Axl receptor tyrosine kinase (hereafter Axl) is a
member of the tyrosine-protein kinase receptor Tyro3 (hereafter
Tyro3), Axl and proto-oncogene tyrosine-protein kinase Mer
(hereafter Mer) (TAM) family of receptor tyrosine kinases (RTKs)
(1). The TAM family is distinguished
from other RTKs by a conserved sequence, KW(I/L)A(I/L)ES, within
the kinase domain and two immunoglobulin (Ig)-like domains plus two
fibronectin type III domains, which comprise nearly the entire
ectodomain of each family member (1,2). In adult
tissues, Tyro3, Axl and Mer are widely distributed (1), and have notable functions in tissue
repair, clearance of apoptotic material and immune regulation
(1,3–5). TAM
receptors were initially considered to be orphan receptors
(6); however, it has since been
revealed that there are diverse ligands for this family of
receptors (6–10). Growth arrest-specific 6 (Gas6) and
protein S were identified to be ligands for TAMs in the 1990s
(6,7).
These proteins are members of the vitamin K-dependent protein
family, and demonstrated significant homology with each other
(8). Gas6 binds to all three TAM RTKs
(Axl>Tyro3>Mer), whereas protein S interacts with Mer and
Tyro3, but not Axl. Previously, tubby, tubby-like protein 1
(Tulp-1) and galectin-3 have been revealed to be ligands for TAM
receptors, and Tulp-1 binds to all three RTKs with differing levels
of affinity, whereas tubby only binds to Mer (9,10).
The name Axl is derived from the Greek word
anexelekto, which means ‘uncontrolled’. The human Axl gene is
located on chromosome 19q13.2 and has 20 exons (11). It was originally identified as a
transforming gene in the cells of patients with chronic myelogenous
leukemia (12) and had transforming
potential when overexpressed in NIH/3T3 fibroblasts (13,14). As
with other RTKs, Axl is composed of an extracellular domain, a
transmembrane domain and an intracellular domain. A soluble Axl has
also been reported (15), which may
possess a diagnostic value for early-stage hepatocellular carcinoma
(16). Axl is ubiquitously expressed
in human tissues (1,17), with notable levels identified in the
kidney (1,18), brain (19), heart (1), testis (1),
skeletal muscle (1), liver (1,20),
endothelial cells (21,22), monocytes/macrophages (23) and platelets (24). This wide expression pattern of Axl
indicates that this protein exerts a notable function in normal
cell function, including cell survival, proliferation, migration
and adhesion (17). However, usually,
more than one TAM receptor is expressed in a given cell type
simultaneously that may be activated by one common ligand; for
example, all three TAM members may be activated by Gas6 (25), thus making it difficult to elucidate
the function of Axl on its own in one cell type.
The activation of RTKs involves ligand binding to
the extracellular domain, which induces receptor dimerization and
subsequent trans-autophosphorylation of the tyrosine
residues within the cytoplasmic domain (2). Axl is activated by the binding of its
ligand Gas6, which was identified as the ligand for Axl in 1995 by
two separate studies (6,7). Prior to that, the Gas6 gene was first
identified as one of several genes to be upregulated in NIH/3T3
fibroblasts under serum starvation-induced growth arrest (26). Gas6 was later revealed to be a common
ligand for Axl, Tyro3 and Mer, with Axl possessing the highest
affinity for Gas6 (3). Gas6 is widely
expressed and has been identified in the lung, heart, kidney,
intestine, endothelial cells, bone marrow, vascular smooth muscle
cells and monocytes and at low levels in the liver and human blood
plasma (27). It has
cell-type-specific functions, including platelet aggregation and
hematopoiesis, proliferation, survival and phagocytosis.
Gas6 possesses an N-terminal region containing a
modified γ-carboxyglutamic acid (Gla) residue, which has the
ability to interact with negatively charged membrane phospholipids
to mediate the binding of Gas6 to apoptotic cells. The Gla domain
is followed by a loop region, four epidermal growth factor
(EGF)-like repeats and a C-terminal sex-hormone-binding globulin
(SHBG)-like structure that is composed of two globular laminin
G-like domains (28). The SHBG domain
binds directly to the Ig domains of Axl, which results in the
formation of a Gas6/Axl complex with a 1:1 ratio (29). The lateral diffusion of these
complexes would then result in the formation of a minimal 2:2
Gas6/Axl signaling complex, which induces activation of Axl
(29). Additionally, the
γ-carboxylation of Gas6 and anionic phospholipids, including the
externalized phosphatidylserine on apoptotic cells and enveloped
viruses, possesses vital functions for the activation of Axl
(3,30,31).
