Introduction
Gastric cancer (GC) is a common type of malignancy
worldwide (1). Although there have
been significant improvements in the clinical management of GC,
chemotherapy remains one of the most important therapeutic
strategies for advanced GC. However, due to the heterogeneity in
the etiology and genetic basis of GC, the efficacy of
chemotherapeutic drugs varies among the different subtypes of
patients. A substantial proportion of patients eventually develop
low chemoresponsiveness to chemotherapeutic drugs, including
cisplatin, and this is one of the main reasons for GC-associated
mortality (2).
Numerous mechanisms have been proposed to explain
the phenomenon of drug resistance in cancer cells. For example, the
enhanced expression of multidrug resistance protein 1
(P-glycoprotein) facilitates drug efflux from cancer cells
(3), and alterations of cell cycle,
autophagy and apoptosis regulators may also serve critical roles in
cellular responsiveness to anticancer drugs (4,5). In recent
decades, research into microRNAs has greatly expanded our
understanding of chemotherapy resistance (6). MicroRNAs are single-stranded, non-coding
RNAs that negatively regulate gene expression by binding to the
3′-untranslated region (UTR) of a specific mRNA. A number of
microRNAs, which are implicated in the processes of DNA damage and
repair, apoptosis regulation, epigenetic regulation and cell cycle
regulation, have been revealed to produce diverse effects on the
response of cells to chemotherapeutic drugs (6). Despite earlier studies demonstrating the
oncogenic role of miR-25 in GCs (7–10), the
exact role of microRNA-25 (miR-25) in cisplatin-resistant GC cells
has not yet been well investigated.
The present study demonstrated that miR-25 was
highly expressed in SGC-7901/DDP cisplatin-resistant GC cells
compared with in the parental cell line, SGC-7901. Overexpression
of miR-25 in the parental cell line led to decreased cisplatin
sensitivity, whereas inhibition of miR-25 in SGC-7901/DDP cells
partially decreased the cisplatin resistance. Subsequently, the
tumor-suppressive transcriptional factor forkhead box O3a (FOXO3a),
which controls a number of genes involved in cell cycle regulation,
was established as a direct target of miR-25. Therefore, to the
best of our knowledge, the present study revealed for the first
time that miR-25 is a major contributor to the cisplatin resistance
of GC cells.
Materials and methods
Cell lines, transfection and drug
treatment
The human GC cell line SGC-7901 and the
cisplatin-resistant variant SGC-7901/DDP were obtained from Nanjing
KeyGen Biotech Co., Ltd. (Nanjing, China). Cells were cultured in
Gibco RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2, and cells
were passaged every other day. To maintain the cisplatin resistance
of SGC-7901/DDP cells, 1 µg/ml cisplatin (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was added to the culture medium. The
cisplatin was dissolved in PBS and applied to cells at a final
concentration of 0.01, 0.1, 1, 10 or 100 µg/ml for 48 h. The mimics
for miR-25 (cat no. miR10000081-1-5) and its inhibitor strand (cat
no. miR20000081-1-5) were synthesized by Guangzhou RiboBio Co.,
Ltd. (Guangzhou, China). Negative controls were also provided by
Guangzhou RiboBio Co., Ltd. Cells were transfected using
Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The concentration of
mimics/inhibitors used for transfection was 100 nM, and the
transfection time was 48 h at 37°C. The small interfering RNA
(siRNA) against human FOXO3a (si-FOXO3a; 5′-ACUCGGGUCCAGCUCCAC-3′)
was also purchased from Guangzhou RiboBio Co., Ltd. The negative
control was also provided by the manufacturer (cat no.
siN05815122147-1-5), and the transfection protocol was identical to
that of the microRNA mimic transfection.
Drug sensitivity assay (MTT
assay)
An MTT assay was used to detect the proportion of
surviving cells following cisplatin treatment. Cells were equally
seeded at 1.5×105/ml at 37°C, grown in 96-well plates,
and transfected with the miR-25 mimic, miR-25 inhibitor or
si-FOXO3a. Subsequently, cells were treated with cisplatin at the
indicated concentrations, as described above, 48 h after
transfection. Cells were allowed to incubate for 48 h, and 20 µl
MTT reagent (5 mg/ml; Sigma-Aldrich; Merck KGaA) was then added to
each well, 4 h prior to the assay being performed. Cisplatin medium
was replaced at this point. Following a 4-h incubation with the MTT
reagent, the formazan in each well was dissolved in dimethyl
sulfoxide, and the absorbance value at 490 nm was detected using a
spectrophotometer.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from the cells was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The RNA was
purified by incubation with 75% ethanol at room temperature for 5
min. First-strand synthesis was performed with 1.5 µg RNA using the
stem-loop primer kit Bulge-Loop™ miRNA qRT-PCR Starter kit (cat no.
C10211-1) provided by Guangzhou RiboBio Co., Ltd., and PCR
amplification of the cDNA was performed using the SYBR Premix Ex
Taq II kit (Takara Biotechnology Co., Ltd., Dalian, China) and
specific primer sets (cat no. miRQ0000081-1-1; Guangzhou RiboBio
Co., Ltd.) for miR-25 and U6 (which was amplified as the internal
control). The thermocycler conditions for the PCR reaction were as
follows: 94°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec, for
35 cycles. The relative expression level of miR-25 was determined
by the 2−ΔΔCq method (11), and the experiments were repeated in
triplicate.
