Introduction
Approximately 1–2% of neoplastic diseases are
located in the central nervous system (1). The majority of these tumors, ~50–60%,
are gliomas that develop more often in Caucasian patients compared
with those of Asian or African origin (2). The incidence of gliomas is 5–11/100,000
in Western Europe, Australia and Northern America (3). The majority of patients are older than
50 years, and males are more often affected than females (4). In Germany, every year ~65 children are
diagnosed with high-grade glioma (HGG) (5). Despite advancements in neurosurgery,
chemotherapy and radiation therapy, the outcome remains poor, with
a median survival subsequent to diagnosis of only 14 months
(6). Glioblastoma multiforme is the
most common type of HGG, accounting for ~50% of cases (7). The tumors may be located in all parts of
the brain, but are mostly located supratentorially within the
cerebral lobes (8).
The histogenetic origin of glioma cells is not clear
yet. Since the beginning of the 20th century, it has been assumed
that the tumor cells develop from precursor glial cells (9,10). An
association with neuroectodermal stem cells appears to be possible
(1).
For glioblastoma, an association between clinical
characteristics, histology and genetics may be observed (11,12). The
malignant progression is associated with an abnormal expression of
proto-oncogenes and tumor-suppressor genes (7,13–15).
The poor outcome following treatment of glioblastoma
is likely associated with the invasive capability of the tumor
cells. Even subsequent to gross total tumor resection, vital tumor
cells may have already migrated into the surrounding brain tissue,
unnoticed during surgical intervention. The infiltrative growth of
glioblastomas is mediated by the interplay of cell invasion, cell
motility and the interaction between matrix proteins and growth
factors (16,17). However, models for glioma cell
migration and invasion are missing. Thus, the adaptation of models
from other types of tumor disease may be a promising method. In
carcinoma, the change from a local carcinoma into an infiltrative
and metastasizing tumor is described by the model of
epithelial-mesenchymal transition (EMT) (18). Under certain circumstances, epithelial
cells may adopt a mesenchymal phenotype (19–22). This
change is marked by the replacement of E-cadherin (CDH1) by
N-cadherin. Another important protein in this process is vimentin
(VIM), which allows the shape of tumor cells to change, thus
enabling these cells to move into the extracellular matrix
(23). Notably, not all cells
undergoing EMT exhibit all the EMT-specific features. Some tumors
exhibit only certain EMT-typical changes or opposed phenotypical
changes in terms of mesenchymal-epithelial transition (24–26).
Several EMT inducers have been described, including
transforming growth factor-β (TGF-β), snail homologs 1 and 2
(SNAI1/SNAI2), twist homolog 1 (TWIST1) and wingless-type mouse
mammary tumor virus integration site family members (WNT) (19–33). The
progression of tumors may also be part of EMT programs (34). The effect of TWIST1, a gene involved
in EMT programs in carcinoma, on the invasion level of glioblastoma
was previously described (35). A
central role of the nuclear factor κ-light-chain-enhancer of
activated B cells (NF-κB) pathway in the induction of EMT has been
revealed in different tumor models (36).
The present study focused on the effect of the
EMT-inducer SNAI1 on the proliferation, migration and invasion of
human glioblastoma cells. SNAI1 belongs to the family of snail
genes. The members of the snail superfamily encode for zinc finger
transcriptions factors and serve an important role during the
development of the mesoderm and transcriptional-repressor functions
(37–40). Another function of SNAI1 is its role
in cell death and cell survival programs (41,42).
SNAI1, as a key EMT-associated gene, is involved in several
pathways, including the TGF-β, WNT, NOTCH and endothelin receptor A
pathways (39,42). In addition, the direct inhibition of
expression of CDH1, the hallmark of EMT, is mediated by members of
the snail superfamily (43).
Materials and methods
Cell culture
The human glioblastoma U87MG (44), T98G (45), LN-18 (46) and U251MG (47) cell lines were donated by Professor M.
Hegi (Department of Clinical Neuroscience, University of Lausanne,
Lausanne, Switzerland) and Dr W. Maes (Laboratory for Thrombosis
Research, Interdisciplinary Research Facility Life Sciences Kulak,
Kortrijk, Belgium), and the human embryonal kidney (HEK) 293T cell
line were obtained from the American Type Culture Collection
(Manassas, VA. Cells were grown in a humidified, 5% CO2
atmosphere at 37°C in Dulbecco's modified Eagle's medium (DMEM; PAA
Laboratories; GE Healthcare Life Sciences, Chalfont, UK) with 10%
fetal calf serum (FCS; Biochrom GmbH, Berlin, Germany), 100 U/ml
penicillin and 100 µg/ml streptomycin (PAA Laboratories; GE
Healthcare Life Sciences) in cell culture flasks. Twice a week, the
cells were washed twice with PBS and detached with a trypsin/EDTA
solution (0.05% trypsin and 0.02% EDTA in PBS; PAA Laboratories; GE
Healthcare Life Sciences). Subsequent to centrifugation at 350 × g
for 7 min, the cells were suspended in DMEM at a ratio of 1:10 in
new cell culture flasks.
Overexpression of SNAI1 in tumor cell
lines
RNA was isolated using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
complementary DNA (cDNA) of SNAI1 from the cell line T98G was
amplified by reverse transcription-polymerase chain reaction
(RT-PCR) using the following PCR primers:
5′-GTTCTTCTGCGCTACTGCTG-3′ (forward) and
5′-GCAGGTATGGAGAGGAAGAGG-3′ (reverse). A total of 2 µl cDNA was
mixed with 2.5 µl 10X buffer (Promega Corporation, Madison, WI,
USA), 1.5 µl MgCl2 (25 mM), 0.2 µl Taq polymerase
(Promega Corporation), 0.5 µl dNTP mix (10 mM; Fermentas; Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA), 0.25 µl
sequence-specific primers (Invitrogen; Thermo Fisher Scientific,
Inc.), 2.5 µl dimethylsulfoxide and 15.3 µl H2O. The
thermocycling conditions were as follows: 95°C for 5 min; 35 cycles
of 94°C for 30 sec, 60°C for 35 sec and 72°C for 45 sec; followed
by 72°C for 5 min and an infinite holding temperature of 4°C. The
PCR product was cloned into the vector pGEM-T Easy (Promega
Corporation). From this vector, SNAI1 was cloned into the
lentiviral vector puc2CL6IN, donated by Professor H. Hanenberg
(Clinic of Pediatrics and Adolescent Medicine III, Essen University
Hospital, Essen, Germany), containing a neomycin resistance
cassette. Sequencing of the vector was performed as previously
described (48). For lentiviral
vector production, the envelope plasmid pczVSV-G and helper plasmid
pCD/NL-BH, both donated by Professor H. Hanenberg, were
co-transfected with puc2CL6IN-SNAI1 or the control vector puc2CL6IN
into HEK293T cells. The resulting lentivirus-containing
supernatants were used to infect U87MG, T98G, LN-18 and U251MG
glioblastoma cells. Three independent transductions were performed.
