Introduction
Hepatocellular carcinoma (HCC) is a fatal disease
(1). Several types of potentially
curative treatments have become available due to the advances in
technology and surgical techniques (2). Despite these advances in the clinical
treatment of HCC, the mortality rate remains high (3). Thus, novel potential therapies for HCC
are urgently required.
As potential therapy targets in malignant cells
microRNA (miRNA/miR) perform important roles in regulating gene
expression (4). Dysregulation of
miRNA is widely involved in human cancer, including HCC (5). A previous study indicated that the
mutation of the miR-122 binding site in the interleukin-1α
3′ untranslated region is associated with the incidence rate of HCC
(6). As a potential tumor suppressor
miRNA, miR-1285-3p has been shown to regulate the expression
of JUN (Jun Proto-Oncogene, AP-1 Transcription Factor
Subunit) in the disease progression of HCC (7). The regulation of feedback between miRNAs
and target genes performs important roles in the progression of HCC
(8). Downregulated miR-148b
has been shown to be a biomarker for the early detection of HCC
(9). Previous miRNA-based HCC gene
therapy studies have demonstrated significant inhibition of miRNA
in HCC cells, indicating a promising alternative to current
therapeutics (10–12). However, the association between miRNA
and their target genes, as well as the influence of these
associations during the process of HCC is rarely studied.
miRNA expression profiling has proven useful in
diagnosing and understanding the development and progression of
several diseases including HCC (13–15). The
use of miRNA expression profiling as a prognostic biomarker for the
detection of HCC is practicable (16). Since chronic hepatitis B virus (HBV)
infection is a significant cause of HCC, the present study
performed a bioinformatics analysis based on the miRNA expression
profile of the HBV infected HCC tissue samples and chronic
Hepatitis B (CHB) tissue samples with no fibrosis from 12 patients
with HCC. The present study may help to explore the potential
disease progression of HCC resulting from CHB and provide
information regarding HCC-related miRNA/marker genes for the gene
therapy of HCC.
Materials and methods
Samples and microarray data
Gene expression profile data GSE67882 was downloaded
from the Gene Expression Omnibus (GEO) database (17) (http://www.ncbi.nlm.nih.gov/geo/) based on the
platform GPL10850; Agilent-021827 Human miRNA Microarray (V3;
Agilent Technologies, Inc., Santa Clara, CA, USA). Since the study
was performed based on the raw microarray data retrieved from the
GEO database, all differential expressed miRNAs meeting the
threshold of adjusted P<0.05 and |log2FC (fold
change)|>0.52 were analyzed. A total of 14 upregulated miRNAs
and 16 downregulated miRNAs were obtained. The present study was
performed by using data from 4 HBV infected HCC tissue samples (HCC
group) and 8 CHB tissue samples with no fibrosis (control group) of
GSE67882 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67882).
The microarray data was downloaded on July 24, 2015, and the
bioinformatic analysis was completed on August 12, 2015.
Data preprocessing and differential
expression analysis
The normalization (robust estimation of
variance-stabilizing and calibrating transformations) of gene
expression profile data was performed using Vsn (18) (http://bioconductor.org/help/search/index.html?q=vsn/)
of Bioconductor in R (v.3.0.0). The Linear Models for Microarray
Data (limma, http://www.bioconductor.org/packages/release/bioc/html/limma.html)
package (19) in Bioconductor was
applied to identify differentially expressed miRNAs by comparing
the expression levels between samples in the HCC group and the
control group. The corresponding P-value of miRNAs subsequent to an
unpaired t-test was defined as the adjusted P-value.
Subsequently, the adjusted P<0.05 and
|log2FC|>0.52 were selected as the threshold values
for screening.
Analysis for the target genes
regulated by differentially expressed miRNAs
Since the inhibition of gene expression following
transcription is realized by the combination of miRNA and mRNA,
differentially expressed miRNAs in HCC samples were further
analyzed to explore the significance of miRNA in liver cancer. In
the present study, two experimental certificate databases including
miRecords (20) and miRWalk (21) were used to explore suitable
miRNA-target gene relations. The relations that existed in at least
one kind of database aforementioned were selected, then the target
genes of differentially expressed miRNAs identified by miRecords or
miRWalk were obtained.
Gene Ontology (GO) annotation and
pathway analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; David Bioinformatics Resource)
(22) is a gene functional
classification tool that provides a comprehensive set of functional
annotation tools for investigators to understand the biological
meaning behind large lists of genes. GO functional enrichment
analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathways (http://www.genome.jp/kegg/pathway.html) enrichment
analysis (23) of DEGs were performed
using DAVID. GO has 3 functional categories: Molecular function
(MF); biological process (BP); and cellular component (CC).
P<0.05 was considered to indicate a statistically significant
difference.
Transcription factor-miRNA association
investigation
Each miRNA may have several target genes, but
outstanding target genes are more likely to assemble transcription
factors (24). Thus, the analysis
between transcription factors and miRNA is beneficial to understand
this pathomechanism. iRegulon can be used to detect transcription
factors, motifs and their optimal sets of direct targets from a set
of genes (25). In the present study,
the iRegulon in Cytoscape software was used to explore the
potential associations between miRNA and transcription factors. The
minimum identity between orthologous genes was 0.05, while the
maximum false discovery rate on motif similarity was 0.001. The
normalized enrichment score (NES) of >4 was considered as the
threshold value for the selection of potential associations.