Cell cycle analysis
Cell cycle distribution was analyzed by flow
cytometry. Briefly, cells were fixed with 70% ethanol at −20°C
overnight. Following rehydration with 1.8 ml PBS on the following
day, cells were treated with 100 µl 100 µg/ml RNase (cat no. RT405;
Tiangen Biotech Co., Ltd., Beijing, China) for 30 min at 37°C, and
stained with 400 µl 50 µg/ml propidium iodide (cat no. st512;
Beyotime Institute of Biotechnology, Shanghai, China) for 30 min at
room temperature. Subsequently, cells were analyzed using a
FACSort™ flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA).
Luciferase activity assay
The putative binding site was searched with miRanda
database (http://www.microrna.org). The wild type
and mutant 3′UTR sequences of FOXO3 that contain the potential
target of miR-25 were synthesized by Shanghai Shenggong Biology
Engineering Technology Service, Ltd. (Shanghai, China). The
predicted binding sequence in wild type 3′UTR (5′-GUGCAAU-3′) was
mutated to 5′-ACAUGGC-3′. The sequences were subcloned into a
pMIR-REPORT miRNA Expression Reporter Vector system (Thermo Fisher
Scientific, Inc.). The pMIR-REPORT-3′UTR constructs were
transfected into HEK293 cells with miR-25 mimics and Renilla
luciferase constructs (Promega Corporation, Madison, WI, USA) at
37°C for 24 h with Lipofectamine® 2000 (Thermo Fisher
Scientific., Inc.). At 24 h after transfection, luciferase activity
was determined by a Dual-Luciferase Reporter Assay system (Promega
Corporation), according to the manufacturer's protocol.
Western blotting
Cells grown in 6-well plates for 24 h at 37°C were
transfected and treated with cisplatin as aforementioned, and then
were analyzed by western blotting. Total protein from the cells was