Selection of transduced cells was performed with 1 mg/ml G418 (PAA
Laboratories; GE Healthcare Life Sciences).
Knockdown of SNAI1 in tumor cell
lines
Potential short hairpin RNA sequences for knockdown
of SNAI1 in tumor cells were identified using the BLOCK-iT™ RNAi
Designer (Invitrogen; Thermo Fisher Scientific, Inc.). The two
oligonucleotides
5′-CGCGTCCCCGCTGCAGGACTCTAATCCATTCAAGAGATGGATTAGAGTCCTGCAGCTTTTTGGAAATT-3′
and
5′-AGGGGCGACGTCCTGAGATTAGGTAAGTTCTCTACCTAATCTCAGGACGTCGAAAAACCTTTAGC-3′
were annealed and cloned in the enhanced green fluorescent protein
(EGFP)-containing lentiviral vector pCL2.THPC, donated by Professor
H. Hanenberg. The production of the lentiviral vectors, infection
and selection of lentivirally transduced U87MG, T98G, LN-18 and
U251MG cell lines was performed as aforementioned. A total of three
independent transductions were performed.
RT-quantitative (q)PCR
Total RNA was isolated from glioblastoma cell lines
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
First strand cDNA synthesis was performed with 2 µg total RNA using
oligo-(dT)12–18 primers (Fermentas; Thermo Fisher
Scientific, Inc.). RT was carried out with Moloney Murine Leukemia
Virus Reverse Transcriptase (Fermentas; Thermo Fisher Scientific,
Inc.), for 1 h at 37°C. A total of 2 µl cDNA was amplified by PCR
[25 (β-actin) or 35 (other targets) cycles of 94°C for 30 sec, 60°C
for 35 sec and 72°C for 45 sec] using Taq polymerase (Promega
Corporation) in a final volume of 25 µl in a Mastercycler personal
(Eppendorf, Hamburg, Germany). All primers were purchased from
Invitrogen; Thermo Fisher Scientific, Inc. The following primers
were used: β-actin (ACTB) forward, 5′-GGCATCGTGATGGACTCCG-3′ and
reverse, 5′-GCTGGAAGGTGGACAGCGA-3′; CDH1 forward,
5′-GCTGGAGATTAATCCGGACA-3′ and reverse, 5′-ACCCACCTCTAAGGCCATCT-3′;
EGFP forward, 5′-ACGTAAACGCCCACAAGTTC-3′ and reverse,
5′-AAGTCGTGCTGCTTCATGTG-3′; lymphoid enhancer-binding factor 1
(LEF1) forward, 5′-AGCACTTTTCTCCAGGGTCA-3′ and reverse,
5′-CCCGTGATGGGATATACAGG-3′; neomycin phosphotransferase II forward,
5′-AGACAATCGGCTGCTCTGAT-3′ and reverse, 5′-AGTGACAACGTCGAGCACAG-3′;
NF-κB1 forward, 5′-CACCTAGCTGCCAAAGAAGG-3′ and reverse,
5′-TCAGCCAGCTGTTTCATGTC-3′; SNAI1 forward,
5′-ACCCCACATCCTTCTCACTG-3′ and reverse, 5′-CCGACAAGTGACAGCCAT-3′;
and TWIST2 forward, 5′-CGAGGAGGAGCTCGAGAGG-3′ and reverse,
5′-CTAGTGGGAGGCGGACAT-3′. RT-qPCR was performed using the Maxima
SYBR-Green qPCR Master Mix kit (Fermentas; Thermo Fisher
Scientific, Inc.) using the following conditions: Denaturation,
94°C for 45 sec; annealing, 62°C for 45 sec; and elongation 72°C
for 60 sec. Each reaction was subjected to melting temperature
analysis to confirm the presence of the expected products. Specific
gene amplification was normalized to ACTB. Target genes and ACTB
were amplified with 40 cycles using a Rotor Gene RG-3000 (Corbett
Life Science; Qiagen GmbH, Hilden, Germany) and Rotor Gene 6
software version 6.1 (build 93; Corbett Life Science; Qiagen GmbH).
Relative expression values were calculated using the
2−ΔΔCq method (49).
Cell proliferation assay
Analysis of cell proliferation was performed with
the MTT Cell Proliferation kit (Roche Applied Science, Mannheim,
Germany) according to the protocol of the manufacturer. The cells
were seeded at a density of 1×103 cells/well in 96-well
plates for 72 h in a humidified, 5% CO2 atmosphere at
37°C. Turnover of MTT was detected using an ELx808 microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA).
Cell migration assay
To analyze tumor cell migration, ThinCert cell
culture inserts with a pore size of 8.0 µm (Greiner Bio-One GmbH,
Frickenhausen, Germany) were used. The tumor cells were washed
twice with PBS, suspended in serum-free medium containing 0.2%
bovine serum albumin (BSA; SERVA Electrophoresis GmbH, Heidelberg,
Germany) and cultured in cell culture flasks overnight in a
humidified, 5% CO2 atmosphere at 37°C. On the next day,
the wells of a 24-well plate were filled with 600 µl medium (DMEM
with 10% FCS, 100 U/ml penicillin and 100 µg/ml streptomycin), and
a ThinCert insert was placed in every well. Subsequently,
2×105 tumor cells in 200 µl serum-free medium (DMEM with
0.2% BSA) were placed in the inserts. After 24 h, the ThinCert
inserts containing the cells were removed and placed in new wells
of a 24-well plate containing 500 µl trypsin/EDTA solution. The
corresponding cell numbers were subsequently determined using a
Neubauer chamber following staining with trypan blue (Invitrogen;
Thermo Fisher Scientific, Inc.).
