Introduction
Breast cancer is one of the most common malignant
neoplasms in women. In total, 5–10% of breast cancer cases are
associated with genetic susceptibility, and the most common breast
cancer susceptibility genes are breast cancer 1 (BRCA1) and
breast cancer 2 (BRCA2) (1).
Differences in mutations of BRCA1 and BRCA2 have been
reported among different ethnic populations. Bergman et al
(2) reported a familial mutation rate
of up to 36% in BRCA1 or BRCA2 in Western Swedish
breast cancer families. In Chinese females with familial breast
cancer the mutation rate of BRCA1 and BRCA2 was ~10%,
of which 50–60% of the mutations have not been previously reported,
and their functions are unknown (3).
The BRCA1 gene is a tumor suppressor gene
that is located on 17q21, with a total length of ~100 kb.
BRCA1 consists of 24 exons and encodes a protein with 1,863
amino acids. BRCA1 has an important role in DNA damage repair, cell
cycle control, protein ubiquitination and chromatin remodeling
(4). BRCA1 includes two important
functional domains, namely really interesting new gene (RING)
domain in the N-terminal and BRCA1 C terminus (BRCT) domain in the
C-terminal. Several studies have shown that the RING domain of
BRCA1 interacts with BRCA1-associated RING domain protein 1
to form a powerful E3 ubiquitin ligase and has a role in tumor
suppression by regulating several signaling transduction pathways
(5,6).
However, Shakya et al (7)
reported that the BRCT domain was the structure with tumor
suppressor function, since point mutations of the BRCT domain
caused rapid formation of tumors in mouse models.
Over 500 different BRCA1 mutations have been
identified throughout its coding region, including nonsense
mutations, missense mutations, frame shift insertions or deletions,
as well as mutations in the untranslated region (8). Among these mutations, frame shift
insertions or deletions or nonsense mutations, resulting in 20–30%
of familial breast cancer cases, are the most deleterious and
usually result in the formation of a truncated protein. For
example, the 185delAG BRCA1 mutation led to the loss of all
known functional domains (9).
Missense mutations, accounting for 5–10% of familial breast cancer
cases, often occur in the coding region of the highly conserved
amino acids (10). Missense mutations
usually only partially affect the function of wild-type
BRCA1 and rarely cause loss of the entire structure or
function, while the clinical significance of these missense
mutations is often uncertain (11).
As a consequence, 10–20% of patients with familial breast cancer
cannot receive any meaningful information from clinical genetic
testing (8,11).
Two missense mutations, G1763V and L1786P were
identified from Chinese females with familiar breast cancer in our
previous study (unpublished data). These two mutations, which are
located in the BRCT domain, have not been previously described in
the database established by the Breast Cancer Information Core. The
present study investigated the function of these two mutations and
revealed that these novel missense mutations did not affect the
tumor suppressor function of the BRCA1 gene.
Materials and methods
Cell line and cell culture
The BRCA1-mutated breast cancer HCC1937 cell
line was obtained from the Cell Bank of Shanghai Institute of Cell
Biology (Shanghai, China). No wild-type BRCA protein was produced
in the HCC1937 cell line. The cells were maintained in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere with 5% CO2.
Construction of mutated plasmids and
transfection
A plasmid containing a full-length BRCA1 cDNA
on a pcDNA3.1 backbone was provided by Dr Genze Shao (Peking
University Health Science Center, Beijing, China), for the
sub-cloning and generation of the mutated expression constructs.
The G1763V and L1786P mutations were generated in this plasmid
using QuikChange II XL Site-Directed Mutagenesis kit (Agilent
Technologies, Inc., Santa Clara, CA, USA). Primer pairs for the
G1763V mutagenesis protocol were as follows: 1763 mutation forward,
5′-AGGACAGAAAGATCTTCAGGGTGCTAGAAATCTG-3′ and reverse,
5′-ACCCTGAAGATCTTTCTGTCCTGGGATTCTCTTG-3′. The primer pairs for the
L1786P mutagenesis protocol were as follows: 1786 mutation forward,
5′-GGAATGGATGGTACAGCcGTGTGGTGCTTCTGTGG-3′ and reverse,
5′-CCACAGAAGCACCACACgGCTGTACCATCCATTCC-3′. Large-scale DNA
preparations were made using the Qiagen Plasmid Maxi Kit (Qiagen,
Inc., Valencia, CA, USA). Each plasmid was sequenced entirely to
verify their identity. For gene transfection, HCC1937 cells were
grown overnight at 37°C and transfected with plasmids using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from transfected HCC1937
cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. Reverse
transcription was performed using a total of 1 µg RNA, oligo (dT)
15 primer, and the ThermoSCRIPT reverse transcription kit
(Invitrogen; Thermo Fisher Scientific, Inc.). RT-qPCR was performed
using SYBR Green PCR Master Mix (Thermo Fisher Scientific, Inc.) on
a ABI7500 Real-Time PCR System (Thermo Fisher Scientific, Inc.).
qPCR was performed using the following conditions: 95°C for 10 min,
followed by 40 cycles of 95°C for 30 sec and 60°C for 1 min.
