Introduction
Extrahepatic cholangiocarcinoma (ECC) is a highly
malignant cancer, representing ~80% of all cholangiocarcinoma
clinical cases. In previous years, the incidence and mortality of
ECC has continued to increase worldwide (1). Although continued advances in surgical
techniques and treatment strategies have been achieved, the 5-year
survival rate for patients who undergo surgical resection has been
reported to be only 20–40% (2,3). The main
reasons for the poor prognosis of ECC are a low rate of early
diagnosis, fast progression and a high rate of recurrence. There is
currently no effective method to improve the diagnosis and
treatment of ECC, and the major cause of the molecular pathogenesis
of oncogenesis and the progression of ECC remains largely
unclear.
Long non-coding RNAs (lncRNAs) are a large class of
ncRNAs. It has been revealed that lncRNAs are involved in the
development and progression of tumors, and that their abnormal
expression is associated with tumor proliferation, apoptosis, the
cell cycle, angiogenesis, recurrence and metastasis in numerous
different types of cancer (4,5). Previous studies have demonstrated the
potential roles of lncRNAs to serve as diagnostic markers and
therapeutic targets for cancers (6–14).
However, the role and mechanism of lncRNAs in ECC remains largely
unknown. Via transcriptome analysis, the present study aimed to
investigate lncRNA and mRNA expression that is up- or downregulated
in ECC tissues compared with paired peritumoral tissues. Additional
bioinformatics analysis and validation studies were performed to
reveal an association between clinical characteristics and lncRNA
expression levels. These analyses and observations indicated that
alterations in lncRNA expression may become a novel biomarker or
therapeutic target for ECC diagnosis and treatment.
Materials and methods
Patients and tissue samples
A total of 42 patients with ECC who underwent
surgical resection at the Second Affiliated Hospital of Harbin
Medical University (between January 2013 and October 2015) were
included in the present study. All patients provided their written
informed consent for inclusion in this study prior to surgery.
Patients who were treated with preoperative radiotherapy or
chemotherapy were excluded. ECC tissues and paired adjacent
non-cancerous tissues were collected and immediately frozen in
liquid nitrogen. Matched non-cancerous tissues were obtained from
regions of at least 3 cm distant from the tumor borders (China
National Genebank v1.00). A total of 3 pairs of samples were used
for microarray analysis, and all samples were subjected to reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
lncRNA and mRNA microarrays
The Agilent human lncRNA + mRNA Array v4.0 was
designed with four identical arrays per slide (4×180 K format),
with each array containing probes interrogating ~41,000 human
lncRNAs and ~34,000 human mRNAs (Agilent Technologies, Inc., Santa
Clara, CA, USA). Those lncRNA and mRNA target sequences were
mergedfrom multiple databases: 23,898 from GENCODE (http://www.gencodegenes.org/)/ENSEMBL(http://www.ensembl.org); 14,353 from Human LincRNA
Catalog (15); 7,760 from RefSeq
(https://www.ncbi.nlm.nih.gov/refseq/); 5,627 from UCSC
(https://genome.ucsc.edu/); 13,701 from ncRNA
Expression Database; 21,488 from LNCipedia; 1,038 from H-InvDB;
3,019 from lncRNAs-a (Enhancer-like); 1,053 from antisense ncRNA
pipeline; and 407 Hox ncRNAs, 962 upstream conserved regions (UCRs)
and 848 lncRNAs from the Chen Ruisheng lab (Institute of
Biophysics, Chinese Academy of Science, Shanghai, China). Each RNA
was detected by probes, and experiments were repeated twice. The
array also contained 4,974 Agilent control probes (Agilent
Technologies, Inc.).
RNA extraction and quality
control
Total RNA was extracted from 42 pairs of frozen ECC
tissues and matched non-cancerous tissues using TRIzol reagent
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. Tissue (50–100 mg) was homogenized with 1
ml TRIzol reagent in a round-bottomed tube using a glass Teflon
homogenizer, and the homogenized sample was incubated for 10 min at
room temperature. The sample was then centrifuged at 12,000 × g for
10 min at 4°C and the cleared supernatant was transferred to a new
tube. Chloroform (0.3 ml per 1 ml TRIzol) was added to the tube,
and the tube was agitated vigorously by hand for 15 sec, and then
incubated for 2–3 min at room temperature. The sample was then
centrifuged at 12,000 × g for 15 min at 4°C. The aqueous phase was
removed to a fresh tube, 0.5 ml of 100% isopropanol per 1 ml TRIzol
was added to the aqueous phase, and the mixture was incubated at
room temperature for 10 min. Subsequently, the sample was
centrifuged at 12,000 × g for 10 min at 4°C, the supernatant was
removed, and the RNA pellet was washed with 75% (v/v) ethyl alcohol
(EtOH) and vortexed. This was followed by centrifugation at 7,500 ×
g for 5 min at 4°C, removal of the supernatant and subsequent
removal of the remaining EtOH by air drying for 5 min. Finally,
diethyl pyrocarbonate water (20–50 µl) was added to resuspend the
RNA pellets in the tube by passing the solution up and down several
times through a pipette tip. The RNA concentrations were assessed
by measuring absorbance at 260 nm using a NanoDrop ND-1000
spectrophotometer (Thermo Fisher Scientific, Inc.).