Matrigel invasion assay
To investigate the invasion capacity of the
different glioblastoma cell lines, a Matrigel invasion chamber (BD
Biosciences, Franklin Lakes, NJ, USA) was used. The cell lines were
harvested and resuspended in DMEM containing 0.1% BSA at a
concentration of 5×104 cells/ml. Matrigel invasion and
BD BioCoat Control inserts (BD Biosciences) were filled with 0.5 ml
DMEM pre-warmed to 37°C containing 0.1% BSA. After 2 h the medium
was removed and the inserts were transferred into 24-well plates
containing DMEM with 10% FCS as a chemoattractant. Immediately
thereafter, 0.5 ml cell suspension was added to the inside of the
insert. The Matrigel invasion and control chambers were incubated
for 24 h in a humidified, 5% CO2 atmosphere at 37°C.
Inserts with invaded cells were stained with Diff-Quick (Medion
Grifols Diagnostics AG, Düdingen, Switzerland). To determine the
number of invaded cells, membranes were cut and placed on a
microscope slide with a drop of immersion oil, and the cells were
counted using an Axiolab light microscope (Zeiss GmbH, Jena,
Germany). To calculate the percentage of invading cells, the mean
number of cells invading into the Matrigel insert membrane was
divided by the mean number of cells migrating through the control
inserts and multiplied by 100.
Statistical analysis
Statistical analysis was performed using Microsoft
Excel 2010 (version 14.0.7128.5000; Microsoft Corporation, Redmont,
WA, USA), Student's t-test was used to analyze differences.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Modulation of EMT-associated genes by