collected using SDS lysis buffer supplemented with protease
inhibitor (Beyotime Institute of Biotechnology, Haimen, China).
Appropriate quantities of cell lysates were denatured by heating in
sample buffer (Beyotime Institute of Biotechnology) at 100°C for 3
min, and then 50 µg protein for each sample was separated by 10%
SDS-PAGE and blotted onto polyvinylidene membranes. Membranes were
blocked with 5% skimmed milk for 1 h at room temperature, followed
by an overnight incubation at 4°C with primary antibodies against
β-actin (cat no. sc-8432; 1:1,000) and cyclin-dependent kinase
(CDK) inhibitor 1B (p27Kip1; cat no. sc-528; 1:300)
(both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
FOXO3a (cat no. 12,829; 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA). The samples were then incubated with the
secondary antibodies [goat anti-rabbit horseradish peroxidase
(HRP); cat no. sc-2004; 1:2,000; goat anti-mouse HRP; cat no.
sc-2005; 1:2,000 (both from Santa Cruz Biotechnology, Inc.)] for 1
h at room temperature. Following a series of washes with PBST (0.5%
Tween-20), protein bands were detected using the SuperSignal West
Pico Chemiluminescent Substrate chemiluminescence visualization kit
(Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
The comparisons were performed using Student's t-test. Two tailed
P<0.05 was considered to indicate a statistically significant
difference. All the experiments were performed ≥3 times.
Results
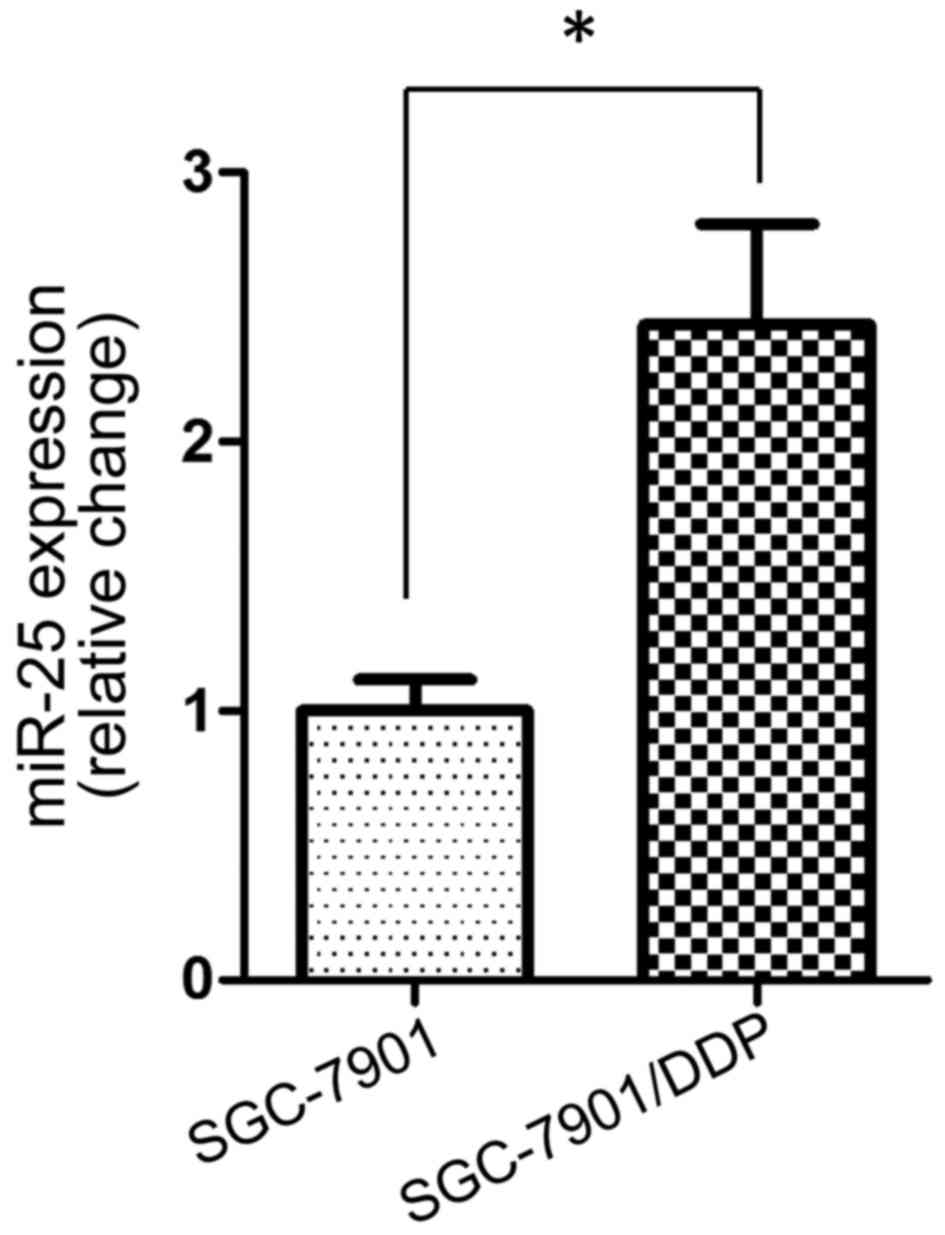
miR-25 expression is upregulated in
the cisplatin-resistant SGC-7901/DDP cell line
To investigate the potential role of miR-25 in
cisplatin resistance in GC cells, the present study first examined