In addition to conventional activation, atypical
activation of Axl has been reported, including activation by
crosstalk between receptors (1).
Meyer et al (32) revealed
that EGF receptor (EGFR) activation associated with Axl and EGF
stimulation may activate Axl through EGFR in triple-negative breast
cancer cells. In non-small cell lung cancer (NSCLC), head and neck
squamous cell carcinoma (HNSCC) (33,34) and
esophageal squamous cell carcinomas (ESCC) cells (34), this physical association between Axl
and EGFR was also observed. This interaction has the potential to
activate EGFR (33,34) and Axl (32,33). The
ability of Axl to form complexes with other RTKs may make certain
cancer types resistant to tyrosine kinase inhibitors (TKIs), as
will be discussed further in the present review.
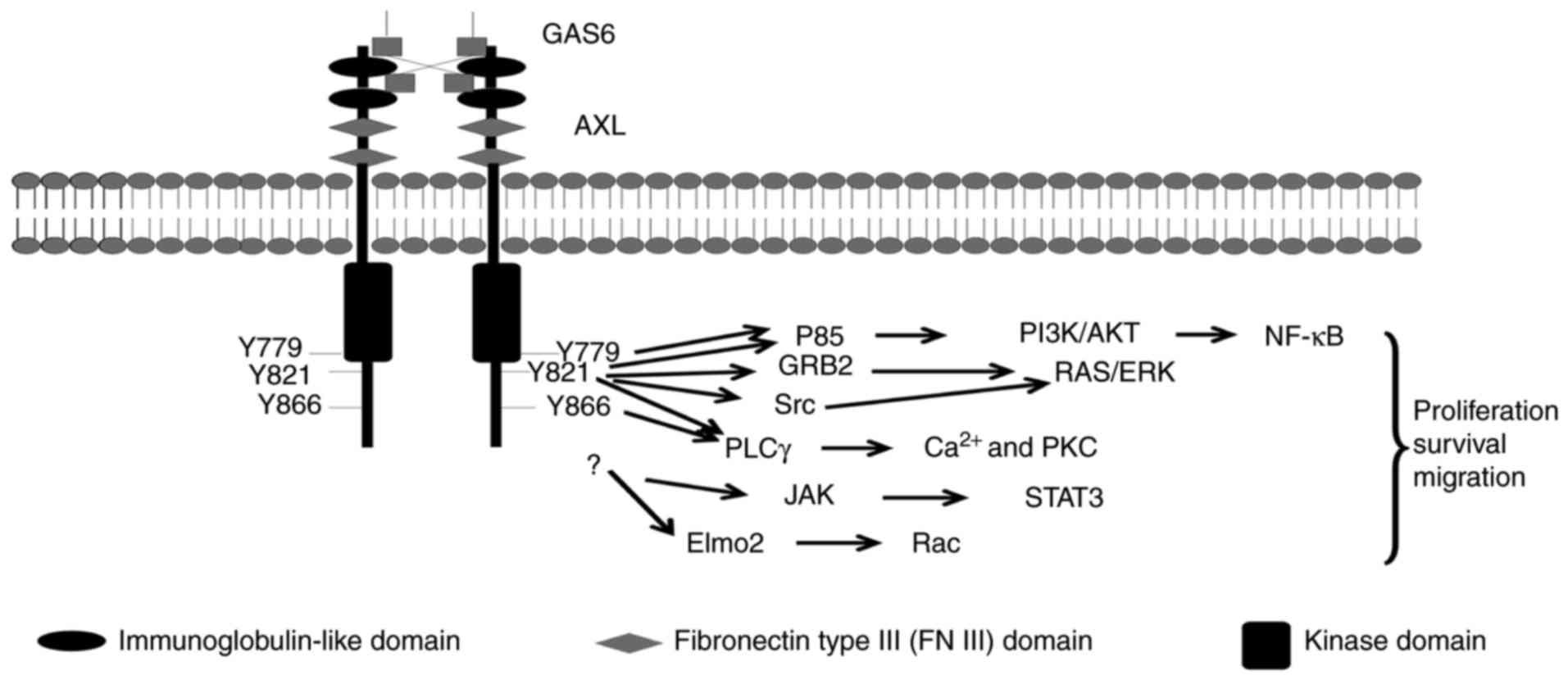
Three tyrosine residues, Y-779, Y-821 and Y-866,
within the C-terminal domain of Axl have been proposed as potential
autophosphorylation sites and putative docking sites for a variety
of signaling proteins (1), including
the p85a and p85b subunits of phosphatidylinositol 3-kinase (PI3K)
(35,36), growth factor receptor-bound protein 2
(25,26), phospholipase Cγ (PLCγ) (36), c-src and lck (36) (Fig. 1).