HCC-associated miRNA
investigation
The Comparative Toxicogenomics Database (CTD) is a
robust, publicly available database that is used to advance
understanding regarding how environmental exposures affect human
health (26). In the present study,
the investigation of HCC-associated miRNA was performed using the
CTD database. The biomarkers of a disease are generally identified
by gene expression traits, abnormalities in gene structure or
mutation which could perform roles in the etiology of a disease.
Thus miRNAs which regulate these genes may be important for
HCC.
Results
Investigation into differentially
expressed miRNAs
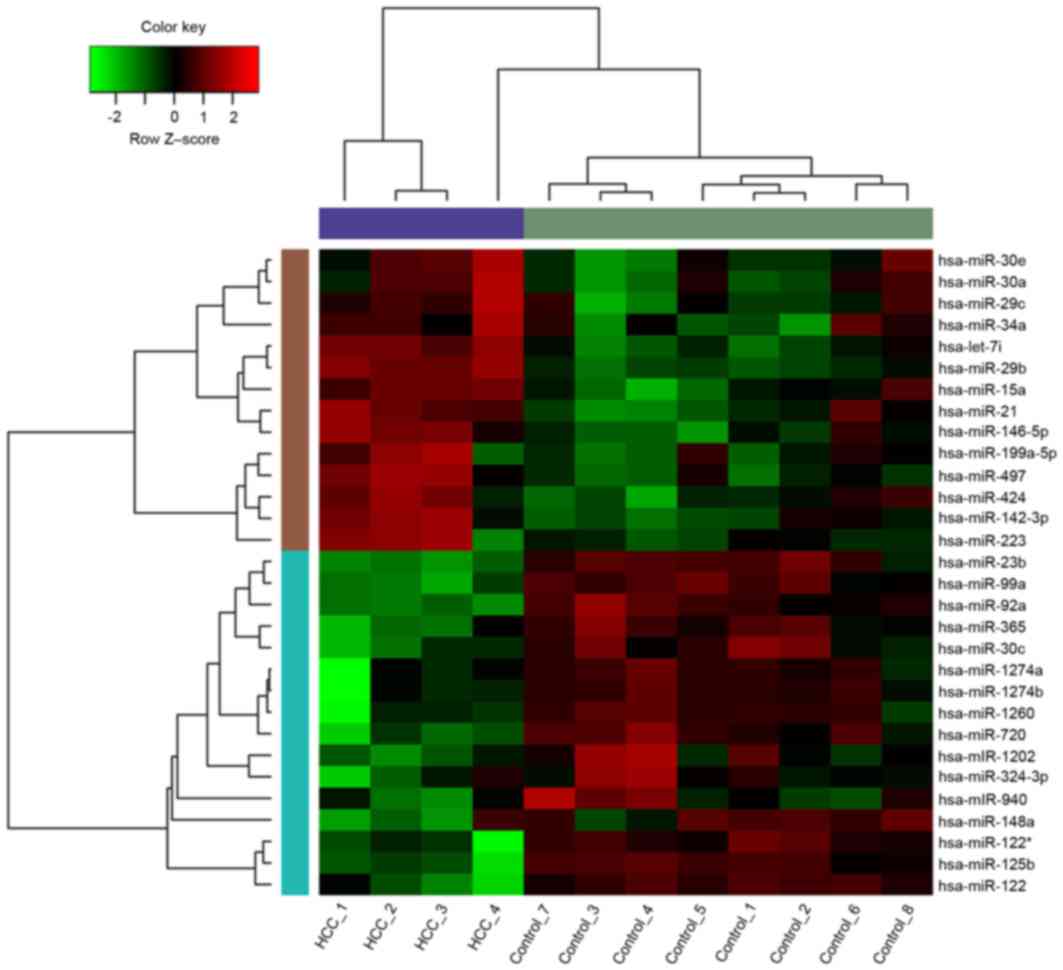
The original data were analyzed and filtered. A
total of 14 upregulated miRNAs and 16 downregulated miRNAs were
obtained with thresholds of P<0.05 and
|log2FC|>0.52. The heat map of differentially
expressed miRNAs is displayed in Fig.
1.
Analysis of differentially expressed
miRNAs and their associated target genes
Based on two experimental certificate databases
(miRecords and miRWalk), a total of 380 suitable miRNA-target gene
relations were obtained. A total of 22 miRNAs including
hsa-miR-15a, hsa-miR-125b, hsa-miR-122, hsa-miR-146b-5p and
hsa-miR-142-3p were revealed to have experimental
certificate target genes.