RT-qPCR
The expression of lncRNAs in ECC and adjacent
non-cancerous tissues was measured by RT-qPCR using SYBR Premix Ex
TaqÔ (Bioneer Corporation, Daejeon, Korea) and using the following
cycling parameters: Initial denaturation at 94°C for 5 min;
followed by 40 cycles of 94°C for 30 sec; 60°C for 30 sec and 72°C
for 30 sec; and 72°C for 5 min. Primers were designed by Sangon
Biotech Co., Ltd. (Shanghai, China). GAPDH was used as a control.
Experiments were performed in triplicate. The median in each
triplicate was used to calculate relative lncRNA concentrations
using the formula: ΔCq=Cqmedian
lncRNAs-Cqmedian GAPDH. Expression fold changes were
calculated using the 2−ΔΔCq method (16). Primer sequences: ENST00000508732
forward, 5′-ACAGAGATAGCGGAAGGACA-3′ and reverse,
5′-AATGGAGGACTGGAGGGATT-3′; ENST00000519319 forward,
5′-AATGGCATGAACCTGGGAGGCG-3′ and reverse,
5′-GGCTTTGGGAAGTGCTTTGGAG-3′; UC022BVT forward,
5′-TGCTAAAGCATCAGAGAAGAGAAG-3′ and reverse,
5′-GGACGTTCAACCTCATTCCC-3′; ENST00000438290 forward,
5′-GAGGGTTAAACCTGGAGAAGGG-3′ and reverse,
5′-GCAAGAAAATGCGAGAAGCCT-3′; ENST00000593604 forward,
5′-CATGAGGACTGAGCGCATGA-3′ and reverse, 5′-TGCAGTTCCTGTAGGTCAGA-3′;
and GAPDH forward, 5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse
5′-AGGGGCCATCCACAGTCTTC-3′.
Microarray analysis
The lncRNA and mRNA microarray data were analyzed
for data summarization, normalization and quality control using the
GeneSpring software version 13.0 (Agilent Technologies, Inc.).
Differentially-expressed lncRNAs and mRNAs were determined based on
P<0.05, following Benjamini-Hochberg correction and a
fold-change difference of ≥2.0. The raw microarray data was
Log2-transformed and median-centered. The hierarchical
clustering with average linkage was performed for genes and samples
using CLUSTER 3.0 software (17).
Finally, tree visualization was performed using Java Treeview
(Stanford University School of Medicine, Stanford, CA, USA).
Construction of the lncRNA-mRNA
co-expression network
The lncRNA-mRNA co-expression network was
constructed based on association analysis between the
differentially-expressed lncRNAs and mRNAs. For each pair of genes,
Pearson's correlation coefficient was calculated and the
significantly correlated pairs were selected to construct the
network. LncRNAs and mRNAs with Pearson's correlation coefficients
>0.99 were selected to draw the network.
Functional enrichment analysis
To investigate the potential functional roles of
lncRNAs, functional enrichment analysis was performed at the gene
ontology (GO; http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) levels using DAVID
Bioinformatics Resources 6.7 (https://david.ncifcrf.gov/). P<0.05 was considered
to indicate statistically significant functional annotations.
Computational predictions of lncRNA
targets
The regulatory roles of lncRNA on target genes were
mediated by cis- and trans-acting mechanisms. The trans-prediction
was conducted using BLAT tools to compare the full sequence of the
lncRNA with the 3′UTR of its co-expression mRNAs, with the default
parameter setting. For cis-acting lncRNAs, the regulatory RNAs were
transcribed from the same locus as that which encodes the target
gene and which was performed by their tight association (Pearson's
association coefficient >0.99) to a group of expressed
protein-coding genes. The lncRNA resided at genomic loci where a
protein-coding gene and an lncRNA gene were within 10 kb of each
other along the genome (18,19); cis therefore refers to same-locus (not
necessarily same-allele) regulatory mechanisms, which include
antisense-mediated regulation by lncRNAs of protein-coding genes
that are encoded in the same locus.