Primers for BRCA1, poly (ADP-ribose) polymerase 1
(PARP) and GAPDH were as follows: BRCA1_F,
5′-AGGTCCAAAGCGAGCAAGAG-3′ and BRCA1_R, 5′-AGGTGCCTCACACATCTGCC-3′;
PARP_F, 5′-CCTAAAGGCTCAGAACGACC-3′ and PARP_R,
5′-AGGAGGGCACCGAACACC-3′; and GAPDH_F, 5′-AGGTCGGAGTCAACGGATTTG-3′
and GAPDH_R, 5′-GTGATGGCATGGACTGTGGT-3′. The results of
BRCA1 and PARP were normalized to GAPDH. Data
were calculated based on 2−ΔCq, where ΔCq=Cq (Target)-Cq
(Reference). Fold change was calculated using the 2−ΔΔCq
method (12).
Protein extraction and
immunoblotting
Proteins were extracted from cells using
radioimmunoprecipitation assay buffer containing complete protease
inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany).
Proteins were separated using 8% SDS-PAGE and transferred to
polyvinylidene fluoride membranes. The membranes were then blocked
with 5% non-fat milk in TBST [15 mM Tris-HCl (pH 7.4), 0.9% NaCl
and 0.05% Tween-20 (pH 7.4)] for 1 h at room temperature., and then
incubated with primary antibody overnight at 4°C using rabbit
monoclonal anti-human BRCA1 antibody (dilution, 1:1,000; A301-377;
Bethyl Laboratories, Montgomery, TX, USA), and goat anti-rabbit
secondary antibody (dilution, 1:5,000; ZDR-5306; ZSGB-BIO, Beijing,
China). Immunoreactive bands were detected using Super Signal West
Femto Chemiluminescent Substrate (Merck KGaA, Darmstadt, Germany).
The aforementioned experiments were performed at least three times
with consistent results.
Cell proliferation assay
The cell proliferation assay was performed using
cell counting kit-8 assay. In total, 2×103 cells were
cultured in 96-well culture plates. The cells were resuspended in
RPMI-1640 medium containing 10% FBS and cultured for 0, 24, 48 or
72 h at 37°C. The number of viable cells was determined by
measuring absorbance at 450 nm using FLUOstar OPTIMA (BMG Labtech,
Offenburg, Germany), according to the manufacturer's instructions.
Each experiment was performed in triplicate.
Laser confocal fluorescence microscopy
for subcellular localization
The cells were fixed in 4% paraformaldehyde for 30
min at room temperature and lysed with 0.2% Triton-X-100 for 15
min. This was followed by the addition of 5% bovine serum albumin
(ZLI-9027, ZSGB-BIO, Beijing, China) to the lysate and incubation
for 30 min at 37°C. Rabbit monoclonal anti-human BRCA1 antibody
(dilution, 1:1,000; A301-377; Bethyl Laboratories) and monoclonal
antibody for histone H2A variant X (γH2AX; dilution, 1:100;
05–636-I; EMD Millipore, Billerica, MA, USA) were added to the
mixture followed by incubation at 4°C overnight. Cell lysate with
0.01 mol/l phosphate-buffered saline instead of the primary
antibody was used as a negative control. Fluorescence-labeled
secondary antibodies (dilution, 1:4,000; ZF-0311, ZF-0316;
ZSGB-BIO, Beijing, China) were added to the mixture, which was then
incubated in the dark at room temperature for 30 min. The mixture
was further incubated with DAPI solution at room temperature for 5
min and then mounted in Tris-buffered glycerol solution. The
subcellular localization of the proteins was determined using
confocal laser scanning microscopy at a magnification of
×12,50.
Flow cytometry analysis of
apoptosis
A total of 3×105 HCC1937 cells were
seeded in 6-wells culture plates in RPMI-1640 medium with 10% FBS.
After 12 h, cells were transfected with the aforementioned plasmids
using Lipofectamine 2000. Subsequent to transfection, cells were
collected at 24, 48 and 72 h and stained with fluorescein
isothiocyanate-conjugated Annexin V (BioVision, Inc., Milpitas, CA,
USA) at room temperature for 30 min, followed by staining with
propidium iodide (PI) 1 min prior to analysis for apoptosis by
FACScan (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis
Cell cycle stage was determined by flow cytometry
using a cell-cycle assay kit (Ab139418, Abcam, Cambridge, UK).