overexpression of SNAI1 in glioblastoma cells
To investigate the effect of SNAI1 on glioblastoma
cells, glioblastoma U87MG, T98G, LN-18 and U251MG cell lines were
transduced with lentiviral vectors allowing the overexpression or
inhibition of SNAI1. Neither SNAI1 overexpression nor inhibition
changed the microscopic appearance (size and shape) of the cells.
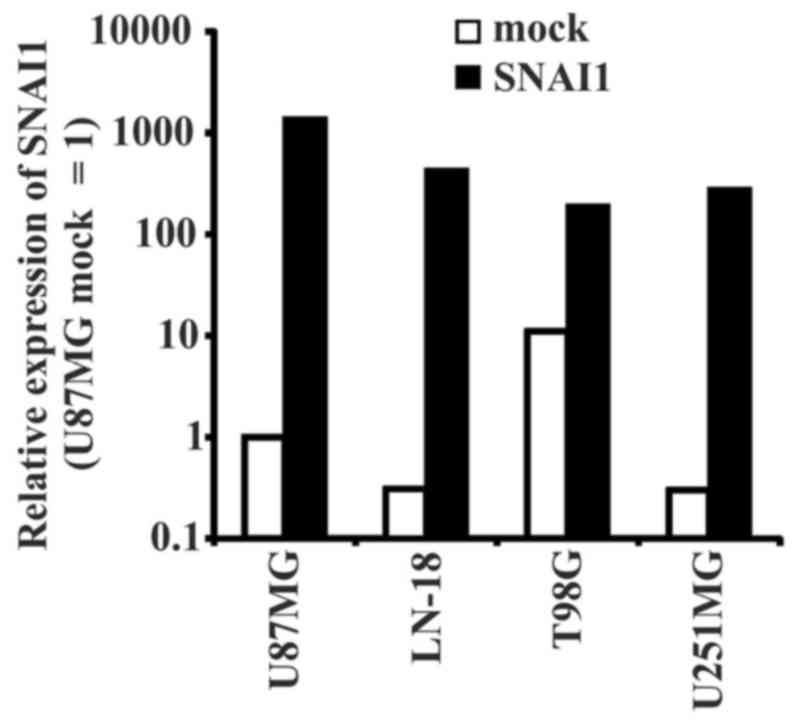
SNAI1 overexpression was confirmed by RT-qPCR analysis (Fig. 1). In all used glioma cell lines, the
expression of SNAI increased following transduction. A
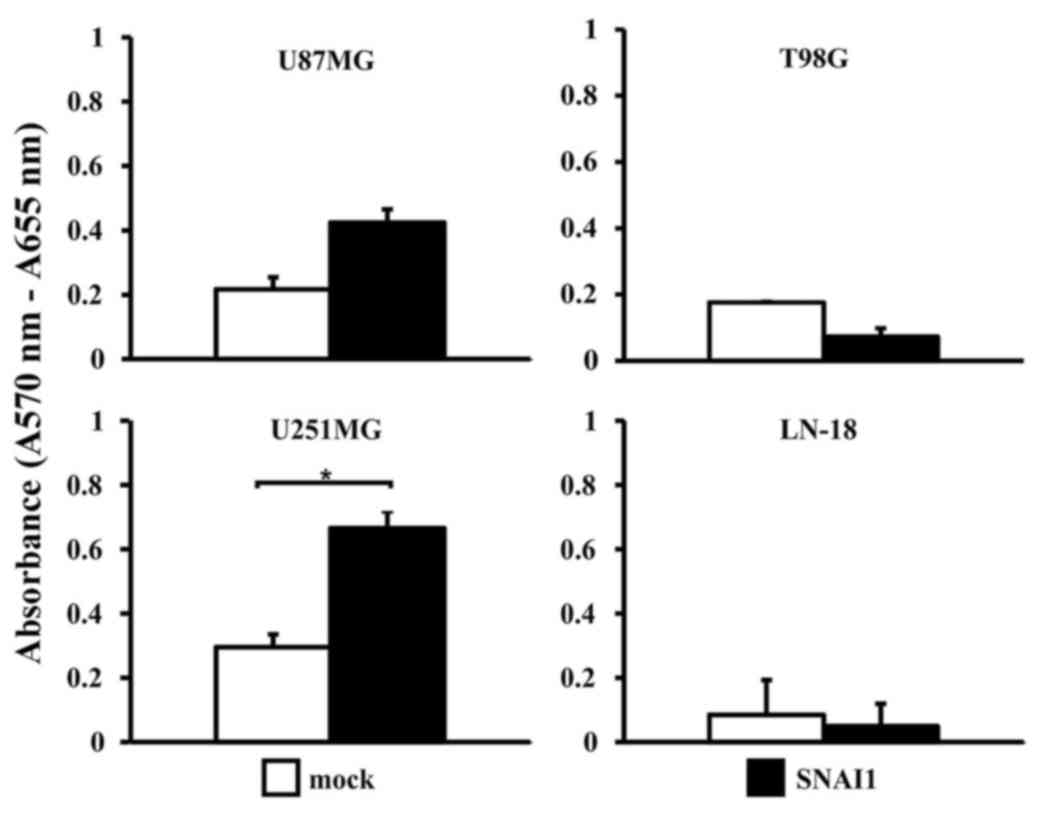
significantly (P<0.05) increased proliferation rate of
SNAI1-overexpressing U251MG cells compared with the vehicle control
(Fig. 2) was observed. The three
other cell lines exhibited no significant proliferative changes
upon SNAI1 overexpression. Furthermore, none of the cell lines
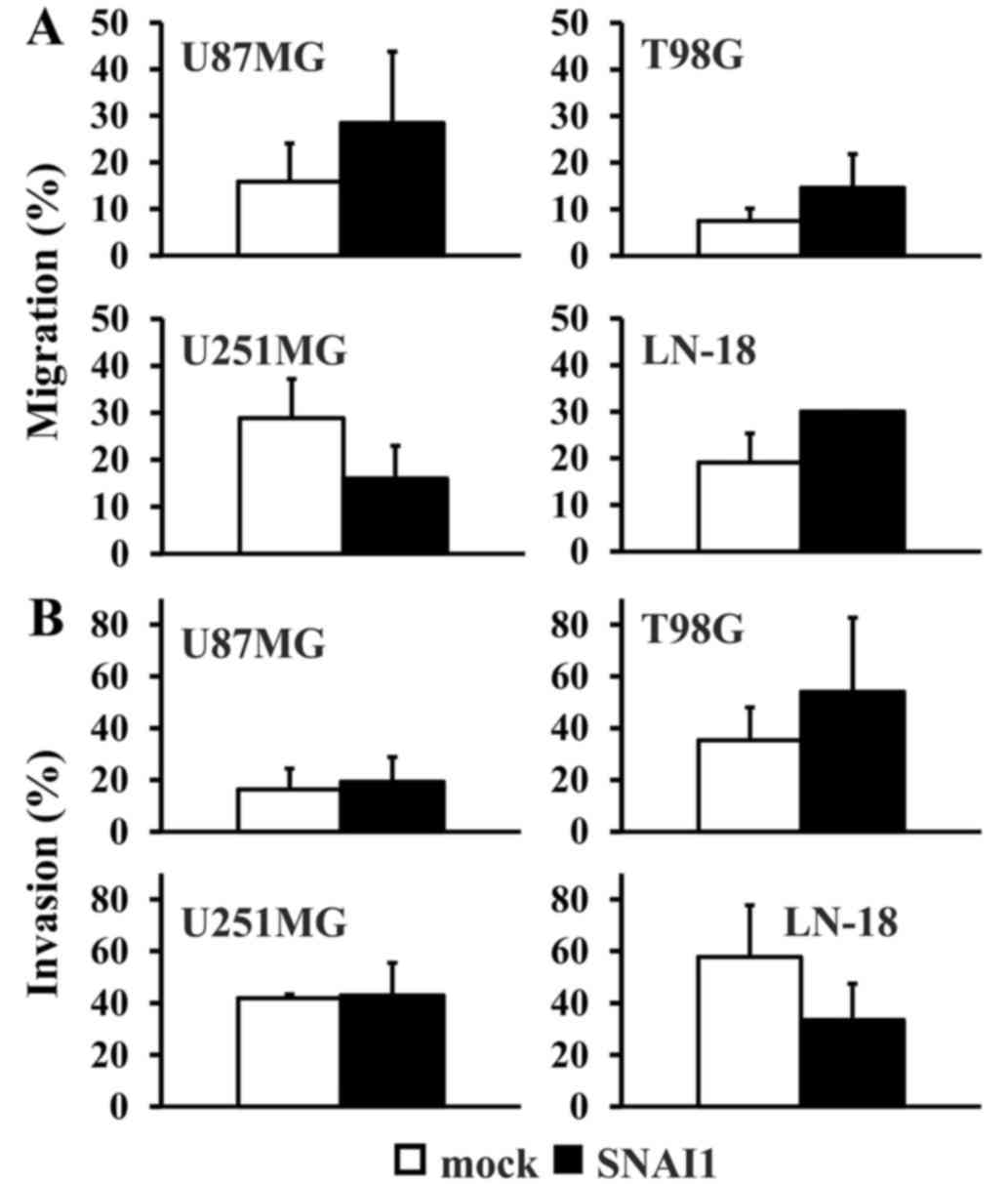
exhibited significant changes on their migration rate, although
there was a tendency towards a relative higher migration rate in
the U87MG, T98G and LN-18 cell lines (Fig. 3A). Similarly, no significant
differences in the invasion rate were detectable when comparing
each cell type to the control (Fig.
3B).
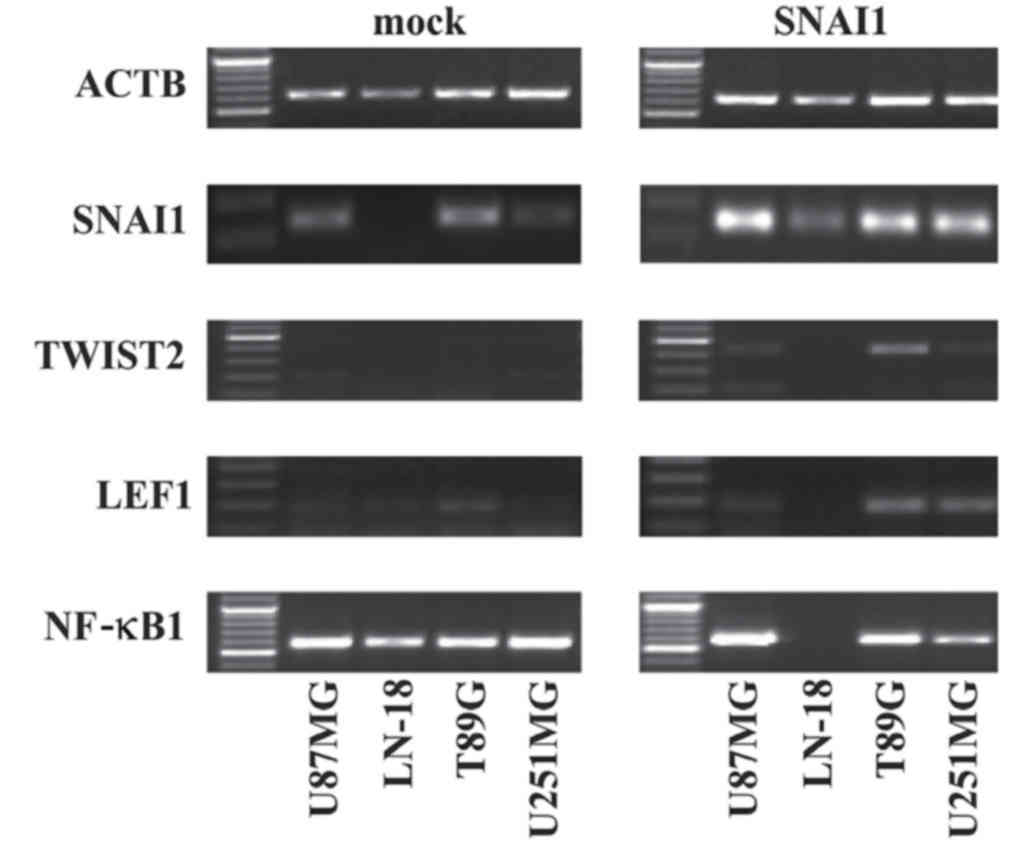
The present study investigated whether EMT target
genes may be affected by the overexpression of SNAI1 in
glioblastoma cells. The regulation of TWIST2 in three of four
glioma cell lines, U87MG, T98G and U251MG, upon SNAI1
overexpression (Fig. 4) was observed.