Statistical analysis
Statistical analysis was performed using SPSS
version 18.0 software (SPSS, Inc., Chicago, IL, USA). Results are
presented as the mean ± standard deviation of three separate
assays. Differences between groups were assessed using the
two-tailed Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Identification of
differentially-expressed lncRNAs and mRNAs in ECC
Differential gene expression analysis in ECC and
adjacent non-cancerous samples was performed to identify
dysregulated lncRNAs and mRNAs in ECC. Among the 41,000 lncRNAs and
34,000 mRNAs transcripts accessed in the present microarray, it was
identified that 268 lncRNAs and 459 mRNAs were differentially
expressed (fold change >2.0) between tumor and adjacent
non-cancerous samples. Among them, 78 lncRNAs and 66 mRNAs were
upregulated (>2-fold in ECC versus adjacent non-cancerous
samples), and 190 lncRNAs and 393 mRNAs were downregulated in ECC
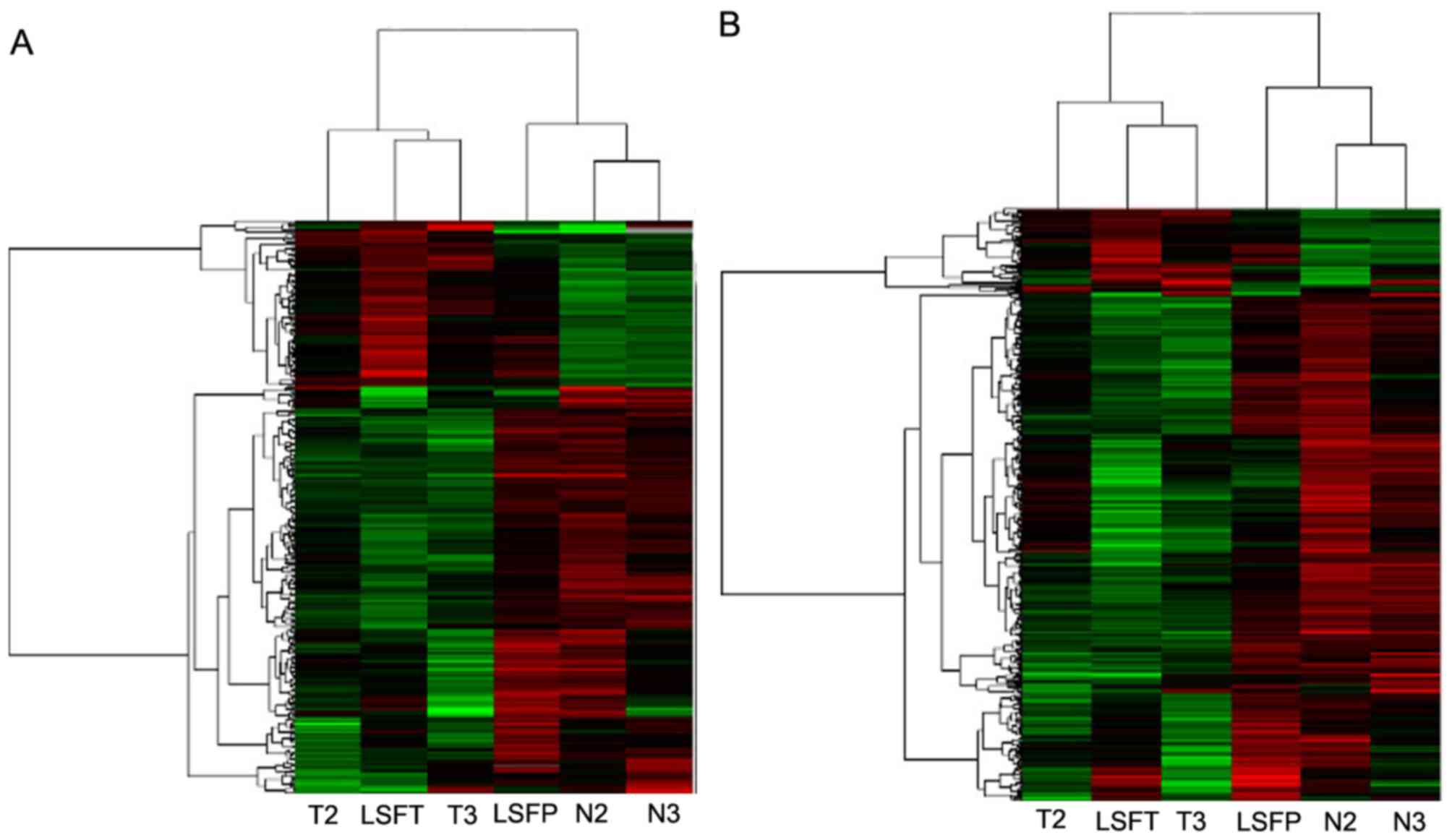
samples. The 10 most differentially-expressed lncRNAs and mRNAs
between ECC and adjacent non-cancerous tissue are listed in
Table I. The data was
Log2-transformed and median-centered by genes using
Adjust Data function of CLUSTER 3.0 software. Hierachial clustering
analysis was then performed, and it was revealed that the
expression profiles of differentially-expressed lncRNAs and mRNAs
were able to distinguish ECC samples from normal tissue samples
(Fig. 1A and B).