Briefly, HCC1937 cells were harvested by centrifugation with 300 ×
g, 5 min at 4°C, washed with PBS and fixed with cold 75% ethanol at
4°C overnight. The fixed cells were then stained with PI and RNaseA
(Ab139418; Abcam, Cambridge, UK), according to the manufacturer's
instruction. Following a 30-min incubation in the dark,
fluorescence-activated cells were sorted in a FACScan flow
cytometer (BD Biosciences, San Jose, CA, USA). The cells were
distinguished as being at the G0/G1, G2/M and S phases of the cell
cycle based on the fluorescence intensity, and the distribution of
the cell-cycle stage was analyzed using ModFit software (BD
Biosciences).
Statistical analysis
Results were expressed as mean ± standard deviation.
One-way analysis of variance was used for statistical comparison
among groups. Multiple comparison between the groups was performed
using Bonferroni method. P<0.05 was considered to indicate a
statistically significant difference.
Results
Bioinformatics analyses showed that
G1763V and L1786P abolish the tumor suppression function of
BRCA1
Two novel BRCA1 missense mutations in the
BRCT domain were identified from the cohort of Chinese women with
familial breast cancer, consisting of G1763V due to p5407 G>T
and L1786P due to p5476 T>C. Protein mutation analysis software,
consisting of PolyPhen (http://genetics.bwh.harvard.edu/pph/), SIFT
(http://sift.jcvi.org/) and Pmut (http://mmb2.pcb.ub.es:8080/PMut/), was used to
predict the impact of these two BRCT mutations on BRCA1 protein and
their possible pathogenicity. PolyPhen software predicted that
G1763V and L1786P both damage the original protein function. SITF