In addition, RT-PCR revealed an increased expression of NF-κB1 in
SNAI1-overexpressing U87MG and T98G cells. On the contrary, the
overexpression of SNAI1 resulted in the decreased expression of
NF-κB1 in LN-18 cells and U251MG cells. LEF1 exhibited low
expression in untreated U87MG, LN-18 and T98G cells. An increased
LEF1 expression was also detected in T98G and U251MG cells upon
SNAI1 overexpression (Fig. 4). No
differences were observed for VIM, which was stably detectable in
U251MG, T98G and U87MG cells. CDH1 was absent from all cell lines
independent from SNAI1 transduction (data not shown; see also
below; Fig. 5A).
Re-induction of CDH1 subsequent to
knockdown of SNAI1 in glioblastoma cells
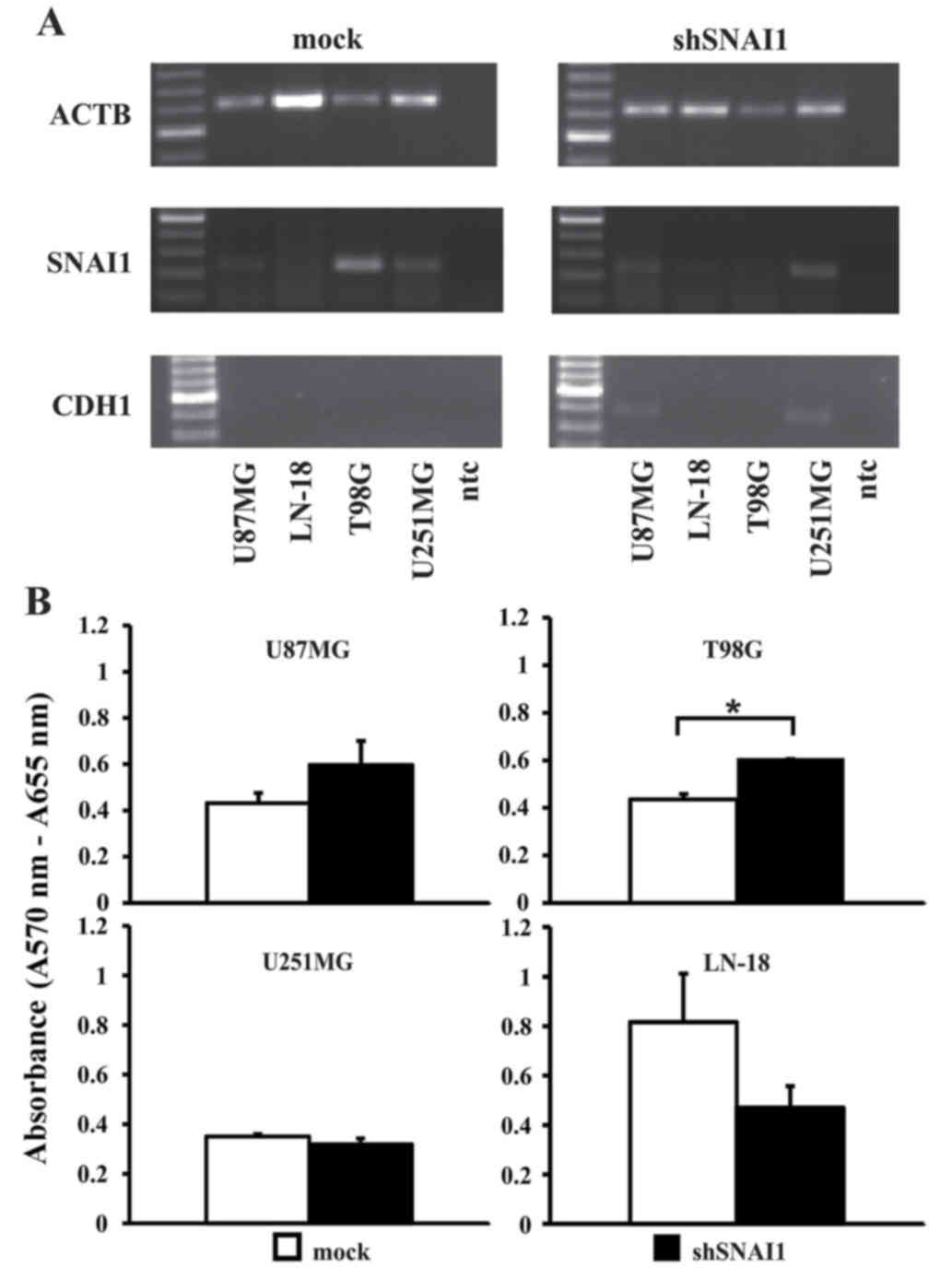
Upon SNAI1 inhibition, as confirmed by RT-PCR
analysis (Fig. 5A), a significantly
higher proliferation rate in the cell line T98G was observed. In
all the other cell lines, no significant effect on cellular
proliferation was observed (Fig. 5B).