the expression levels of miR-25 in the SGC-7901 cell line and its
cisplatin-resistant variant SGC-7901/DDP. RT-qPCR demonstrated that
miR-25 had a significantly higher expression level in SGC-7901/DDP
cells compared with in the parental SGC-7901 cell line (P<0.05;
Fig. 1).
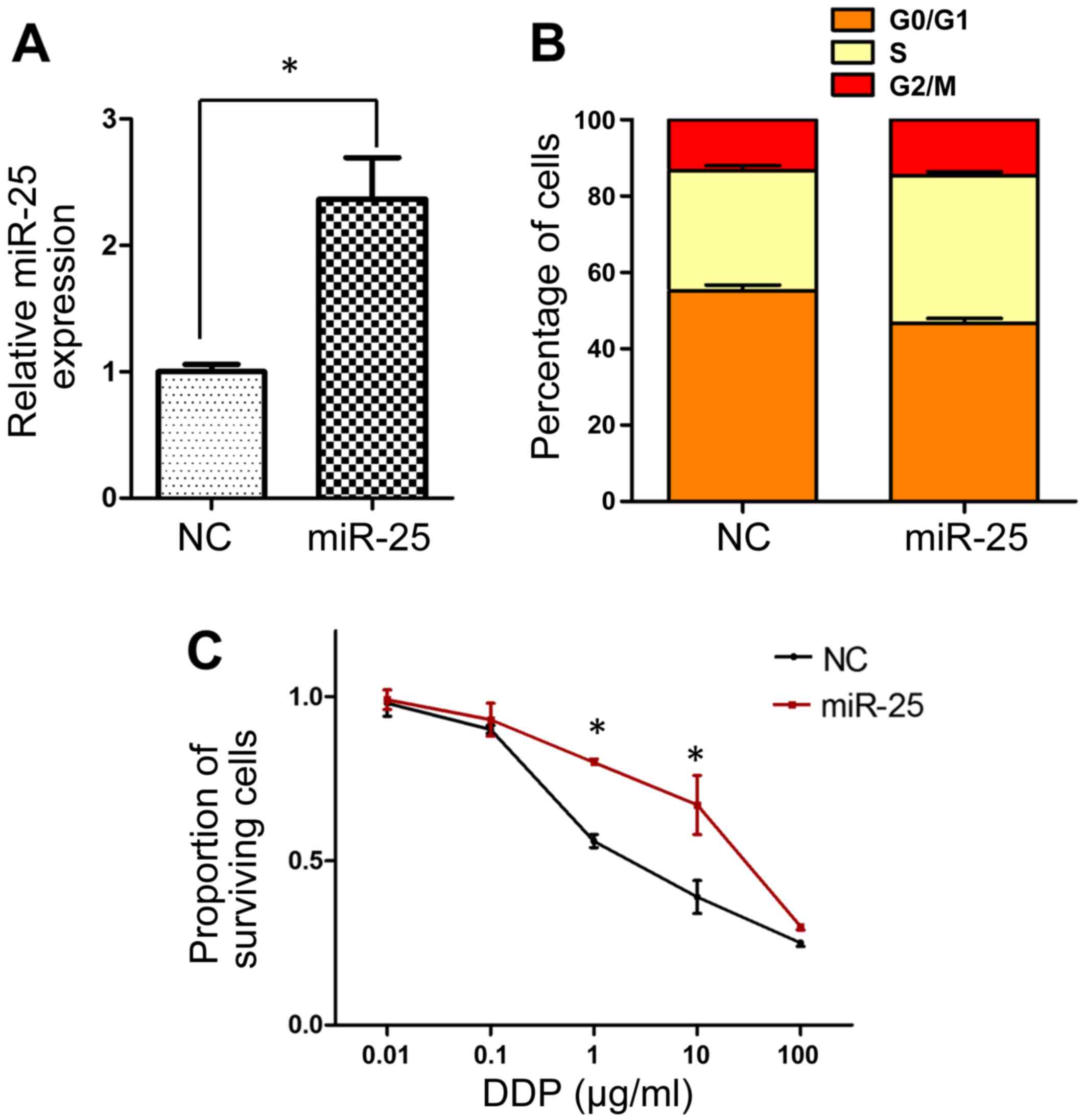
Overexpression of miR-25 decreases the
sensitivity of SGC-7901 cells to cisplatin
The observed upregulation of miR-25 expression in
SGC-7901/DDP cells prompted the hypothesis that miR-25 may serve an
important role in the development of cisplatin resistance, and this
was investigated by modulating miR-25 expression levels in SGC-7901
cells by transfection with miR-25 mimics. The transfection efficacy
was verified by RT-qPCR, which demonstrated significant
upregulation of miR-25 in the mimic-transfected cells (P<0.05;
Fig. 2A). Subsequently, flow
cytometric analysis revealed fewer cells in the
G0/G1 cell cycle phase in mimic-transfected
compared with negative control cells, indicating enhanced cell
cycle progression concomitant with increased miR-25 levels
(Fig. 2B). Furthermore, an MTT assay
revealed that transfection with miR-25 mimics resulted in
significantly decreased sensitivity of SGC-7901 cells to cisplatin
at doses of 1–10 µg/ml (P<0.05; Fig.
2C).
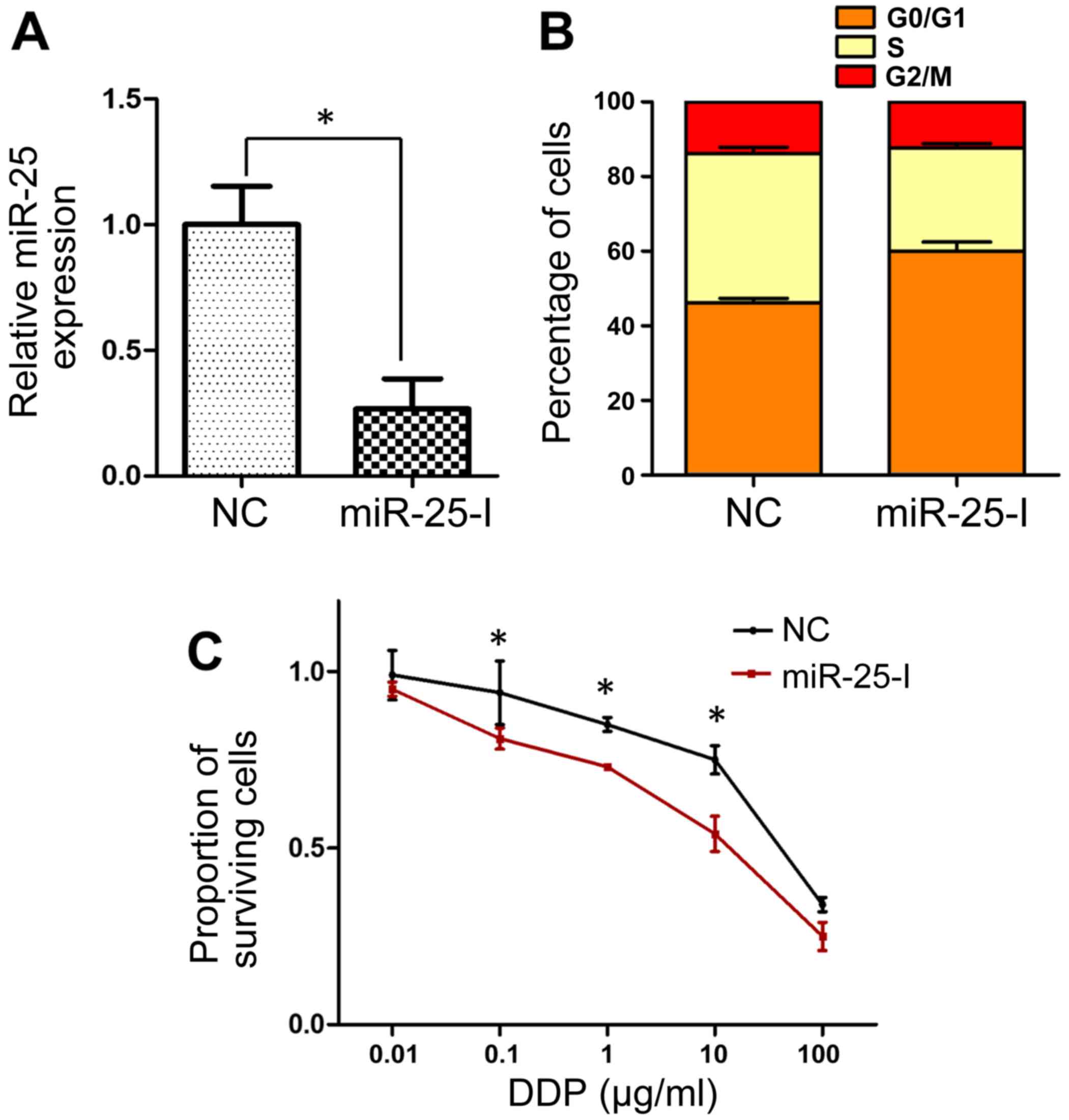
Inhibition of miR-25 reverses the
cisplatin resistance of SGC-7901/DDP cells
miR-25 expression was significantly inhibited in
SGC-7901/DDP cells via transfection with an miR-25 inhibitor
(P<0.05; Fig. 3A). In contrast to
miR-25 overexpression, the inhibition of miR-25 resulted in cell
cycle arrest, with a greater proportion of cells in the
G0/G1 phase in inhibitor-transfected cells
than in the negative control group (Fig.