and Pmut software predicted that both mutations were deleterious
(Table I).
 | Table I.Bioinformatics analysis of the two
breast cancer 1 mutations. |
Table I.
Bioinformatics analysis of the two
breast cancer 1 mutations.
| Nucleotide
change | Protein change | Mutation type | SIFT | PolyPhen | Pmut |
|---|
| 5407 G>T | G1763V | Germline | Damage | Possible
damaging | Pathological |
| 5476 T>C | L1786P | Germline | Damage | Possible
damaging | Pathological |
G1763V and L1786P mutants were
expressed in the BRCA1-deficient HCC1937 cell line
In order to test and verify the function of G1763V
and L1786P mutations, mutation plasmids were constructed from
pcDNA3.1-BRCA1 (termed wt BRCA1), and the expression of wild and
mutant BRCA1 was examined in the BRCA1-deficiency breast
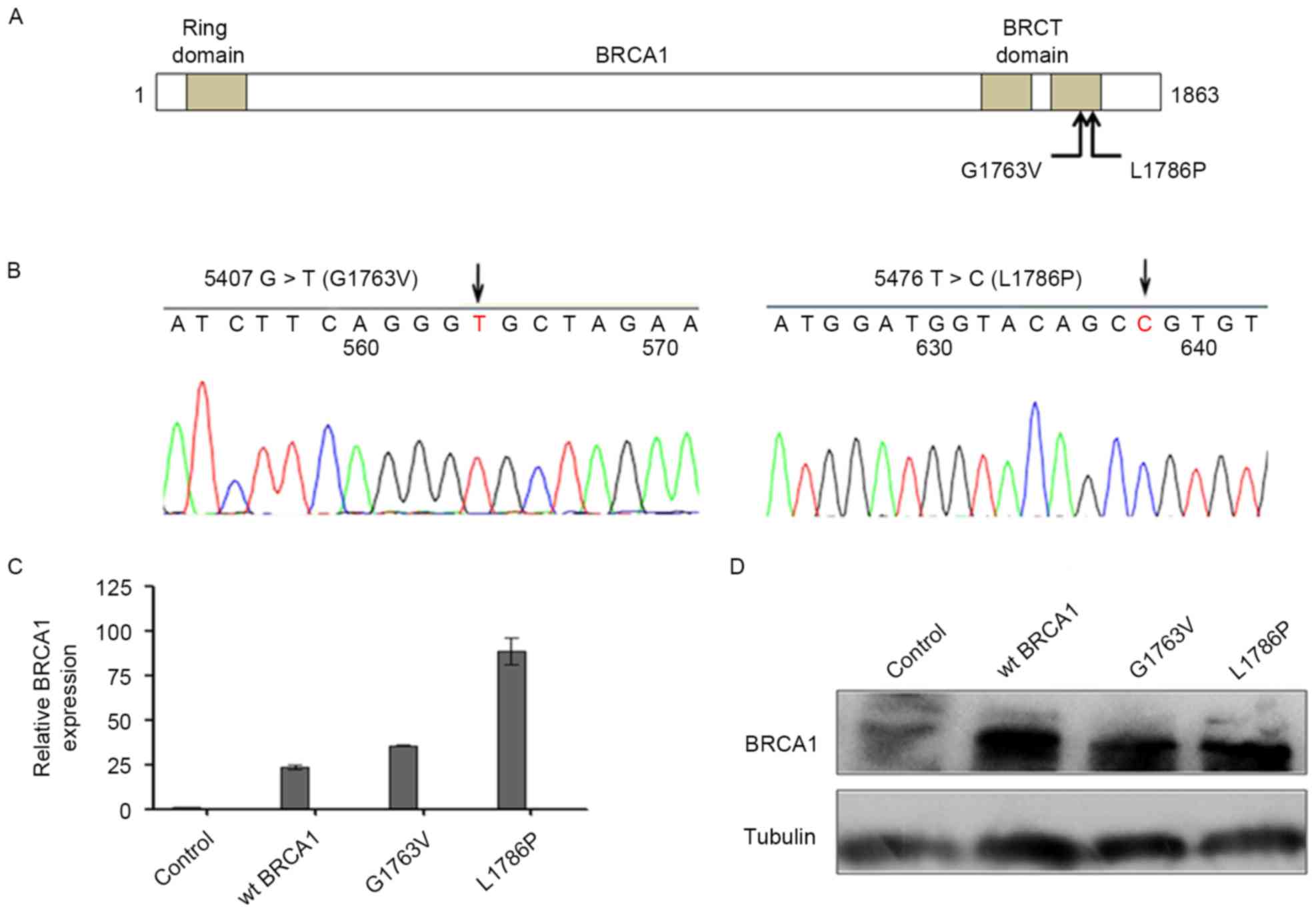
cancer HCC1937 cell line. Fig. 1A
shows the schematic diagram of the location of the mutants. The
sequences of the mutant constructs were verified by sequencing
(Fig. 1B). BRCA1
overexpression was observed both at the mRNA level and protein
level in these three groups compared with the control group 24 h
subsequent to transfection with wt BRCA1 and two mutants (Fig. 1C and D).
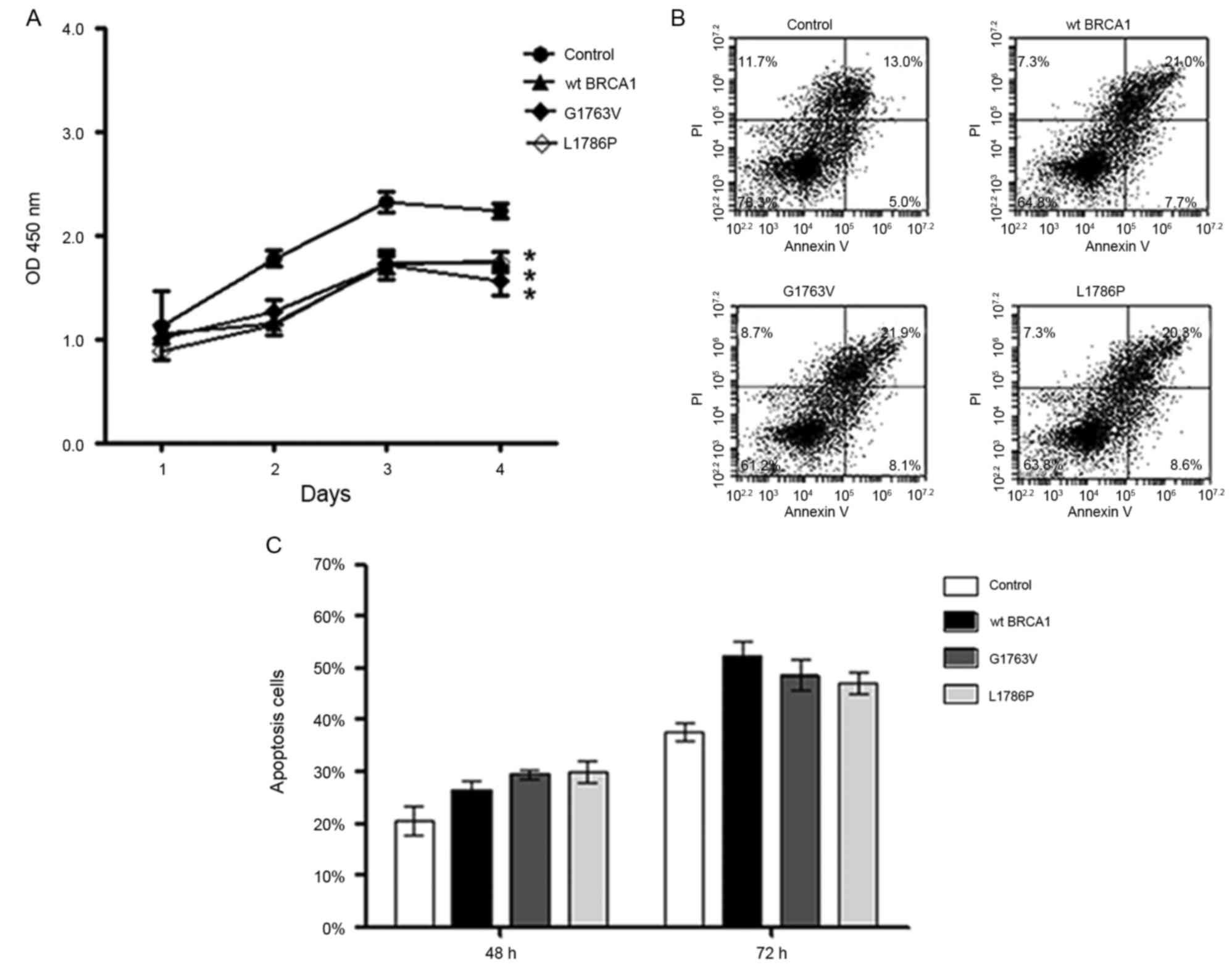
G1763V and L1786P mutants reduced cell
growth and increased cell apoptosis in vitro
Subsequent to successfully constructing the
transient transfection cell model, the effect of BRCA1
mutants on breast cancer cell growth and apoptosis was assessed
in vitro. Cell proliferation assay using HCC1937 cells
showed that the wild type BRCA1 and both of the two mutants
significantly reduced the growth of breast cancer cells compared