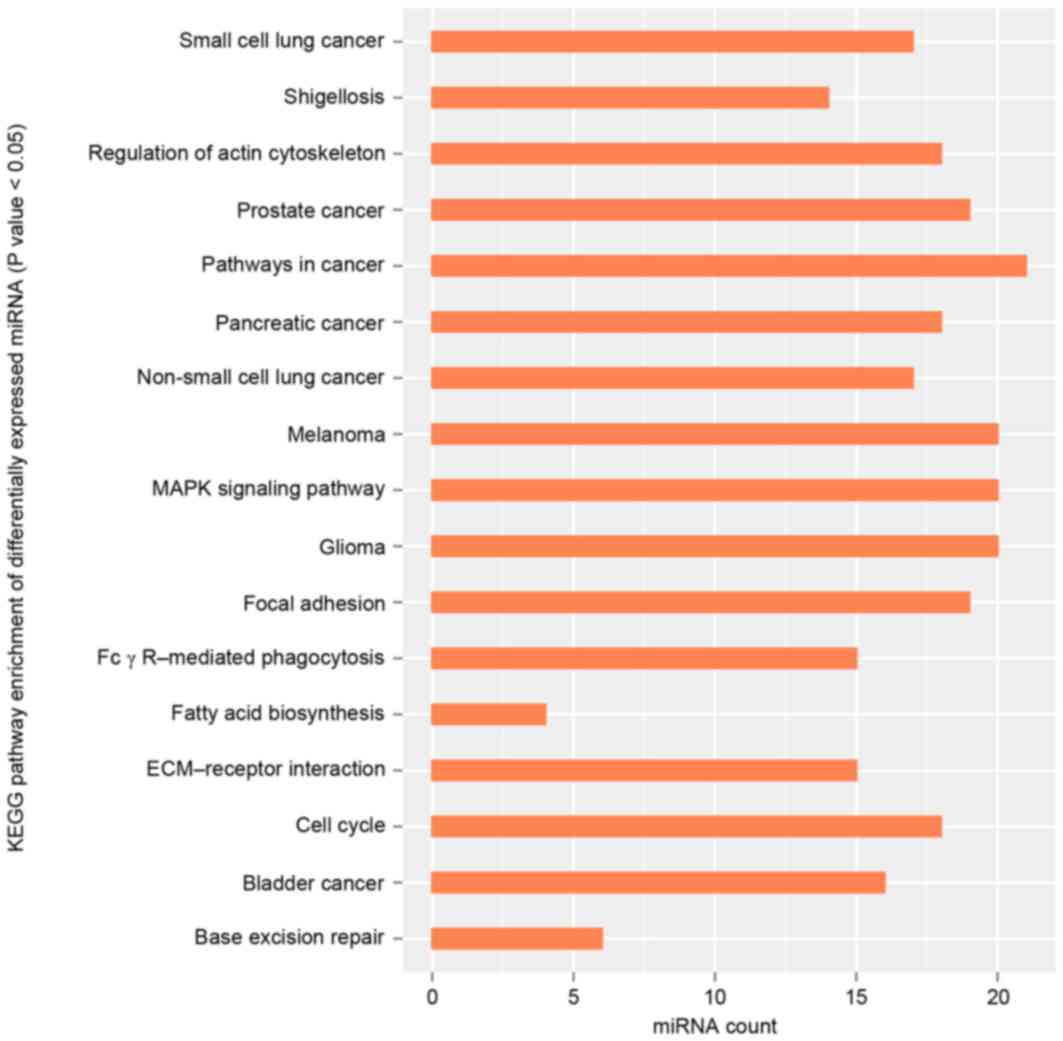
GO and KEGG pathway analysis were used to gain
further insights into the pathways significantly involved by
miRNAs. GO functional enrichment analysis demonstrated that
differentially expressed miRNAs, including hsa-miR-15a and
hsa-miR-125b, were mainly involved in the positive
regulation of histone H3-K9 methylation (GO, 0051574; BP),
fibrillar center (GO, 0001650; CC) and mismatched DNA binding (GO,
0030983; MF; Table I). KEGG pathway
analysis in the present study demonstrated that differentially
expressed miRNAs were mainly enriched in the pathways in cancer
map05200 (http://www.genome.jp/dbget-bin/www_bget?pathway:map05200;
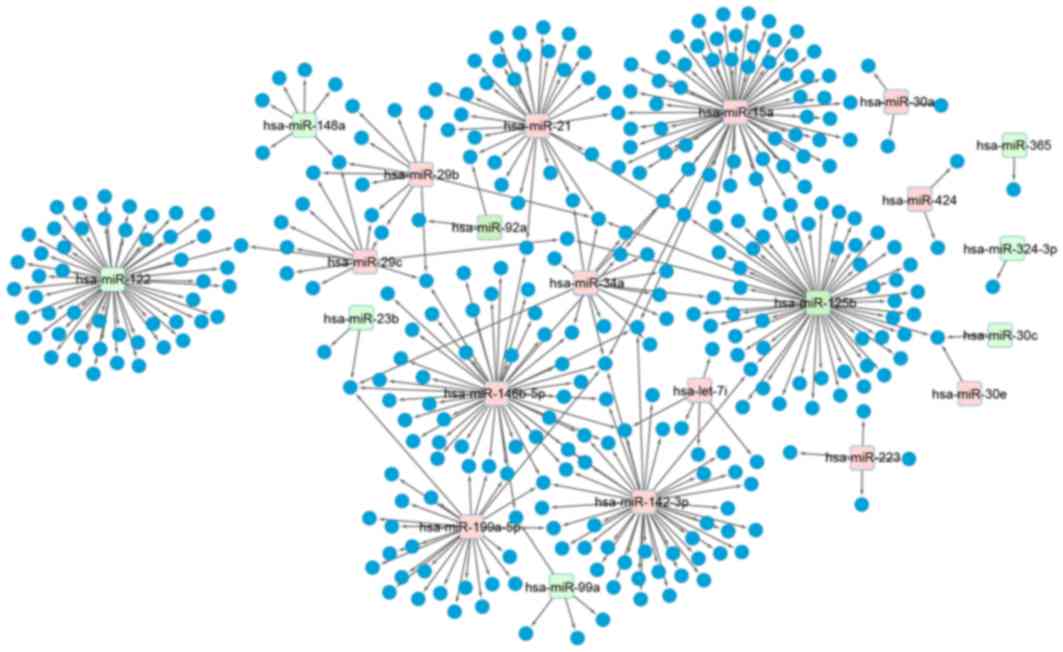
Fig. 2). Based on the experimental
certificate miRNA-target gene databases aforementioned, the
miRNA-target gene regulatory network was constructed using
Cytoscape software. The results demonstrate that there were 13
significantly upregulated miRNAs and 9 significantly downregulated
miRNAs (Fig. 3).