3B). Analysis of cisplatin sensitivity by MTT assay revealed
that cells transfected with miR-25 inhibitor exhibited
significantly decreased cell viability following treatment with
cisplatin at doses of 0.1–10 µg/ml (Fig.
3C), which suggested that miR-25 inhibition may reverse the
cisplatin resistance of SGC-7901/DDP cells.
FOXO3a serves as a target of
miR-25
Since microRNAs function to block gene expression by
inexact base-pair matching with the 3′UTR of the target mRNA, an
online database search was performed in the present study to
investigate the mechanisms underlying the effects of miR-25. A
possible interaction between miR-25 and the tumor suppressive gene
FOXO3 was identified (Fig. 4A).
Subsequently, a luciferase activity assay revealed that
transfection of HEK293 cells with miR-25 mimics decreased the
luciferase activity of the reporter containing the wild-type 3′UTR
of FOXO3a (Fig. 4B); however, the
luciferase activity of the reporter carrying the mutant 3′UTR was
not affected by miR-25 mimics, which suggested that this
interaction is specific. In addition, western blotting demonstrated
that the overexpression of miR-25 in SGC-7901 cells led to
decreased protein levels of FOXO3a and, consistently,
downregulation of p27Kip1, the transcriptional target of FOXO3a
(Fig. 4C-E). Furthermore, FOXO3a
protein level was significantly decreased in SGC-7901/DDP cells
compared with SGC-7901 cells (Fig. 4F and
G). Collectively, these results indicated that FOXO3a is a
direct target of miR-25 in GC cells.
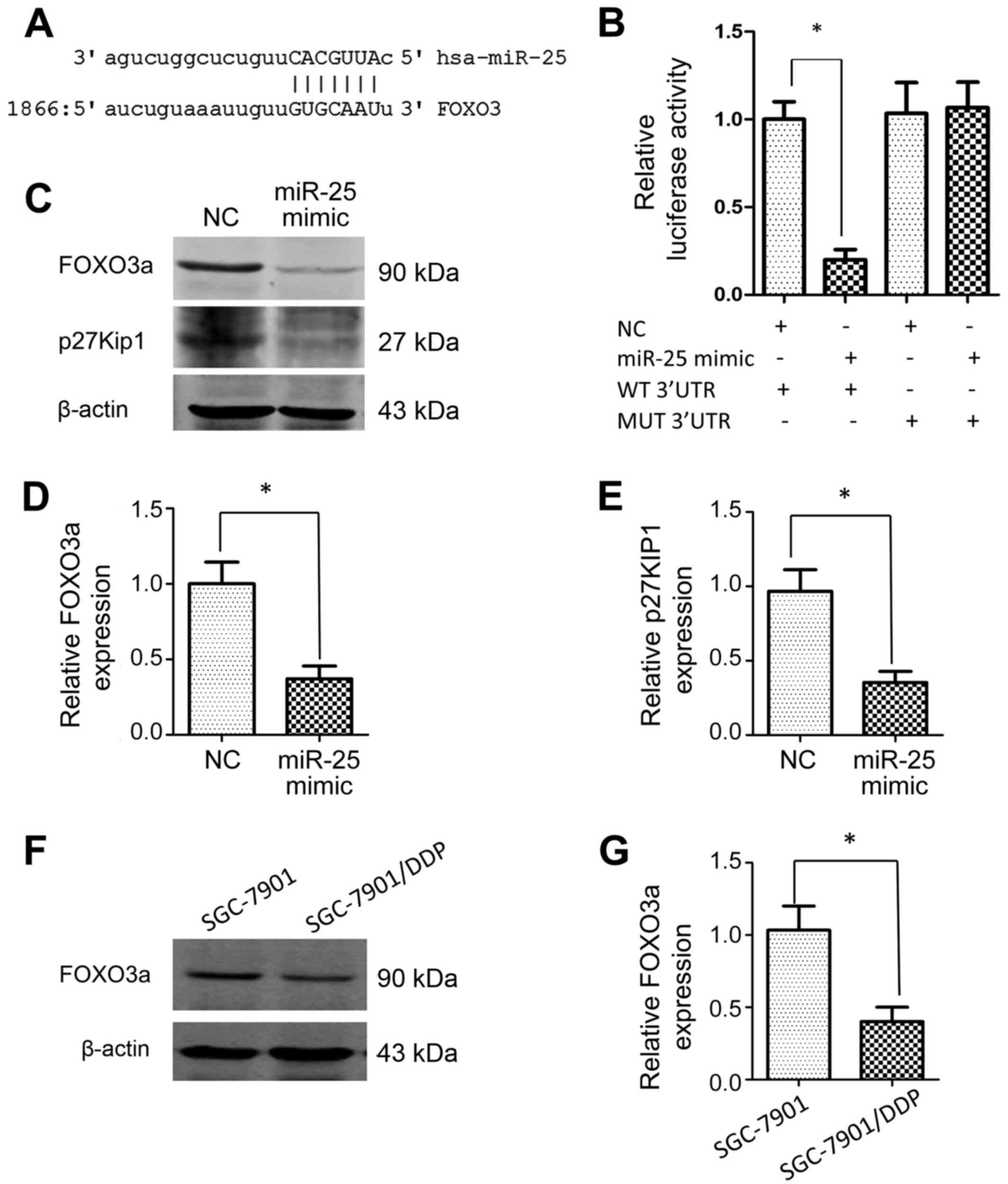 | Figure 4.FOXO3a is a target of miR-25. (A) The
predicted binding site, as determined using the miRanda database,
is shown. (B) The relative luciferase activity levels of the
reporters containing WT or MUT 3′UTR sequences of FOXO3a were
measured following miR-25 transfection in HEK293 cells. (C) Western
blotting was used to assess the effect of miR-25 mimics on the
expression levels of FOXO3a and p27Kip1 in SGC-7901 cells. (D and
E) Graphs showing the quantification of the integrated protein band
densities following western blotting (as shown in C). (F) Western
blotting was used to assess the expression level of FOXO3a in the
SGC-7901 cell line and its DDP-resistant variant, SGC-7901/DDP. (G)