with the control group (P<0.05; Fig.
2A). Additionally, flow cytometric analysis showed that the
wild type BRCA1 and both of the mutants significantly increased
cell apoptosis compared with the control group (Fig. 2B and C) 72 h subsequent to
transfection. These data indicated that G1763V and L1786P have
similar tumor suppressor function to wild-type BRCA1 in
vitro.
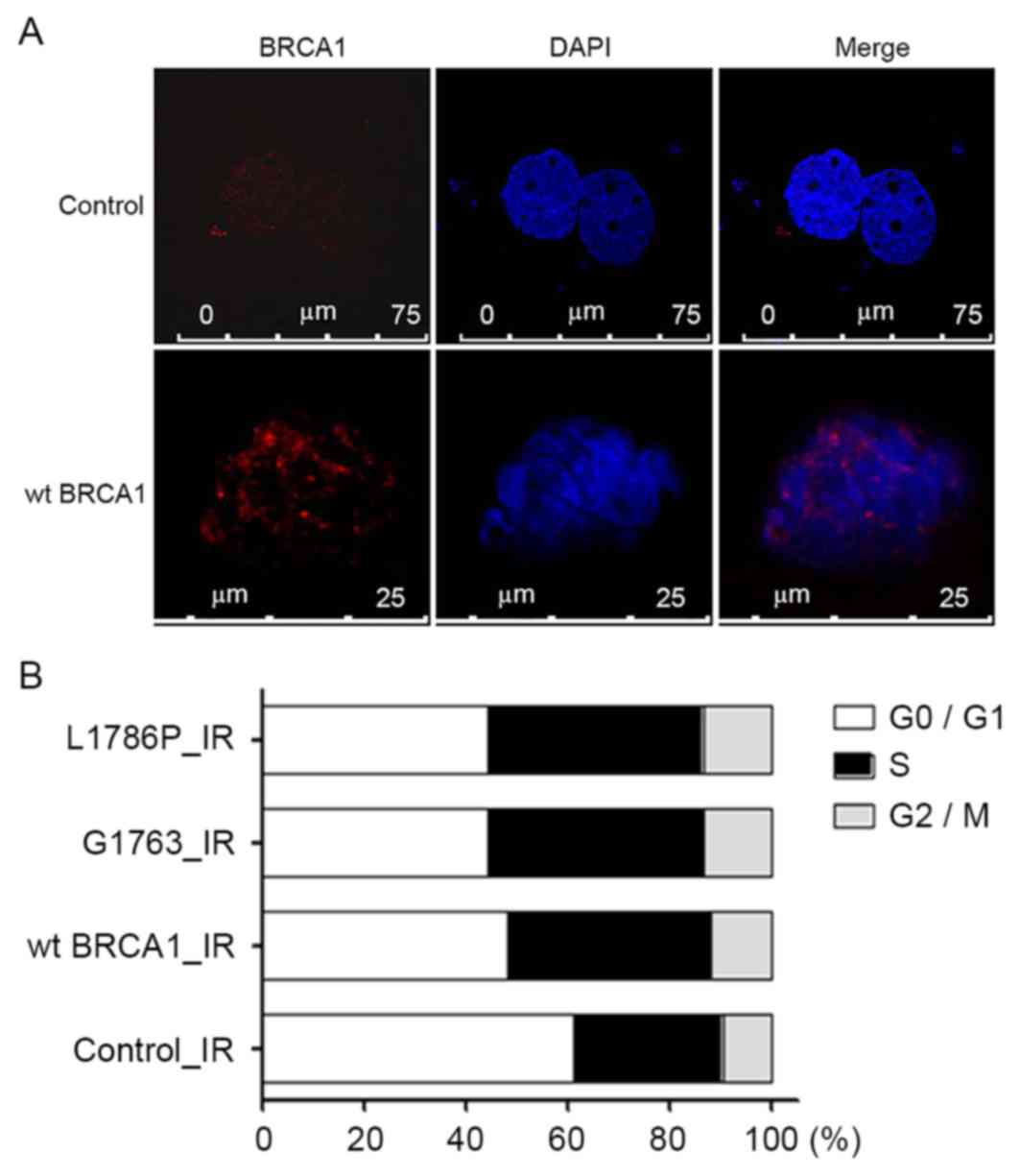
G1763V and L1786P mutants caused
S-phase arrest after irradiation
In order to investigate the function of these two
mutants following DNA damage, a DNA damage model was created using
γ radiation. Phosphorylation of γH2AX is the most sensitive marker
that can be used to examine the DNA damage and the subsequent DNA
repair. It was found that γH2AX foci increased significantly in the
irradiated group when compared with the groups without irradiation
(Fig. 3A). After 8-Gy irradiation,
the percentages of cells in the S-phase were 42.1, 42.1 and 40.0%
in the wild-type BRCA1, G1763V and L1786P cells,
respectively, which were increased compared with the control group
with 29.2% (control vs. wt BRCA1, control vs. G1763V, control vs.
L1786P; P<0.05; Fig. 3B).
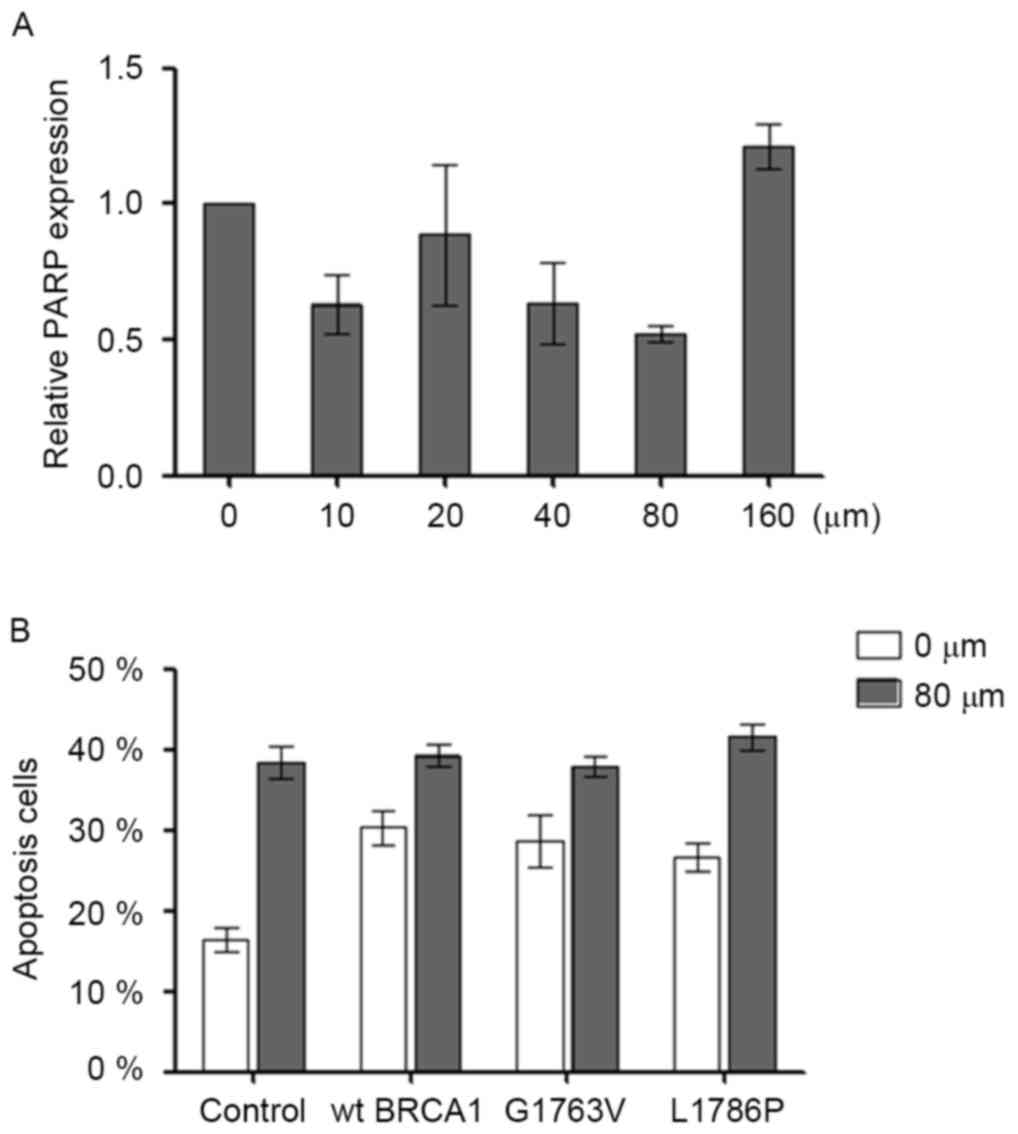
G1763V and L1786P mutants did not
sensitize cells to the PARP1 inhibitor Olaparib
In order to test the optimal concentration of
Olaparib, HCC1937 cells were cultured in the presence of 0, 20, 40,
80 or 160 µM Olaparib. After 48 h, it was observed that 80 µM
Olaparib produced the maximum suppression of PARP expression by 50%
(Fig. 4A). Olaparib (80 µM) was then
added into the cell cultures when the transfected cells were