 | Table I.List of 10 differentially-expressed
lncRNAs in ECC identified using a microarray screening in ECC and
adjacent non-cancerous tissues (fold change, >2.0;
P<0.05). |
Table I.
List of 10 differentially-expressed
lncRNAs in ECC identified using a microarray screening in ECC and
adjacent non-cancerous tissues (fold change, >2.0;
P<0.05).
| lncRNAs | Chr | Strand | Genomic
coordinates | Expression | Fold-change |
|---|
|
ENST00000508732.2 | 15 | − |
95822513–95870329 | Upregulated | 21.486 |
| TCONS_00004225 | 2 | − |
43199538–43228604 | Downregulated | 7.547 |
|
ENST00000438290.1 | 13 | + |
94712716–94716246 | Downregulated | 6.386 |
|
ENST00000423943.1 | 1 | + |
159931014–159948851 | Downregulated | 5.770 |
| TCONS_00014813 | 8 | + |
102326509–102328921 | Upregulated | 5.134 |
|
ENST00000515485.1 | 4 | + |
165675216–165722606 | Downregulated | 4.172 |
|
ENST00000437097.1 | 9 | + |
128329858–128335302 | Upregulated | 4.079 |
| TCONS_00008571 | 4 | − |
128015586–128017878 | Downregulated | 4.043 |
|
ENST00000606993.1 | 1 | + |
1104737–1105723 | Downregulated | 4.042 |
|
ENST00000550334.1 | 12 | − |
72255681–72271991 | Downregulated | 3.954 |
Functional analysis of
differentially-expressed lncRNAs
To reveal the potential roles of lncRNAs in ECC, the
association between lncRNAs and mRNAs was investigated, and a
coding-non-coding gene co-expression (CNC) network was constructed
by examining the association between the expression values of
lncRNAs and those of the mRNAs. A total of 270 network nodes were
associated with 5,788 network pairs of co-expressed lncRNAs and
mRNA. The number of positively-associated pairs was greater than
the number of negatively-associated pairs. The CNC network
indicated that mRNAs may be associated with one or numerous
lncRNAs. Similarly, lncRNAs may be associated with one or numerous
mRNAs. XLOC_002797 had 38 neighbors, whereas collagen a-3
(type VI) mRNA had 29 neighbors. The CNC networkrevealed the
inter-regulation of lncRNAs and mRNAs in ECC.
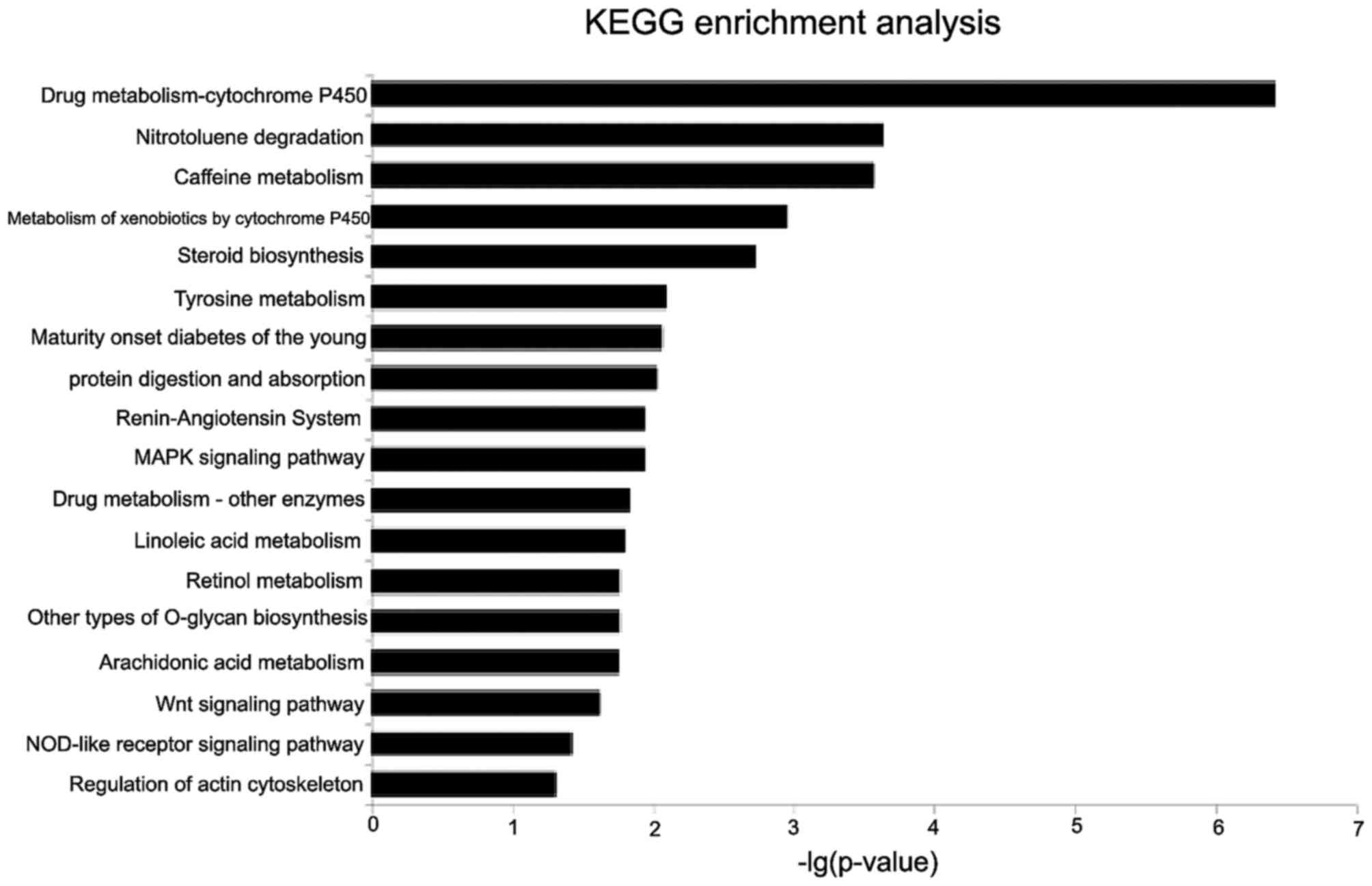
GO and KEGG function enrichment analysis was then
performed for mRNAs co-expressed with lncRNAs to identify
biological processes and signaling pathways affected by