Graphs showing the quantification of the integrated protein band
densities following western blot analysis of FOXO3a (as shown in
F). β-actin was used as the loading control for all western blot
analyses. *P<0.05, n=3 in each group. FOXO3a, forkhead box O3a;
miR-25, microRNA-25; WT, wild-type; MUT, mutant; 3′UTR, 3′
untranslated region; NC, negative control; p27Kip1,
cyclin-dependent kinase inhibitor 1B. |
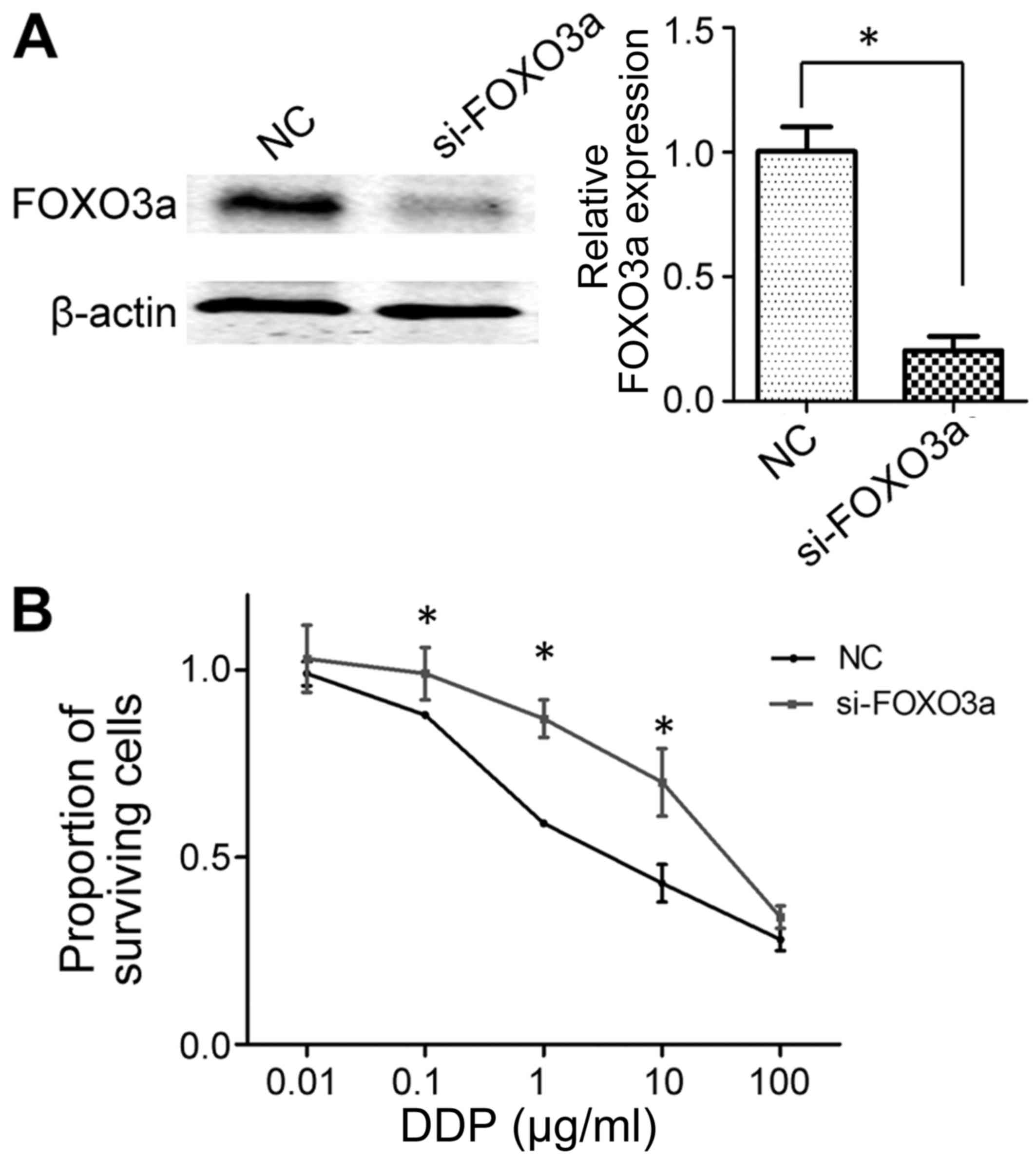
Knockdown of FOXO3a decreases the
cisplatin sensitivity of SGC-7901 cells
Following the siRNA-mediated knockdown of FOXO3a,
western blot analysis confirmed that si-FOXO3a was sufficient to
inhibit FOXO3a expression in SGC-7901 cells (Fig. 5A). Subsequently, an MTT assay was used
to determine the sensitivity of SGC-7901 cells to cisplatin. As
presented in Fig. 5B, cell viability
was significantly higher in FOXO3a-knockdown cells compared with
the negative control group following treatment with cisplatin at
doses of 0.1–10 µg/ml, indicating decreased sensitivity. This
result was similar to the data from the miR-25 mimic transfection,
in that the overexpression of miR-25 and the knockdown of FOXO3a
each decreased the sensitivity of GC cells to cisplatin.
Discussion
Cisplatin is one of the predominant first-line
chemotherapeutic drugs used for the treatment of GC in clinical
practice. Despite the high sensitivity of patients with GC to
cisplatin at initial administration, a substantial number of
patients develop drug resistance, which is one of the major causes
of treatment failure and GC relapse (2). Therefore, solving the clinical problem
of acquired drug resistance is of considerable importance for the
successful treatment of recurrent GC. To date, the molecular
mechanism of the drug resistance have not been fully elucidated.
Previous studies established numerous molecular models for
cisplatin resistance, including low efficiency in drug
transportation, increased DNA damage repair response and
suppression of cell cycle inhibitors and apoptosis signaling
(4,5).
Epigenetic regulation is important in the
pathogenesis and progression of GC, and recent evidence has
indicated a central role of microRNAs in the regulation of drug
resistance in GC (6). A number of
studies have demonstrated that various microRNAs have altered
