Biol Chem. 280:6369–6379.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kosaka M, Kang MR, Yang G and Li LC:
Targeted p21 (WAF1/CIP1) activation by RNAa inhibits hepatocellular
carcinoma cells. Nucleic Acid Ther. 22:335–343. 2012.PubMed/NCBI
|
|
12
|
Tian D, Zhu M, Chen WS, Li JS, Wu RL and
Wang X: Role of glycogen synthase kinase 3 in squamous
differentiation induced by cigarette smoke in porcine
tracheobronchial epithelial cells. Food Chem Toxicol. 44:1590–1566.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Geng H, Zhao L, Liang Z, Zhang Z, Xie D,
Bi L, Wang Y, Zhang T, Cheng L, Yu D and Zhong C: ERK5 positively
regulates cigarette smoke-induced urocystic epithelial-mesenchymal
transition in SV40 immortalized human urothelial cells. Oncol Rep.
34:1581–1588. 2015.PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hecht SS: Progress and challenges in
selected areas of tobacco carcinogenesis. Chem Res Toxicol.
21:160–171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen LM, Nergard JC, Ni L, Rosser CJ and
Chai KX: Long-term exposure to cigarette smoke extract induces
hypomethylation at the RUNX3 and IGF2-H19 loci in immortalized
human urothelial cells. PLoS One. 8:e655132013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang W, Cui S, Ma J, Lu Q, Kong C, Liu T
and Sun Z: Cigarette smoking extract causes hypermethylation and
inactivation of WWOX gene in T-24 human bladder cancer cells.
Neoplasma. 59:216–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sebolt-Leopold JS and Herrera R: Targeting
the mitogen-activated protein kinase cascade to treat cancer. Nat
Rev Cancer. 4:937–947. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mitra AP and Cote RJ: Molecular
pathogenesis and diagnostics of bladder cancer. Annu Rev Pathol.
4:251–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kohno M and Pouyssegur J: Pharmacological
inhibitors of the ERK signaling pathway: Application as anticancer
drugs. Prog Cell Cycle Res. 5:219–224. 2003.PubMed/NCBI
|
|
22
|
Zhao L, Geng H, Liang ZF, Zhang ZQ, Zhang
T, Yu DX and Zhong CY: Benzidine induces epithelial-mesenchymal
transition in human uroepithelial cells through ERK1/2 pathway.
Biochem Biophys Res Commun. 459:643–649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu S, Wang F, Yan L, Zhang L, Song Y, Xi
S, Jia J and Sun G: Oxidative stress and MAPK involved into ATF2
expression in immortalized human urothelial cells treated by
arsenic. Arch Toxicol. 87:981–989. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hess J, Angel P and Schorpp-Kistner M:
AP-1 subunits: Quarrel and harmony among siblings. J Cell Sci.
117:5965–5973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tiniakos DG, Mellon K, Anderson JJ,
Robinson MC, Neal DE and Horne CH: c-jun oncogene expression in
transitional cell carcinoma of the urinary bladder. Br J Urol.
74:757–761. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Crawford JM: The origins of bladder
cancer. Lab Invest. 88:686–693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown JR, Nigh E, Lee RJ, Ye H, Thompson
MA, Saudou F, Pestell RG and Greenberg ME: Fos family members
induce cell cycle entry by activating cyclinD1. Mol Cell Biol.
18:5609–5619. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seiler R, Thalmann GN, Rotzer D, Perren A
and Fleischmann A: CCND1/CyclinD1 status in metastasizing bladder
cancer: A prognosticator and predictor of chemotherapeutic
response. Mod Pathol. 27:87–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gillardon F, Moll I and Uhlmann E:
Inhibition of c-Fos expression in the UV-irradiated epidermis by
topical application of antisense oligodeoxynucleotides suppresses
activation of proliferating cell nuclear antigen. Carcinogenesis.
16:1853–1856. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deng QF, Sun X, Liang ZF, Zhang ZQ, Yu DX
and Zhong CY: Cigarette smoke extract induces the proliferation of
normal human urothelial cells through the NF-κB pathway. Oncol Rep.
35:2665–2672. 2016.PubMed/NCBI
|
|
32
|
Xia S, Kang J, Jiang Y, Huang D, Wang S
and Pang B: Simvastatin promotes alveolar epithelial cell
proliferation and attenuates cigarette smoke-induced emphysema in
rats. Mol Med Rep. 12:5903–5910. 2015.PubMed/NCBI
|
|
33
|
Waga S, Hannon GJ, Beach D and Stillman B:
The p21 inhibitor of cyclin-dependent kinases controls DNA
replication by interaction with PCNA. Nature. 369:574–578. 1994.
View Article : Google Scholar : PubMed/NCBI
|