differentially-expressed lncRNAs. GO analysis revealed that the
differentially-expressed mRNAs between ECC and adjacent
non-cancerous tissue were significantly enriched in cellular
response to ultraviolet-A rays, the sensory perception of pain, the
creatinine metabolic process and protein processing. KEGG analysis
indicated that the deregulated mRNAs between ECC and adjacent
non-cancerous tissue were mainly involved in drug
metabolism-cytochrome P450, nitrotoluene degradation, caffeine
metabolism, the mitogen-activated protein kinase (MAPK) signaling
pathway, the peroxisome proliferator-activated receptor (PPAR)
signaling pathway, protein digestion and absorption, the Wnt
signaling pathway and the nucleotide oligomerization domain-like
receptor signaling pathway (P<0.05, following multiple testing
correction; Fig. 2).
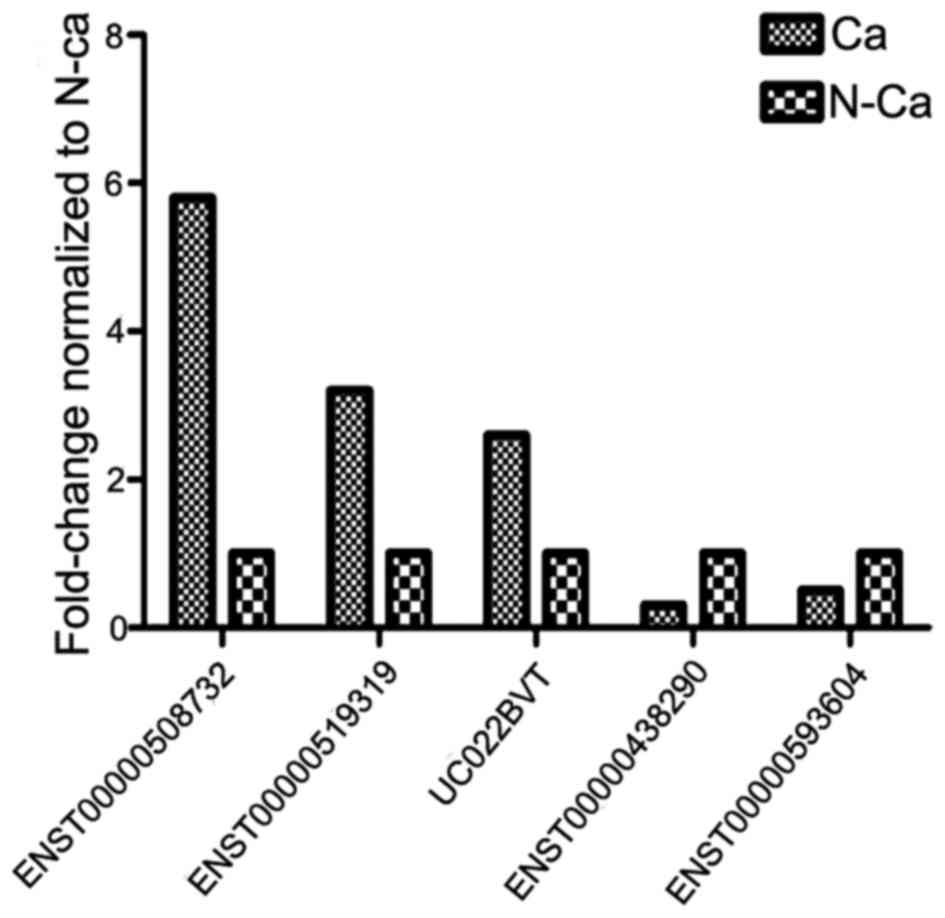
Validation of microarray results by
RT-qPCR
In order to confirm aberrant lncRNA expression, 5 of
the differentially-expressed lncRNAs in ECC were randomly selected
for subsequent analysis to ensure the validity of microarray
results using RT-qPCR in 42 pairs of ECC and adjacent non-cancerous
tissues. The results revealed that ENST00000508732, ENST00000519319
and UC022BVT were upregulated, while ENST00000438290 and
ENST00000593604 were downregulated in ECC compared with adjacent
non-cancerous tissue (Fig. 3).
Overall, the present results demonstrated an association between
RT-qPCR and microarray findings.
Discussion
lncRNAs are important regulators of gene expression
during biological information processing and major cellular
pathways, including proliferation, differentiation and apoptosis in
living cells. Therefore, lncRNAs are involved in carcinogenesis or
the antitumor effects of numerous human malignancies (20). However, thus far, knowledge about
lncRNA expression in ECC is largely unknown.
A number of the differentially expressed lncRNAs and
mRNAs that were identified in the present study are known to
perform important roles in ECC. Differential expression of 268
lncRNA transcripts (defined as expression differences >2-fold)
was observed. The expression was successfully validated for