expression profiles in GC, and that they are involved in GC
carcinogenesis. For example, miR-532-5p targets runt-related
transcription factor 3 in GC, thereby serving an oncogenic role
(12); and miR-429, by targeting ZEB
proteins, is able to regulate the invasiveness of GC cells
(13). Additionally, a recent study
by Zhao et al (13) revealed
that miR-181a acts to sensitize cells to cisplatin treatment. In
the present study, miR-25 was shown to be significantly upregulated
in the established cisplatin-resistant GC cell line SGC-7901/DDP
compared with the cisplatin-sensitive parental cell line SGC-7901.
Overexpression of miR-25 in the parental GC cell line led to
acquisition of the cisplatin-resistance phenotype, whereas
inhibition of miR-25 in the cisplatin-resistant cell line
resensitized cells to cisplatin and increased cisplatin-induced
cell death. Previous studies have identified the oncogenic
potential of miR-25 in GC: Gong et al (7), Zhao et al (9) and Li et al (10) revealed that miR-25 promoted
proliferation and cell invasiveness by inhibiting various tumor
suppressors. The results of the present study support the findings
of these previous studies, and demonstrate the involvement of
miR-25 in the cisplatin responsiveness of GC cells, thereby
expanding current knowledge on the effects of miR-25.
In the present study, FOXO3a was identified as a
novel functional target of miR-25. FOXO3a is a critical
transcriptional factor in the processes of autophagy, cell cycle
progression and apoptosis (14,15), and
the functioning of FOXO3a is associated with GC. A series of genes
critical for regulating cell survival are under the control of
FOXO3a. For example, p27Kip1, which is a negative regulator of the
cell cycle that inhibits cell cycle progression, is a
well-documented transcriptional target of FOXO3a (16,17).
p27Kip1 is able to prevent the activation of cyclin-CDK complexes
(18,19), which are essential for mitotic cell
cycle transition. The present study revealed that the expression of
p27Kip1 was also decreased when miR-25 was overexpressed, which is
consistent with the cell cycle data obtained from the flow
cytometric analysis; inhibition of miR-25 in cisplatin-resistant GC
cells induced a significant G0/G1 cell cycle
arrest, and p27Kip1 may be involved in this process.