adherent. Olaparib increased cell apoptosis in HCC1937 (control 0
µM vs. control 80 µM; P<0.05). However, no synergistic effect
between the Olaparib and BRCA1 mutation was noted on cell
apoptosis (Fig. 4B).
Discussion
BRCA1 is one of the best-known tumor
suppressor genes in breast cancer (8,13).
Numerous mutations of BRCA1 have been reported (14–17), but
the function of many of these mutations has not been studied. The
present study identified two novel missense mutations of the BRCT
domain from a cohort of Chinese Han patients with familial breast
cancer, and explored the function and clinical significance of the
mutations.
The key function of BRCA1 is to suppress
tumor formation in breast and ovarian tissues. Using a mouse model
of hereditary breast cancer, Shakya et al (7) found that the tumor suppression function
of BRCA1 is dependent on the ability of the BRCT domain to
bind to its phospho-ligands. The BRCT motif of BRCA1 forms a
phospho-recognition domain that preferentially binds to the
phosphorylated isoforms of repair proteins, Abraxas/CCDC98,
BACH1/FancJ and CtIP, and thus has a critical role in tumor
suppression (18,19). Numerous tumor-associated BRCA1
alleles have frame shift/nonsense mutations that eliminate one or
both BRCT motifs. According to the bioinformatics analyses, it was
speculated that G1763V and L1786P may damage the tumor suppressor
function of BRCA1. Notably, it was found that these two
mutants had a similar tumor suppression function to wild-type
BRCA1, in terms of reducing proliferation and inducing
apoptosis in breast cancer cells.
PARP1 is a nuclear protein that rapidly binds to
single-stranded DNA breaks to facilitate DNA repair (20). Inhibitors of PARP efficiently shrink