Loss of CDH1 is a central step during EMT, accompanying the
establishment of a highly infiltrative and metastasizing tumor
phenotype (50). Thus, it was notable
to observe that two out of four cell lines (U87MG and U251MG;
Fig. 5A) regained CDH1 expression
upon inhibition of SNAI1. No changes in the expression of VIM were
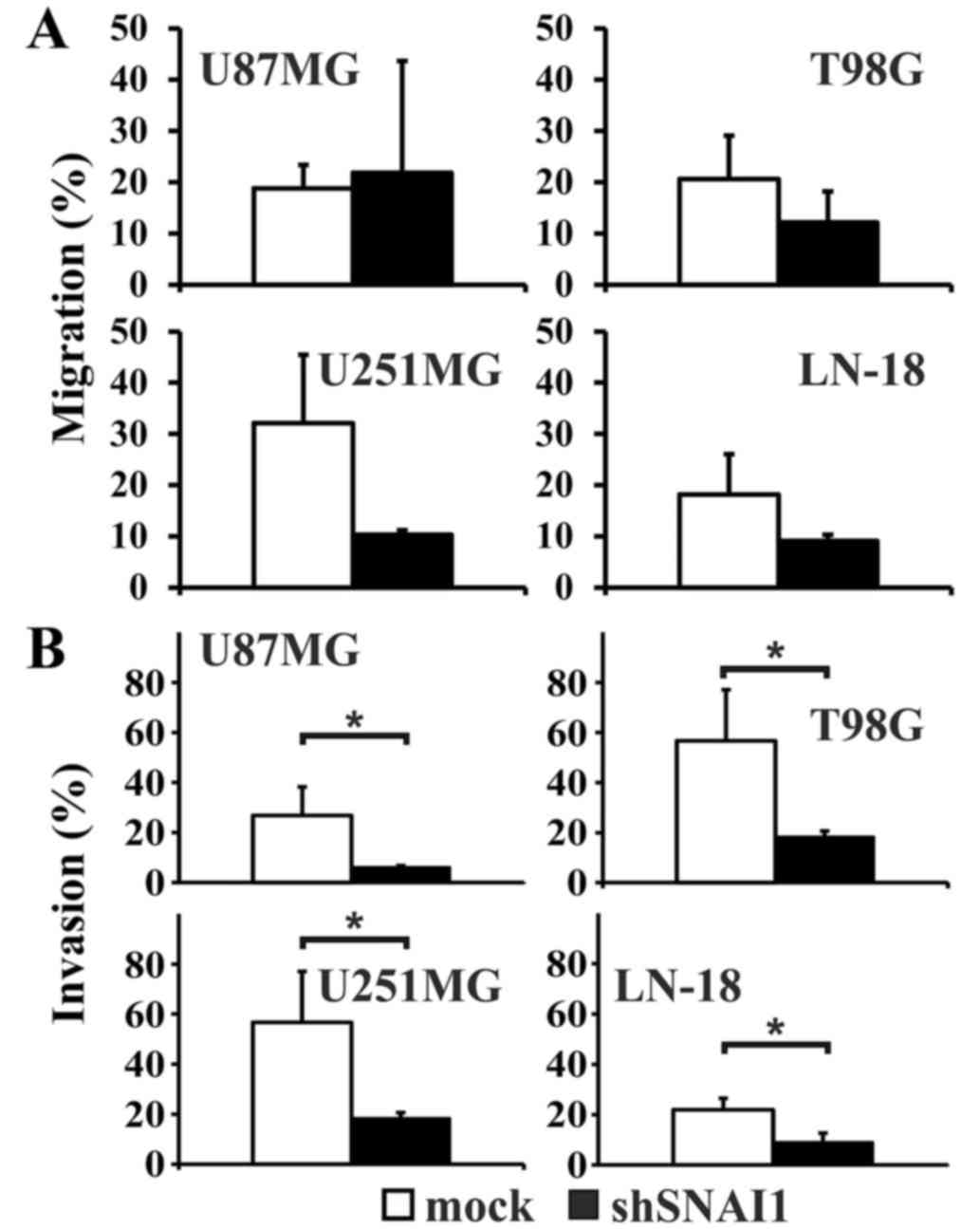
observed (data not shown). The migration rate appeared relatively
decreased compared with the mock transfected cells in the cell
lines T98G, LN-18 and U251MG, but the differences did not reach
significance (Fig. 6A). In U87MG
cells, the migration rate was enhanced. However, the invasion
capability significantly decreased in all four human glioma cell
lines subsequent to SNAI1 inhibition (Fig. 6B).
Discussion
EMT represents a well-established model explaining
the switch from a local into an infiltrating and metastasizing
carcinoma phenotype on a signal transduction level (50). The present study investigated if this
model may also be adaptable to human glioblastoma cells, which are
characterized by extensively infiltrating tumor cells (16,17).
A key gene of EMT is SNAI, which is involved in
different signaling pathways that downregulate CDH1 expression as a
hallmark of EMT (50). A baseline
SNAI1 expression in all investigated four human glioma cell lines,
U87MG, T98G, U251MG and LN-18, was observed in the present study.
High SNAI1 baseline expression may contribute to the infiltrative
and migratory phenotype of glioblastoma cells, similar to
EMT-associated functions of SNAI in carcinoma cells (51). Growth inhibition of U87MG cells
subsequent to knockdown of SNAI1 has been described (37). The present study did not observe
inhibition of proliferation subsequent to SNAI1 knockdown in any of
the investigated cell lines. This divergence may be a consequence
of experimental differences. For instance, viral transduction was
used for genetic engineering, whereas in the study by Han et
al (37), transfection of small
interfering RNA was performed. In addition, proliferation was not
investigated directly in the present study. Han et al
(37) used a viability assay that
depends on an intact respiratory chain, whereas the present study
used an assay that depends on intact glycolysis. The effect of
SNAI1 on the proliferation of glioma cells requires additional
investigation.
The SNAI baseline expression may also be responsible
for the observation of the present study that the additional
increase in SNAI expression subsequent to lentiviral gene transfer
did not result in significantly increased cell migration or
invasion in any investigated cell line, although there was at least
a tendency towards increased cell migration subsequent to SNAI
overexpression in the majority of the cell lines.
RT-PCR analyses of EMT target genes in the four
glioblastoma cell lines also exhibited heterogeneous results. Mock
transfected cell lines displayed no baseline expression of TWIST2,
but the overexpression of SNAI1 induced a high expression of TWIST2
in three of four cell lines. TWIST2 is known as a direct inducer of
EMT (52,53). In addition, there is evidence that
TWIST1 may act as a potential oncogene in gliomas exhibiting an
increased expression of TWIST1 during the transformation from
low-grade glioma to HGG (54). There
appear to be different signaling pathways involved in TWIST
activation upon the induction of SNAI expression in the various
glioblastoma cell models used in the present study. In U87MG and
T98G cells, the activation of TGF-β-dependent signaling pathways
appeared to be involved, as these cells exhibited increased NF-κB1
expression upon induction of SNAI expression; NF-κB1 is one of the
key genes of the TGF-β pathway, but may also be involved in other
pathways (55). However, in LN-18 and
U251MG cells, NF-κB1-associated signaling did not appear to be
driving TWIST expression subsequent to SNAI gene transfer. In these
cell lines, NF-κB1 expression decreased subsequent to the
overexpression of SNAI1.