 | Table I.Results of GO functional enrichment
analysis of differentially expressed miRNAs. |
Table I.
Results of GO functional enrichment
analysis of differentially expressed miRNAs.
| GO ID | Term | Count | P-value |
|---|
| BP |
|
|
|
|
0051574 | Positive regulation
of histone H3-K9 methylation | 13 |
6.04×10−7 |
|
0051570 | Regulation of
histone H3-K9 methylation | 13 |
6.48×10−6 |
|
0031061 | Negative regulation
of histone methylation | 12 |
3.76×10−5 |
|
0031062 | Positive regulation
of histone methylation | 13 |
5.34×10−5 |
|
0010001 | Glial cell
differentiation | 21 |
7.58×10−5 |
| CC |
|
|
|
|
0001650 | Fibrillar
center | 6 |
1.32×10−4 |
|
0035098 | ESC/E(Z)
complex | 12 |
8.27×10−4 |
|
0005869 | Dynactin
complex | 5 |
1.09×10−3 |
|
0044452 | Nucleolar part | 7 |
4.18×10−3 |
|
0000791 | Euchromatin | 8 |
8.40×10−3 |
| MF |
|
|
|
|
0030983 | Mismatched DNA
binding | 7 |
1.84×10−4 |
|
0008263 | Pyrimidine-specific
mismatch base pair | 6 |
2.74×10−4 |
|
| DNA N-glycosylase
activity |
|
|
|
0043398 | HLH domain
binding | 9 |
3.92×10−4 |
|
0035198 | miRNA binding | 13 |
4.56×10−4 |
|
0000700 | Mismatch base pair
DNA N-glycosylase activity | 6 |
6.17×10−4 |
Transcription factor-miRNA relations