upregulated (ENST00000508732, ENST00000519319 and UC022BVT) and
downregulated (ENST00000438290 and ENST00000593604) lncRNAs using
qPCR in 42 pairs of ECC and adjacent non-cancerous frozen specimen.
Thus, the specificity of the microarray results was confirmed.
If expression differences of lncRNAs are validated
by independent researchers, these lncRNAs may represent diagnostic
biomarkers or therapeutic targets in ECC. A previous study reported
that lncRNA may be a potential diagnostic and prognostic biomarker
for intrahepatic cholangiocarcinoma (ICC), using lncRNA and mRNA
microarrays, and also considered that the expression of lncRNA and
mRNA may predict the survival of patients with ICC (21), although, as is commonlyunderstood, the
embryogenesis, anatomy and biological behavior of ECC and ICC
differ (22). Identification of
diagnostic biomarkers or therapeutic targets in bodily fluids may
assist to improve patient outcome and understanding of the
molecular mechanismof cancer progression, for example, lncRNA
PCA3 in urine is used as a diagnostic biomarker for prostate
cancer (23). CertainlncRNAs,
including H19 (imprinted maternally-expressed transcript) and
FENDRR (adjacent non-coding developmental regulatory RNA), have
been revealed to be differentially expressed in certain tumors
(24,25); this dysregulationwas also observedin
ECC tissues by microarray detection in the present study. The role
and mechanism of certain known lncRNAs in ECC require additional
investigations.
However, little is known on the function of lncRNAs
and how to research them. Therefore, microarrays of lncRNAs and
mRNAs may assist to elucidate this through certain bioinformatics
methods, including CNC network and target gene predictions (cis and
trans). The present results may provide clues for additional basic
studies (Table II). The theory of
target gene predictions may reveal that lncRNA functions via
lncRNA-mRNA-protein interactions (26). The majority of these proteins code
genes that function in the splicing, binding, transport,
localization, transcription, translation and processing of RNA,
according to GO function prediction.
| Table II.Target prediction from lncRNAs to
mRNAs. |
Table II.