In T98G and U251MG cells, the WNT signaling pathway
appears to be activated subsequent to SNAI gene transfer. In the
present study, increased expression of TWIST and LEF1 as target
genes of the canonical WNT pathway was observed. However, the
present study did not additionally corroborate if the activation of
WNT signaling led to TWIST expression or vice versa. The
involvement of WNT and TWIST in EMT is well known, and it has been
shown that TWIST may activate canonical WNT signaling in carcinoma
(56).
However, the involvement of SNAI1 in EMT-like
pathways appeared much clearer following inhibition of SNAI
expression compared with overexpression of SNAI1. Subsequent to the
inhibition of SNAI1 by lentiviral knockdown, re-expression of CDH1
in U87MG and U251MG cells was observed. Since in carcinoma cells
the downregulation of CDH1 expression represents the hallmark of
EMT induction, the finding of the re-induction of CDH1 expression
subsequent to SNAI inhibition may provide indirect evidence that
there may be an EMT-like phenotype in human glioblastoma. CDH1 was
not expressed in wild-type or SNAI1-overexpressing glioblastoma
cells, but was induced in half of the cell lines subsequent to the
knockdown of SNAI1. This observation is in agreement with the
significantly lower cellular invasion capabilities observed
subsequent to the inhibition of SNAI expression.
Cell lines are suitable models for glioblastoma
research in vitro (57,58).
However, cell culture-dependent effects may also be observed with
respect to the behavior of tumor cells, and certain widely used
tumor cell lines do not exhibit the typical glioblastoma growth
pattern in xenotransplantation models (59). Therefore, the analysis of SNAI1 and
interacting factors in patient-derived material is required.
Taken together, the data of the present study may
indicate that EMT-like processes similar to those in carcinoma may
also occur in human glioblastoma, thereby potentially contributing
to their aggressive and invasive phenotype. However, systematic
investigation of the key EMT genes and associated signaling
pathways is required to obtain a more comprehensive overview, as
the signaling network may be the same as that in carcinoma or there
may be differences. In addition, an analysis of the involved
factors at the protein level will increase the level of information
about the relevant pathways.
Acknowledgements
The authors would like to thank Professor Monika
Hegi (Department of Clinical Neuroscience, University of Lausanne,
Lausanne, Switzerland) and Dr Wim Maes (Laboratory for Thrombosis
Research, Interdisciplinary Research Facility Life Sciences Kulak,
Kortrijk, Belgium) for providing the glioblastoma cell lines, as
well as Professor Helmut Hanenberg (Clinic of Pediatrics and
Adolescent Medicine III, Essen University Hospital, Essen, Germany)
for providing the vectors pczVSV-G, pCD/NL-BH, pCL2.THPC, pcl2.THPC
and puc2CL6IN.
References
|
1
|
Kleihues P, Louis DN, Scheithauer BW,
Rorke LB, Reifenberger G, Burger PC and Cavenee WK: The WHO
classification of tumours of the nervous system. J Neuropathol Exp
Neurol. 61:215–225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Legler JM, Ries LA, Smith MA, Warren JL,
Heineman EF, Kaplan RS and Linet MS: Cancer surveillance series
(corrected): Brain and other central nervous system cancers: Recent
trends in incidence and mortality. J Natl Cancer Inst.
91:1382–1390. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kramm C, Rausche U, Butenhoff S, Kühnöl C,
Kunze C, Kortmann R, Wolff J and van Gool S: Hochmaligne Gliome im
Kindes-und Jugendalter. Monatschr Kinderheilkd. 156:1201–1207.
2008. View Article : Google Scholar
|
|
6
|
Stupp R and Weber DC: The role of radio-
and chemotherapy in glioblastoma. Onkologie. 28:315–317.
2005.PubMed/NCBI
|
|
7
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Larjavaara S, Mäntylä R, Salminen T,
Haapasalo H, Raitanen J, Jääskeläinen J and Auvinen A: Incidence of
gliomas by anatomic location. Neuro Oncol. 9:319–325. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith MT, Ludwig CL, Godfrey AD and
Armbrustmacher VW: Grading of oligodendrogliomas. Cancer.
52:2107–2114. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bailey P and Cushing H: Microchemical
color reactions as an Aid to the Identification and classification
of brain tumours. Proc Natl Acad Sci USA. 11:82–84. 1925.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Collins VP: Gene amplification in human
gliomas. Glia. 15:289–296. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dirks PB and Rutka JT: Current concepts in
neuro-oncology: The cell cycle - a review. Neurosurgery.
40:1000–1015. 1997. View Article : Google Scholar
|
|
13
|
Ohgaki H, Dessen P, Jourde B, Horstmann S,
Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM,
Maiorka PC, et al: Genetic pathways to glioblastoma: A
population-based study. Cancer Res. 64:6892–6899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmidt MC, Antweiler S, Urban N, Mueller
W, Kuklik A, Meyer-Puttlitz B, Wiestler OD, Louis DN, Fimmers R and
von Deimling A: Impact of genotype and morphology on the prognosis
of glioblastoma. J Neuropathol Exp Neurol. 61:321–328. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nozaki M, Tada M, Kobayashi H, Zhang CL,
Sawamura Y, Abe H, Ishii N and Van Meir EG: Roles of the functional
loss of p53 and other genes in astrocytoma tumorigenesis and
progression. Neuro Oncol. 1:124–137. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rao JS: Molecular mechanisms of glioma
invasiveness: The role of proteases. Nat Rev Cancer. 3:489–501.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maria BL, Eskin TA and Quisling RG:
Brainstem and other malignant gliomas: II. Possible mechanisms of
brain infiltration by tumour cells. J Child Neurol. 8:292–305.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: Parallels between
normal development and tumour progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Townsend TA, Wrana JL, Davis GE and
Barnett JV: Transforming growth factor-beta-stimulated endocardial
cell transformation is dependent on Par6c regulation of RhoA. J
Biol Chem. 283:13834–13841. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Greenburg G and Hay ED: Epithelia
suspended in collagen gels can lose polarity and express
characteristics of migrating mesenchymal cells. J Cell Biol.