analysis
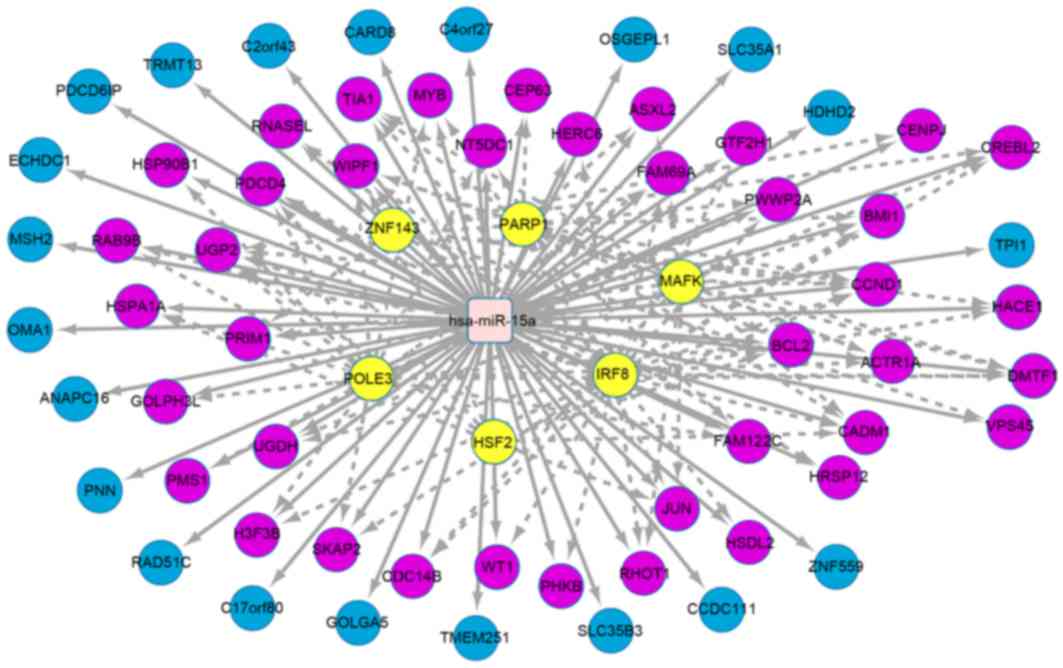
The iRegulon in Cytoscape software was used to
explore the potential association between miRNA and transcription
factors. With NES >4, the 3 miRNAs with the highest number of
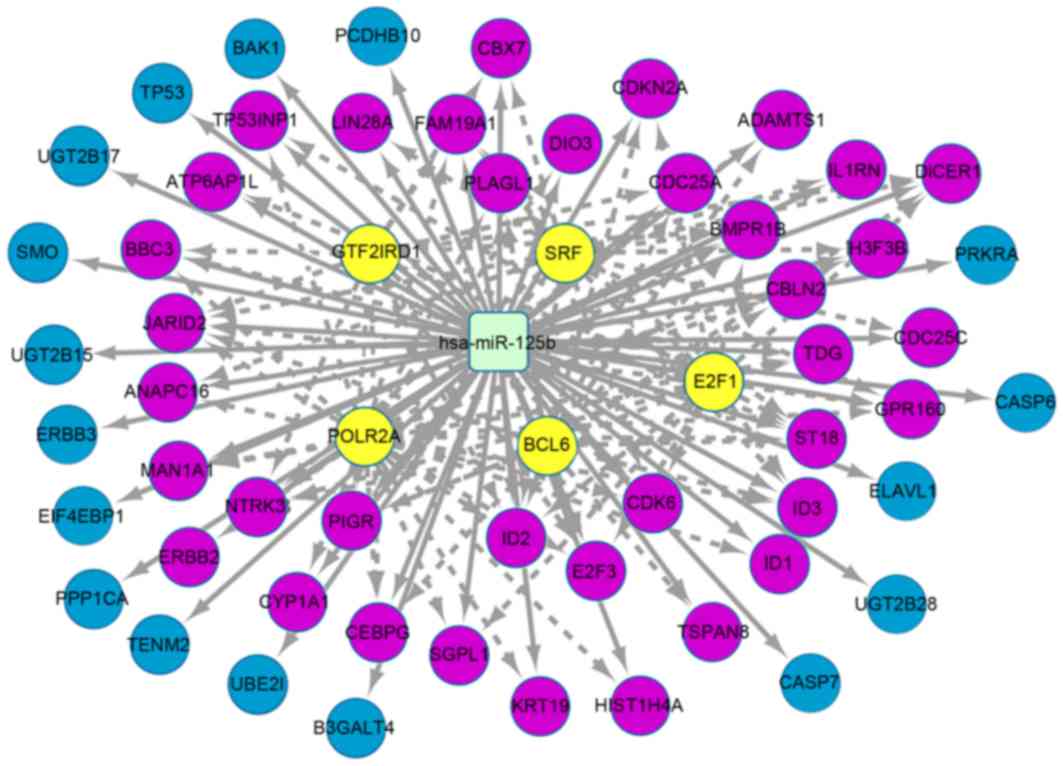
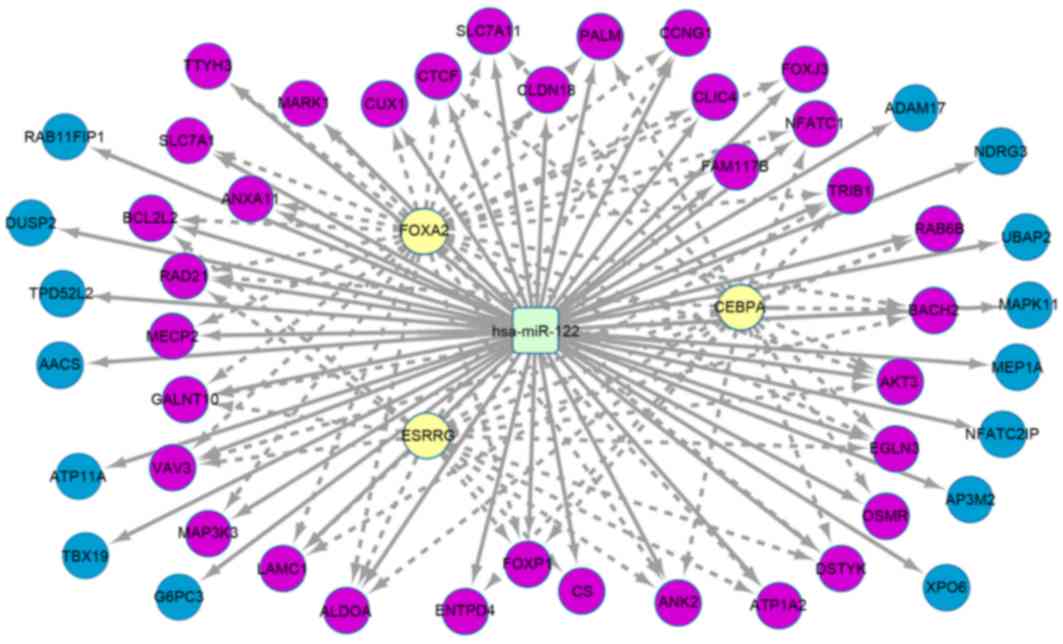
target genes were hsa-miR-15a (Fig. 4), hsa-miR-125b (Fig. 5) and hsa-miR-122 (Fig. 6). There were 61 target genes that were
upregulated by hsa-miR-15a, including transcription factors
such as zinc finger protein 143, poly (ADP-ribose) polymerase 1,
V-maf musculoaponeurotic fibrosarcoma oncogene homolog K, DNA
polymerase epsilon subunit 3, heat shock factor protein 2 and
interferon regulatory factor 8. A total of 54 target genes were
downregulated by hsa-miR-125b, including transcription
factors such as general transcription factor II–I repeat
domain-containing protein 1, serum response factor, polymerase
(RNA) II (DNA directed) polypeptide A, B-cell lymphoma 6 protein
and transcription factor E2F1. A total of 49 target genes were
downregulated by hsa-miR-122, including transcription
factors such as forkhead box protein A2, CCAAT/enhancer-binding
protein α and estrogen-associated receptor γ.