Previous clinical evidence has demonstrated that
high expression levels of FOXO3a were commonly observed in less
aggressive types of GC, and were associated with a good prognosis
in patients with GC (20,21). These findings are corroborated by
in vitro data from the present study, which indicated low
FOXO3a expression levels in cells with the cisplatin-resistance
phenotype. Notably, cell cycle regulation by FOXO3a may not be the
only process that contributes to cisplatin resistance; FOXO3a
regulates a network of genes that is crucial in numerous cellular
processes, including apoptosis, cell cycle regulation and autophagy
(14). The additive effects of these
FOXO3a-regulated processes may also promote the drug-resistance
phenotype. Thus, the present study revealed the essential role of
FOXO3a in the cisplatin resistance of GC cells, and this conclusion
is in accord with previous findings.
As multiple different genes may be targeted by a
single microRNA, FOXO3a may not be the only target of miR-25, and
it is possible that other mechanisms may underlie miR-25-induced
cisplatin resistance. Recent studies demonstrated that a number of
tumor suppressive genes, including F-box and WD repeat
domain-containing 7, large tumor suppressor kinase 2, and
transducer of ERBB2-1, can be targeted by miR-25 in GC (7,8,10). Thus, these proteins are possibly
involved in the acquisition of cisplatin resistance. However, the
findings of the present study reinforce the importance of miR-25 in
the development and progression of GC.
In conclusion, the present findings demonstrated
that miR-25 is upregulated in cisplatin-resistant GC cells, and
represses FOXO3a expression to promote cell cycle progression. The
inhibition of miR-25 resulted in cell cycle arrest and enhanced
chemotherapeutic sensitivity of GC cells to cisplatin, which may be
a novel approach to reversing drug resistance in clinical
practice.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Silberman H: Perioperative adjunctive
treatment in the management of operable gastric cancer. J Surg
Oncol. 90(174–186): 186–187. 2005.
|
|
3
|
Arnason T and Harkness T: Development,
Maintenance and Reversal of Multiple Drug Resistance: At the
Crossroads of TFPI1, ABC Transporters and HIF1. Cancers (Basel).
7:2063–2082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dehghanzadeh R, Jadidi-Niaragh F, Gharibi
T and Yousefi M: MicroRNA-induced drug resistance in gastric
cancer. Biomed Pharmacother. 74:191–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gong J, Cui Z, Li L, Ma Q, Wang Q, Gao Y
and Sun H: MicroRNA-25 promotes gastric cancer proliferation,
invasion and migration by directly targeting F-box and WD-40 Domain
Protein 7, FBXW7. Tumour Biol. 36:7831–7840. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang M, Wang X, Li W and Cui Y: miR-107
and miR-25 simultaneously target LATS2 and regulate proliferation
and invasion of gastric adenocarcinoma (GAC) cells. Biochem Biophys
Res Commun. 460:806–812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao H, Wang Y, Yang L, Jiang R and Li W:
MiR-25 promotes gastric cancer cells growth and motility by
targeting RECK. Mol Cell Biochem. 385:207–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li BS, Zuo QF, Zhao YL, Xiao B, Zhuang Y,
Mao XH, Wu C, Yang SM, Zeng H, Zou QM and Guo G: MicroRNA-25
promotes gastric cancer migration, invasion and proliferation by
directly targeting transducer of ERBB2, 1 and correlates with poor
survival. Oncogene. 34:2556–2565. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Zhang Y, Liu Z, Zhang X and Jia J:
miRNA-532-5p functions as an oncogenic microRNA in human gastric
cancer by directly targeting RUNX3. J Cell Mol Med. 20:95–103.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu W, An J, Li K and Hou H: MiR-429
regulates gastric cancer cell invasiveness through ZEB proteins.
Tumour Biol. 2015.(Epub ahead of print).
|
|
14
|
Nho RS and Hergert P: FoxO3a and disease
progression. World J Biol Chem. 5:346–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang H and Tindall DJ: FOXO factors: A
matter of life and death. Future Oncol. 2:83–89. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dijkers PF, Medema RH, Pals C, Banerji L,
Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW,
Koenderman L and Coffer PJ: Forkhead transcription factor FKHR-L1
modulates cytokine-dependent transcriptional regulation of p27
(KIP1). Mol Cell Biol. 20:9138–9148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rathbone CR, Booth FW and Lees SJ: FoxO3a
preferentially induces p27Kip1 expression while
impairing muscle precursor cell-cycle progression. Muscle Nerve.
37:84–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ray A, James MK, Larochelle S, Fisher RP
and Blain SW: p27Kip1 inhibits cyclin D-cyclin-dependent
kinase 4 by two independent modes. Mol Cell Biol. 29:986–999. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Borriello A, Cucciolla V, Oliva A, Zappia
V and Ragione F Della: p27Kip1 metabolism: A fascinating
labyrinth. Cell cycle. 6:1053–1061. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park SH, Jang KY, Kim MJ, Yoon S, Jo Y,
Kwon SM, Kim KM, Kwon KS, Kim CY and Woo HG: Tumor suppressive
effect of PARP1 and FOXO3A in gastric cancers and its clinical
implications. Oncotarget. 6:44819–44831. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu S, Yu Y, Sun Y, Wang X, Luo R, Zhao N,
Zhang W, Li Q, Cui Y, Wang Y, et al: Activation of FOXO3a suggests
good prognosis of patients with radically resected gastric cancer.
Int J Clin Exp Pathol. 8:2963–2970. 2015.PubMed/NCBI
|