breast, ovarian or prostate tumors in patients carrying hereditary
mutations in the homologous recombination genes BRCA1 or
BRCA2 (21–24). Using the BRCA1-deficient HCC1937
breast cancer cell line, the present results showed that G1763V and
L1786P mutants were not sensitive to the inhibition of PARP
inhibitor Olaparib. This data further indicated that these two
mutations had no deleterious function.
These two mutants have no effect on the suppressor
function of BRCA1, the reason for which requires additional
investigation. Using the protein structure software, it was found
that the G1763V and L1786P missense mutations were far away from
the interaction site between BRCA1 and its phosphor-ligands
(data not shown), which may at least partially explain why these
two mutants have no deleterious effect on BRCA1
function.
In summary, the present study identified two novel
BRCA1 missense mutations in the BRCT domain, G1763V and
L1786P, and these two mutations did not affect the tumor suppressor
function of BRCA1. It was concluded that not all
BRCA1 missense mutations are pathogenic and that any new
BRCA1 mutation should be assessed for its effect on the
tumor suppressor function of BRCA1.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81202106). The
authors thank Dr Genze Shao (Department of Cell Biology, Peking
University Health Science Center, Beijing, China) for providing the
plasmid containing full-length BRCA1, and Dr Shuxing Zhang
(Department of Experimental Therapeutics, Division of Cancer
Medicine, MD Anderson Cancer Center, Houston, USA) for assistance
with protein structure analysis.