HCC-associated miRNA analysis
A total of 11 differentially expressed miRNA that
regulated the marker genes of HCC were identified (Table II). Among them, hsa-miR-146-5p
(n=13), hsa-miR-15a (n=5) and hsa-miR-125b (n=4) were
the miRNAs that regulated the highest number of marker genes of
HCC. A further investigation was made on other HCC-associated genes
(not the marker genes but recorded in databases) in the CTD
database. The results indicated that a total of 49 target genes of
miR-122 were HCC-associated genes.
| Table II.Differentially expressed miRNAs that
regulated the marker genes of hepatic cell carcinoma (HCC). |
Table II.
Differentially expressed miRNAs that
regulated the marker genes of hepatic cell carcinoma (HCC).
| miRNA | Marker gene |
|---|
|
hsa-miR-99a | MTOR,
IGF1R |
|
hsa-miR-424 | KDR |
|
hsa-miR-34a | CCND1, MYC,
MET |
|
hsa-miR-23b | MET |
|
hsa-miR-21 | PPARA, E2F1,
BTG2 |
|
hsa-miR-199a-5p | CCND1, MET,
FASN |
|
hsa-miR-15a | UGDH, BCL2,
CCND1, HSDL2, JUN |
|
hsa-miR-148a | CDKN1B |
|
hsa-miR-146b-5p | JUN, IL6, FOS,
MET, TNF, SOCS1, CCND1, MYC, BRAF, MMP9, IGF1R, BCL2, FASN |
|
hsa-miR-142-3p | APCS, KRAS, JUN,
CCND1 |
|
hsa-miR-125b | ADAMTS1, TP53,
CDKN2A, CYP1A1 |
Discussion
HCC is one of the most common types of human cancer
worldwide (1). However, the molecular
mechanisms behind the pathogenesis and progression of HCC remain
unclear. The present study performed a bioinformatics analysis on
the miRNA expression profile of HCC. By screening the
differentially expressed miRNAs, a total of 14 upregulated miRNAs
and 16 downregulated miRNAs between HCC samples and control samples
were obtained. These differentially expressed miRNAs were mainly
involved in BP like positive regulation of histone H3-K9
methylation, and were observed in KEGG pathways in cancer map05200.
A total of 3 outstanding regulatory networks of miRNA
(hsa-miR-15a, hsa-miR-125b and hsa-miR-122) were
further investigated. A total of 11 differentially expressed miRNAs
that regulated the candidate marker genes of HCC were explored.
In the present study, GO enrichment analysis
demonstrated that the target genes that were regulated by the
differentially expressed miRNA, such as hsa-miR-15a and
hsa-miR-125b, were mainly enriched in functions such as
histone H3-K9 methylation. DNA methylation and histone H3-K9
modifications perform important roles in the process of tumor
formation by the dysregulation of target genes (27). A previous study demonstrated that
H3-K9 methylation contributed to chromosomal targeting of polycomb
group proteins (28). DNA methylation
profiling for serum DNA of patients with HCC revealed a potential
association between HCC and DNA methylation (29). Peng et al (30) indicated that heterochromatin
(essential for chromosome organization and inheritance) stability
requires regulators of histone H3-K9 methylation, which indicates
that regulation of histone H3-K9 methylation is vital for
cancer-associated gene expression. Thus, the present study
speculated that histone H3-K9 modifications may relate to the
progression of HCC. KEGG pathway analysis in the present study
revealed that target genes of differentially expressed miRNAs were
mainly enriched in multiple cellular processes related to cancer
and MAPK signaling. Daroqui et al (31) proved that MAPK signaling promotes cell
invasiveness and in vivo tumor progression via actin
remodeling in response to transforming growth factor-β. Tumor
necrosis factor has been revealed to activate the cyclic adenosine
monophosphate response element-binding protein through the p38
MAPK/mitogen and stress activated protein kinase 1 signaling
pathway (32). MAPK signaling and
pathways in cancer had a close association with HCC processes, and
so the differentially expressed miRNA that relate with the MAPK
signaling pathway and cancer-related pathways (as determined by
KEGG analysis) may take part in the development of HCC.