Target prediction from lncRNAs to
mRNAs.
| lncRNA | mRNA | Correlation | P-value | Cis-regulation |
Trans-regulation |
|---|
| p14589 | A_P186342 | 0.99929 |
2.387×10−2 |
| miRNA
sequestration |
| p29152 | A_P186342 | −0.99977 |
1.353×10−2 |
| miRNA
sequestration |
| p21976 | A_P0002916 | 0.99992 |
7.638×10−3 | Sense |
|
| p17770 | A_P328023 | −0.99848 |
3.508×10−2 |
| miRNA
sequestration |
| p20598 | A_P132317 | −0.99908 |
2.722×10−2 |
| miRNA
sequestration |
| p6091 | A_P3414127 | 0.99851 |
3.469×10−2 |
| miRNA
sequestration |
| p9680 | A_P0001828 | 0.99999 |
6.390×10−5 | Sense |
|
| p41956 | A_P3221253 | −0.99772 |
4.293×10−2 |
| miRNA
sequestration |
It has also been reported that lncRNAs may act as a
microRNA sponge by binding specific microRNAs and thereby
disrupting microRNA regulation of mRNA 3′UTRs (27). In the present study, 39 lncRNAswere
predicted to target mRNAs by comparingsequences of lncRNAs with the
3′UTR of mRNAs. The miRTarget2 algorithm (28), starBase(http://starbase.sysu.edu.cn/) (29) and miRcode (www.mircode.org/) (30)
were used to predict miRNA seeds within the validated lncRNA
transcripts, which may assist in constructing the lncRNA-miRNA-mRNA
axis. Prensner et al showed that prostate cancer-associated
transcript 1 is able to abrogate the downregulation of cMyc by
downregulating the expression of miR-34a in prostate cancer
(4).
KEGG analysis has revealed that drug
metabolism-cytochrome P450 (31),
nitrotoluene degradation and caffeine metabolism induced by
N-acetyltransferase (NAT) 1 and NAT2 (32) have significant association with the
genesis and development of ECC, with the exception of classical
proliferation and apoptotic pathways (such as the PPAR and MAPK
pathways).
In summary, the present study revealed that
dysregulation of ~4% of the lncRNA transcripts occurs in ECC, and
altered lncRNA expression may modulate fundamental cellular
processes. lncRNA profiles were able to accurately distinguish
between ECC and adjacent non-cancer tissue. Thus, lncRNAs may be
used as biomarkers and therapeutic targets for patients with
ECC.
References
|
1
|
Khan SA, Emadossadaty S, Ladep NG, Thomas
HC, Elliott P, Taylor-Robinson SD and Toledano MB: Rising trends in
cholangiocarcinoma: Is the ICD classification system misleading us?
J Hepatol. 56:848–54. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeOliveira ML, Cunningham SC, Cameron JL,
Kamangar F, Winter JM, Lillemoe KD, Choti MA, Yeo CJ and Schulick
RD: Cholangiocarcinoma: Thirty-one-year experience with 564
patients at a single institution. Ann Surg. 245:755–762. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Gaag NA, Kloek JJ, de Bakker JK,
Musters B, Geskus RB, Busch OR, Bosma A, Gouma DJ and van Gulik TM:
Survival analysis and prognostic nomogram for patients undergoing
resection of extrahepatic cholangiocarcinoma. Ann Oncol.
23:2642–2649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun M, Jin FY, Xia R, Kong R, Li JH, Xu
TP, Liu YW, Zhang EB, Liu XH and De W: Decreased expression of long
noncoding RNA GAS5 indicates a poor prognosis and promotes cell
proliferation in gastric cancer. BMC Cancer. 14:3192014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Prensner JR, Chen W, Han S, Iyer MK, Cao
Q, Kothari V, Evans JR, Knudsen KE, Paulsen MT, Ljungman M, et al:
The long non-coding RNA PCAT-1 promotes prostate cancer cell
proliferation through cMyc. Neoplasia. 16:900–908. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian
J, Zhang X and Fang JY: A long non-coding RNA signature to improve
prognosis prediction of colorectal cancer. Oncotarget. 5:2230–2242.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun J, Chen X, Wang Z, Guo M, Shi H, Wang
X, Cheng L and Zhou M: A potential prognostic long non-coding RNA
signature to predict metastasis-free survival of breast cancer
patients. Sci Rep. 5:165532015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou M, Guo M, He D, Wang X, Cui Y, Yang
H, Hao D and Sun J: A potential signature of eight long non-coding
RNAs predicts survival in patients with non-small cell lung cancer.