References
|
1
|
Welcsh PL, Owens KN and King MC: Insights
into the functions of BRCA1 and BRCA2. Trends Genet. 16:69–74.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bergman A, Flodin A, Engwall Y, Arkblad
EL, Berg K and Nordling M, Martinsson T, Wahlström J, Karlsson P
and Nordling M: A high frequency of germline BRCA1/2 mutations in
western Sweden detected with complementary screening techniques.
Fam Cancer. 4:89–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Pei R, Pang Z, Ouyang T, Li J,
Wang T, Fan Z, Fan T, Lin B and Xie Y: Prevalence and
characterization of BRCA1 and BRCA2 germline mutations in Chinese
women with familial breast cancer. Breast Cancer Res Treat.
132:421–428. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mullan PB, Quinn JE and Harkin DP: The
role of BRCA1 in transcriptional regulation and cell cycle control.
Oncogene. 25:5854–5863. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baer R and Ludwig T: The BRCA1/BARD1
heterodimer, a tumor suppressor complex with ubiquitin E3 ligase
activity. Curr Opin Genet Dev. 12:86–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moynahan ME and Jasin M: Mitotic
homologous recombination maintains genomic stability and suppresses
tumorigenesis. Nat Rev Mol Cell Biol. 11:196–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shakya R, Reid LJ, Reczek CR, Cole F, Egli
D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al: BRCA1
tumor suppression depends on BRCT phosphoprotein binding, but not
its E3 ligase activity. Science. 334:525–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Linger RJ and Kruk PA: BRCA1 16 years
later: Risk-associated BRCA1 mutations and their functional
implications. FEBS J. 277:3086–3096. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Konishi H, Mohseni M, Tamaki A, Garay JP,
Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, et
al: Mutation of a single allele of the cancer susceptibility gene
BRCA1 leads to genomic instability in human breast epithelial
cells. Proc Natl Acad Sci USA. 108:17773–17778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Figge MA and Blankenship L: Missense
mutations in the BRCT domain of BRCA-1 from high-risk women
frequently perturb strongly hydrophobic amino acids conserved among
mammals. Cancer Epidemiol Biomarkers Prev. 13:1037–1041.
2004.PubMed/NCBI
|
|
11
|
Easton DF, Deffenbaugh AM, Pruss D, Frye
C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen
ES, Couch FJ and Goldgar DE: A systematic genetic assessment of
1,433 sequence variants of unknown clinical significance in the
BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet.
81:873–883. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foulkes WD and Shuen AY: In brief: BRCA1
and BRCA2. J Pathol. 230:347–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pohlreich P, Zikan M, Stribrna J, Kleibl
Z, Janatova M, Kotlas J, Zidovska J, Novotny J, Petruzelka L, Szabo
C and Matous B: High proportion of recurrent germline mutations in
the BRCA1 gene in breast and ovarian cancer patients from the
Prague area. Breast Cancer Res. 7:R728–R736. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Walsh T, Casadei S, Coats KH, Swisher E,
Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, et
al: Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in
families at high risk of breast cancer. JAMA. 295:1379–1388. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kwong A, Ng EK, Tang EY, Wong CL, Law FB,
Leung CP, Chan A, Cheung MT, To MY, Ma ES, et al: A novel de novo
BRCA1 mutation in a Chinese woman with early onset breast cancer.
Fam Cancer. 10:233–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neamatzadeh H, Shiryazdi SM and Kalantar
SM: BRCA1 and BRCA2 mutations in Iranian breast cancer patients: A
systematic review. J Res Med Sci. 20:284–293. 2015.PubMed/NCBI
|
|
18
|
Moynahan ME and Jasin M: Mitotic
homologous recombination maintains genomic stability and suppresses
tumorigenesis. Nat Rev Mol Cell Biol. 11:196–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huen MS, Sy SM and Chen J: BRCA1 and its
toolbox for the maintenance of genome integrity. Nat Rev Mol Cell
Biol. 11:138–148. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Satoh MS and Lindahl T: Role of
poly(ADP-ribose) formation in DNA repair. Nature. 356:356–358.
1992. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guha M: PARP inhibitors stumble in breast
cancer. Nat Biotechnol. 29:373–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JM, Ledermann JA and Kohn EC: PARP
Inhibitors for BRCA1/2 mutation-associated and BRCA-like
malignancies. Ann Oncol. 25:32–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bixel K and Hays JL: Olaparib in the
management of ovarian cancer. Pharmgenomics Pers Med. 8:127–135.
2015.PubMed/NCBI
|