It is considered that the regulation of
transcription factors and miRNAs is essential for the appropriate
execution of biological events and developmental processes
(33). By incorporating prior
information from predicted regulatory interactions with parallel
miRNA/mRNA expression datasets, Peng et al (34) indicated that the transcription
factor-miRNA co-regulatory based on feed-forward loops served an
important role in the development of prostate cancer. In the
present study, the regulatory networks of 3 outstanding miRNAs
(hsa-miR-15a, hsa-miR-125b and hsa-miR-122) were
obtained. This result demonstrates that these transcription factors
had close associations with the 3 miRNAs in the co-regulation of
target genes, which further indicated the complex regulatory roles
of them during the development of HCC. Thus, it is speculated that
hsa-miR-15a, hsa-miR-125b and hsa-miR-122 may be the
miRNAs to be investigated for their role in HCC gene therapy.
Human miR-146b-5p is an miRNA which is
located on chromosome 10q24.3 (35).
miR-146b-5p is frequently downregulated in solid tumors,
including prostate cancer, pancreatic cancer and glioblastoma
(36). In the present study,
HCC-associated miRNA analysis revealed that there were 11
differentially expressed miRNAs that regulate the marker genes of
HCC including miR-146b-5p, which had the highest number of
marker genes. Furthermore, the HCC-associated gene (not recorded
marker genes) investigation based on the CTD database revealed that
miR-122 had the highest number of HCC-associated genes. A
previous study indicated that miR-122 negatively correlates
with liver fibrosis as detected by histology and FibroScan software
(37). In animal models,
miR-122 has been proven to be a marker of liver cell damage,
inflammation and regeneration in acetaminophen-induced liver injury
(38). In the present study,
miR-122 was revealed to be closely associated with HCC, thus
the target gene of miR-122 may be the potential novel marker
gene for HCC. Therefore, it was speculated that hsa-miR-15a,
hsa-miR-125b, hsa-miR-122 and hsa-miR-146b-5p may
perform important roles in the progression of HCC, and may be new
biomarkers for gene therapy of HCC. However, there were limitations
to the present this study, such as lack of experimental
verification and joint analysis of expression profile data. An
investigation based on experimental verification is required in
future studies.
In conclusion, miRNAs such as hsa-miR-15a,
hsa-miR-125b, hsa-miR-122 and hsa-miR-146b-5p may be new
biomarkers for gene therapy of HCC. Histone H3-K9 methylation and
miRNAs identified through KEGG analysis to be involved in cancer
formation may be closely associated with the development of
HCC.
Acknowledgements
The present project was supported by the social
development fund of Nantong (grant no. HS2014063) and The 12th
Talent Summit of Top Six Industries in Jiangsu Province (grant no.
WSW-080).
References
|
1
|
Chen KW, Ou TM, Hsu CW, Horng CT, Lee CC,
Tsai YY, Tsai CC, Liou YS, Yang CC, Hsueh CW, et al: Current
systemic treatment of hepatocellular carcinoma: A review of the
literature. World J Hepatol. 7:1412–1420. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin S, Hoffmann K and Schemmer P:
Treatment of hepatocellular carcinoma: A systematic review. Liver
Cancer. 1:144–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cabrera R and Nelson DR: Review article:
The management of hepatocellular carcinoma. Aliment Pharmacol Ther.
31:461–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang HH, Wei PL, Hung CS, Wu CT, Wang W,
Huang MT and Chang YJ: MicroRNA-200a/b influenced the therapeutic
effects of curcumin in hepatocellular carcinoma (HCC) cells. Tumour
Biol. 34:3209–3218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cao W, Guo Q, Zhang T, Zhong D and Yu Q:
Prognostic value of microRNAs in acute myocardial infarction: A
systematic review and meta-analysis. Int J Cardiol. 189:79–84.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao Y, He Y, Ding J, Wu K, Hu B, Liu Y, Wu
Y, Guo B, Shen Y, Landi D, et al: An insertion/deletion
polymorphism at miRNA-122-binding site in the interleukin-1alpha 3′
untranslated region confers risk for hepatocellular carcinoma.
Carcinogenesis. 30:2064–2069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J, Yan J, Zhou C, Ma Q, Jin Q and Yang
Z: miR-1285-3p acts as a potential tumor suppressor miRNA via
downregulating JUN expression in hepatocellular carcinoma. Tumour
Biol. 36:219–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang Z, Zhang Y and Wang L: A feedback
inhibition between miRNA-127 and TGFβ/c-Jun cascade in HCC cell
migration via MMP13. PLoS One. 8:e652562013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ziari K, Zarea M, Gity M, Fayyaz AF,
Yahaghi E, Darian EK and Hashemian AM: Downregulation of miR-148b
as biomarker for early detection of hepatocellular carcinoma and
may serve as a prognostic marker. Tumour Biol. 37:5765–5768. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang X and Jia Z: Construction of
HCC-targeting artificial miRNAs using natural miRNA precursors. Exp
Ther Med. 6:209–215. 2013.PubMed/NCBI
|
|
11
|
Borel F, Konstantinova P and Jansen PL:
Diagnostic and therapeutic potential of miRNA signatures in
patients with hepatocellular carcinoma. J Hepatol. 56:1371–1383.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang N, Ekanem NR, Sakyi CA and Ray SD:
Hepatocellular carcinoma and microRNA: New perspectives on
therapeutics and diagnostics. Adv Drug Deliv Rev. 81:62–74. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murakami Y, Tanahashi T, Okada R, Toyoda
H, Kumada T, Enomoto M, Tamori A, Kawada N, Taguchi YH and Azuma T:
Comparison of hepatocellular carcinoma miRNA expression profiling
as evaluated by next generation sequencing and microarray. PLoS
One. 9:e1063142014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie Y, Yao Q, Butt AM, Guo J, Tian Z, Bao
X, Li H, Meng Q and Lu J: Expression profiling of serum
microRNA-101 in HBV-associated chronic hepatitis, liver cirrhosis,
and hepatocellular carcinoma. Cancer Biol Ther. 15:1248–1255. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Lee AT, Ma JZ, Wang J, Ren J, Yang
Y, Tantoso E, Li KB, Ooi LL, Tan P and Lee CG: Profiling microRNA
expression in hepatocellular carcinoma reveals microRNA-224
up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific
target. J Biol Chem. 283:13205–13215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei L, Lian B, Zhang Y, Li W, Gu J, He X
and Xie L: Application of microRNA and mRNA expression profiling on
prognostic biomarker discovery for hepatocellular carcinoma. BMC
Genomics. 15:(Suppl 1). S132014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huber W, Von Heydebreck A, Sültmann H,
Poustka A and Vingron M: Variance stabilization applied to
microarray data calibration and to the quantification of
differential expression. Bioinformatics. 18:(Suppl 1). S96–S104.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:(Database Issue). D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopaedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in Drosophila. Genome Biol.