J Transl Med. 13:2312015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou M, Wang X, Shi H, Cheng L, Wang Z,
Zhao H, Yang L and Sun J: Characterization of long non-coding
RNA-associated ceRNA network to reveal potential prognostic lncRNA
biomarkers in human ovarian cancer. Oncotarget. 7:12598–12611.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou M, Zhao H, Wang Z, Cheng L, Yang L,
Shi H, Yang H and Sun J: Identification and validation of potential
prognostic lncRNA biomarkers for predicting survival in patients
with multiple myeloma. J Exp Clin Cancer Res. 34:1022015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng N, Li X, Zhao C, Ren S, Chen X, Cai
W, Zhao M, Zhang Y, Li J, Wang Q and Zhou C: Microarray expression
profile of long non-coding RNAs in EGFR-TKIs resistance of human
non-small cell lung cancer. Oncol Rep. 33:833–839. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li C, Liang G, Yao W, Sui J, Shen X, Zhang
Y, Ma S, Ye Y, Zhang Z, Zhang W, et al: Differential expression
profiles of long non-coding RNAs reveal potential biomarkers for
identification of human gastric cancer. Oncol Rep. 35:1529–1540.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo J, Xu L, Jiang Y, Zhuo D, Zhang S, Wu
L, Xu H and Huang Y: Expression profile of long non-coding RNAs in
colorectal cancer: A microarray analysis. Oncol Rep. 35:2035–2044.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu JJ, Ye LC, Ding JX, Feng WW, Jin HY,
Zhang Y, Li Q and Hua KQ: Expression and clinical significance of
estrogen-regulated long non-coding RNAs in estrogen receptor
α-positive ovarian cancer progression. Oncol Rep. 31:1613–1622.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ørom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q,
et al: Long noncoding RNAs with enhancer-like function in human
cells. Cell. 143:46–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jia H, Osak M, Bogu GK, Stanton LW,
Johnson R and Lipovich L: Genome-wide computational identification
and manual annotation of human long noncoding RNA genes. RNA.
16:1478–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun J, Zhou M, Mao ZT, Hao DP, Wang ZZ and
Li CX: Systematic analysis of genomic organization and structure of
long non-coding RNAs in the human genome. FEBS Lett. 587:976–982.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martens-Uzunova ES, Bottcher R, Croce CM,
Jenster G, Visakorpi T and Calin GA: Long noncoding RNA in
prostate, bladder, and kidney cancer. Eur Urol. 65:1140–1151. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Xie H, Ling Q, Lu D, Lv Z, Zhuang
R, Liu Z, Wei X, Zhou L, Xu X and Zheng S: Coding-noncoding gene
expression in intrahepatic cholangiocarcinoma. Transl Res.
168:107–121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
ICD-O-Cs (International Classification of
Diseases-Oncology-Czech edition). Cesk Patol. 19:228–250. 1983.(In
Czech). PubMed/NCBI
|
|
23
|
Bradley LA, Palomaki GE, Gutman S, Samson
D and Aronson N: Comparative effectiveness review: Prostate cancer
antigen 3 testing for the diagnosis and management of prostate
cancer. J Urol. 190:389–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Raveh E, Matouk IJ, Gilon M and Hochberg
A: The H19 Long non-coding RNA in cancer initiation, progression
and metastasis-a proposed unifying theory. Mol Cancer. 14:1842015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu TP, Huang MD, Xia R, Liu XX, Sun M, Yin
L, Chen WM, Han L, Zhang EB, Kong R, et al: Decreased expression of
the long non-coding RNA FENDRR is associated with poor
prognosis in gastric cancer and FENDRR regulates gastric
cancer cell metastasis by affecting fibronectin1 expression. J
Hematol Oncol. 7:632014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu J, Fu H, Wu Y and Zheng X: Function of
lncRNAs and approaches to lncRNA-protein interactions. Sci China
Life Sci. 56:876–885. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X and El Naqa IM: Prediction of both
conserved and nonconserved microRNA targets in animals.
Bioinformatics. 24:325–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction
networks from large-scale CLIP-Seq data. Nucleic Acids Res.
42:(Database Issue). D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jeggari A, Marks DS and Larsson E:
miRcode: A map of putative microRNA target sites in the long
non-coding transcriptome. Bioinformatics. 28:2062–2063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yongvanit P, Phanomsri E, Namwat N, Kampan
J, Tassaneeyakul W, Loilome W, Puapairoj A and Khuntikeo N: Hepatic
cytochrome P450 2A6 and 2E1 status in peri-tumor tissues of
patients with Opisthorchis viverrini-associated cholangiocarcinoma.
Parasitol Int. 61:162–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prawan A, Kukongviriyapan V, Tassaneeyakul
W, Pairojkul C and Bhudhisawasdi V: Association between genetic
polymorphisms of CYP1A2, arylamine N-acetyltransferase 1 and 2 and
susceptibility to cholangiocarcinoma. Eur J Cancer Prev.
14:245–250. 2005. View Article : Google Scholar : PubMed/NCBI
|