95:333–339. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumour metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Savanger P: Leaving the neighbourhood:
Molecular mechanisms involved during epithelial-mesenchymal
transition. Bioessays. 23:912–923. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boyer B, Vallès AM and Edme N: Induction
and regulation of epithelial-mesenchymal transition. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chaffer CL, Thompson EW and Williams ED:
Mesenchymal to epithelial transition in development and disease.
Cells Tissues Organs. 185:7–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ozdamar B, Bose R, Barrios-Rodiles M, Wang
HR, Zhang Y and Wrana JL: Regulation of the polarity protein Par6
by TGFbeta receptors controls epithelial cell plasticity. Science.
307:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ikenouchi J, Matsuda M, Furuse M and
Tsukita S: Regulation of tight junctions during the
epithelium-mesenchyme transition: Direct repression of the gene
expression of claudins/occludin by Snail. J Cell Sci.
116:1959–1967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumour
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Savagner P, Yamada KM and Thiery JP: The
zinc-finger protein slug causes desmosome dissociation, an initial
and necessary step for growth factor-induced epithelial-mesenchymal
transition. J Cell Biol. 137:1403–1419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumour metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim K, Lu Z and Hay ED: Direct evidence
for a role of beta-catenin/LEF-1 signaling pathway in induction of
EMT. Cell Biol Int. 26:463–476. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Logullo AF, Nonogaki S, Pasini FS, Osório
CA, Soares FA and Brentani MM: Concomitant expression of
epithelial-mesenchymal transition biomarkers in breast ductal
carcinoma: Association with progression. Oncol Rep. 23:313–320.
2010.PubMed/NCBI
|
|
35
|
Mikheeva SA, Mikheev AM, Petit A, Beyer R,
Oxford RG, Khorasani L, Maxwell JP, Glackin CA, Wakimoto H,
González-Herrero I, et al: TWIST1 promotes invasion through
mesenchymal change in human glioblastoma. Mol Cancer. 9:1942010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu QC, Gao RY, Wu W and Qin HL:
Epithelial-mesenchymal transition and its role in the pathogenesis
of colorectal cancer. Asian Pac J Cancer Prev. 14:2689–2698. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han SP, Kim JH, Han ME, Sim HE, Kim KS,
Yoon S, Baek SY, Kim BS and Oh SO: SNAI1 is involved in the
proliferation and migration of glioblastoma cells. Cell Mol
Neurobiol. 31:489–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Paznekas WA, Okajima K, Schertzer M, Wood
S and Jabs EW: Genomic organization, expression and chromosome
location of the human SNAIL gene (SNAI1) and a related processed
pseudogene (SNAI1P). Genomics. 62:42–49. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Inukai T, Inoue A, Kurosawa H, Goi K,
Shinjyo T, Ozawa K, Mao M, Inaba T and Look AT: SLUG, a
ces-1-related zinc finger transcription factor gene with
antiapoptotic activity, is a downstream target of the E2A-HLF
oncoprotein. Mol Cell. 4:343–352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rosanò L, Cianfrocca R, Spinella F, Di
Castro V, Nicotra MR, Lucidi A, Ferrandina G, Natali PG and Bagnato
A: Acquisition of chemoresistance and EMT phenotype is linked with
activation of the endothelin A receptor pathway in ovarian
carcinoma cells. Clin Cancer Res. 17:2350–2360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Beckman G, Beckman L, Pontén J and
Westermark B: G-6-PD and PGM phenotypes of 16 continuous human
tumor cell lines. Evidence against cross-contamination and
contamination by HeLa cells. Hum Hered. 21:238–241. 1971.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stein GH: T98G: An anchorage-independent
human tumor cell line that exhibits stationary phase G1 arrest in
vitro. J Cell Physiol. 99:43–54. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Diserens AC, de Tribolet N, Martin-Achard
A, Gaide AC, Schnegg JF and Carrel S: Characterization of an
established human malignant glioma cell line: LN-18. Acta
Neuropathol. 53:21–28. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pontén J and Macintyre EH: Long term
culture of normal and neoplastic human glia. Acta Pathol Microbiol
Scand. 74:465–486. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Neumann I, Foell JL, Bremer M, Volkmer I,
Korholz D, Burdach S and Staege MS: Retinoic acid enhances
sensitivity of neuroblastoma cells for imatinib mesylate. Pediatr
Blood Cancer. 55:464–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Casas E, Kim J, Bendesky A, Ohno-Machado
L, Wolfe CJ and Yang J: Snail2 is an essential mediator of
Twist1-induced epithelial mesenchymal transition and metastasis.
Cancer Res. 71:245–254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li Y, Wang W, Wang W, Yang R, Wang T, Su
T, Weng D, Tao T, Li W, Ma D, et al: Correlation of TWIST2
up-regulation and epithelial-mesenchymal transition during
tumorigenesis and progression of cervical carcinoma. Gynecol Oncol.
124:112–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang
Z, Yang CJ, Yuan L and Ouyang G: Twist2 contributes to breast
cancer progression by promoting an epithelial-mesenchymal
transition and cancer stem-like cell self-renewal. Oncogene.
30:4707–4720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Elias MC, Tozer KR, Silber JR, Mikheeva S,
Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA,
et al: TWIST is expressed in human gliomas and promotes invasion.
Neoplasia. 7:824–837. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Maier HJ, Schmidt-Strassburger U, Huber
MA, Wiedemann EM, Beug H and Wirth T: NF-kappaB promotes
epithelial-mesenchymal transition, migration and invasion of
pancreatic carcinoma cells. Cancer Lett. 295:214–228. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Luo GQ, Li JH, Wen JF, Zhou YH, Hu YB and
Zhou JH: Effect and mechanism of the Twist gene on invasion and
metastasis of gastric carcinoma cells. World J Gastroenterol.
14:2487–2493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wick W, Weller M, Weiler M, Batchelor T,
Yung AW and Platten M: Pathway inhibition: Emerging molecular
targets for treating glioblastoma. Neuro Oncol. 13:566–579. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Niclou SP, Fack F and Rajcevic U: Glioma
proteomics: Status and perspectives. J Proteomics. 73:1823–1838.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Frosina G: Development of therapeutics for
high grade gliomas using orthotopic rodent models. Curr Med Chem.
20:3272–3299. 2013. View Article : Google Scholar : PubMed/NCBI
|