5:R12003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Janky R, Verfaillie A, Imrichová H, Van de
Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K,
Sanchez M Naval, Potier D, et al: iRegulon: From a gene list to a
gene regulatory network using large motif and track collections.
PLoS Comput Biol. 10:e10037312014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davis AP, Grondin CJ, Lennon-Hopkins K,
Saraceni-Richards C, Sciaky D, King BL, Wiegers TC and Mattingly
CJ: The comparative toxicogenomics database's 10th year
anniversary: Update 2015. Nucleic Acids Res. 43:(Database Issue).
D914–D920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kitamoto S, Yamada N, Yokoyama S, Houjou
I, Higashi M, Goto M, Batra SK and Yonezawa S: DNA methylation and
histone H3-K9 modifications contribute to MUC17 expression.
Glycobiology. 21:247–256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sewalt RG, Lachner M, Vargas M, Hamer KM,
den Blaauwen JL, Hendrix T, Melcher M, Schweizer D, Jenuwein T and
Otte AP: Selective interactions between vertebrate polycomb
homologs and the SUV39H1 histone lysine methyltransferase suggest
that histone H3-K9 methylation contributes to chromosomal targeting
of Polycomb group proteins. Mol Cell Biol. 22:5539–5553. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tu T, Shackel NA and McCaughan G: ‘Testing
your methyl’: DNA methylation profiling of serum DNA of HCC
patients. Hepatol Int. 7:785–787. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peng JC and Karpen GH: Heterochromatic
genome stability requires regulators of histone H3 K9 methylation.
PLoS Genet. 5:e10004352009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Daroqui MC, Vazquez P, Bal de Kier Joffé
E, Bakin AV and Puricelli LI: TGF-β autocrine pathway and MAPK
signaling promote cell invasiveness and in vivo mammary
adenocarcinoma tumor progression. Oncol Rep. 28:567–575.
2012.PubMed/NCBI
|
|
32
|
Gustin JA, Pincheira R, Mayo LD, Ozes ON,
Kessler KM, Baerwald MR, Korgaonkar CK and Donner DB: Tumor
necrosis factor activates CRE-binding protein through a p38
MAPK/MSK1 signaling pathway in endothelial cells. Am J Physiol Cell
Physiol. 286:C547–C555. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang S, Li W, Lian B, Liu X, Zhang Y, Dai
E, Yu X, Meng F, Jiang W and Li X: TMREC: A database of
transcription factor and MiRNA regulatory cascades in human
diseases. PLoS One. 10:e01252222015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng C, Wang M, Shen Y, Feng H and Li A:
Reconstruction and analysis of transcription factor-miRNA
co-regulatory feed-forward loops in human cancers using
filter-wrapper feature selection. PLoS One. 8:e781972013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Katakowski M, Zheng X, Jiang F, Rogers T,
Szalad A and Chopp M: MiR-146b-5p suppresses EGFR expression and
reduces in vitro migration and invasion of glioma. Cancer Invest.
28:1024–1030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Wang Y, Yu L, Sun C, Cheng D, Yu S,
Wang Q, Yan Y, Kang C, Jin S, et al: miR-146b-5p inhibits glioma
migration and invasion by targeting MMP16. Cancer Lett.
339:260–269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Halász T, Horváth G, Pár G, Werling K,
Kiss A, Schaff Z and Lendvai G: miR-122 negatively correlates with
liver fibrosis as detected by histology and FibroScan. World J
Gastroenterol. 21:7814–7823. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park HK, Jo W, Choi HJ, Jang S, Ryu JE,
Lee HJ, Lee H, Kim H, Yu ES and Son WC: Time-course changes in the
expression levels of miR-122, −155 and −21 as markers of liver cell
damage, inflammation, and regeneration in acetaminophen-induced
liver injury in rats. J Vet Sci. 17:45–51. 2016. View Article : Google Scholar : PubMed/NCBI
|