Additionally, the engulfment and cell motility (Elmo) scaffold
protein has been reported to directly interact with Axl, and serves
vital functions in Axl-induced breast cancer invasion (37). Notably, mutation of the three tyrosine
residues did not abrogate the Axl-Elmo2 association, indicating
that other docking sites may exist (37).
Activation of Axl regulates a number of signal
transduction pathways, depending on the cell types in question,
primarily PI3K/protein kinase B (Akt) and mitogen-activated protein
kinase (MEK)/extracellular-signal-regulated kinase (ERK), nuclear
factor-κB (NF-κB), signal transducer and activator of transcription
3 (STAT3) (17,38,39)
[references (1,3,17,40) contain further information on Axl
signaling]. The activation of these pathways elicits different
responses in different cells, including cell proliferation,
migration or survival (3,17) (Fig.
1).
The expression of Axl and Gas6 in normal lung cells
is not well documented. Studies aimed at detecting Axl expression
in lung cancer cells demonstrated that Axl is not expressed in
normal lung alveolar cells or the bronchial epithelium adjacent to
a tumor (41,42). Axl is expressed in lung airway
macrophages, but not interstitial macrophages and other lung
leukocytes under homeostatic conditions and is constitutively
ligated to Gas6 (43). Axl was
believed to serve notable functions in mediating immune homeostasis
through the clearance of apoptotic cells during inflammation
(43) and promoting an antiviral
response through maintaining the appropriate production of type I
interferon (44). Axl and Gas6 are
also expressed in the blood endothelial cells of lung (45), in which they maintaining the integrity
of the vasculature and vascular remodeling under pathological
conditions (27).
Axl is expressed in NSCLC, with expression rates
varying from ~33.0 to ~93.2% detected by different groups (Table I). This inconsistency may be caused by
different Axl antibodies used, and different evaluation methods.
The varying clinicopathological characteristics of patients with
NSCLC studied may also contribute to this inconsistence. These
studies suggest a number of patients with NSCLC express Axl, and it
may be associated with a poorer prognosis. Consistently, patients
with NSCLC that exhibit high Axl mRNA expression had a shorter
disease-free survival time than patients exhibiting low Axl mRNA
expression (46).
Similar to other RTKs, Axl overexpression may
provide survival and growth advantages to tumor cells (3). In NSCLC cell lines, the small
interfering RNA (siRNA)-mediated downregulation of Axl results in
decreased cell growth in vitro and in xenograft mouse models
(41,47), in addition to the suppression of Akt
and ERK activation (34). Similarly,
the proliferation of H226 cells, which express moderate levels of
Axl, may be suppressed by the anti-Axl monoclonal antibody MAb173
or the specific Axl inhibitor R428 (33). These studies indicate that Axl is
involved in maintaining the proliferation of NSCLC cells.
EMT is a process by which epithelial cells lose
their cell polarity and cell-cell adhesion, and gain mesenchymal
cell-like migratory and invasive properties (48). Axl is recognized as having vital
functions in NSCLC EMT. Firstly, Axl is a marker for the
mesenchymal phenotype in NSCLC. From the RNA-sequencing data of 643
cancer cell lines, including NSCLC, Axl expression was markedly
associated with a mesenchymal phenotype (49). It was further revealed that in 45
NSCLC cell lines, higher Axl protein expression tends to be
associated with a higher protein expression of vimentin, a
mesenchymal marker (49). Similarly,
Axl expression was higher in NSCLC mesenchymal cancer cells than in
epithelial cancer cells, based on the mRNA expression profile of 54
NSCLC cell lines and the protein expression data of 49 patients
with NSCLC (50). Furthermore, in the
transforming growth factor β-induced EMT model, Axl is upregulated,
similar to vimentin (49). In
addition, Axl aids the maintenance of the EMT state in NSCLC cells.
A549 and H460 are mesenchymal NSCLC cells (50); in these cell lines, Axl downregulation
results in the increased expression of E-cadherin and decreased
expression of vimentin and N-cadherin, which are features of the
reverse of EMT (47).
Patients with cancer that have solid tumors
primarily succumb to mortality due to metastatic lesions rather
than from the primary tumors (51).
Axl has been implicated in metastasis in multiple tumor types
(3). In patients with NSCLC, Axl
expression is associated with lymph node metastasis (52–54).
Cisplatin- and gefitinib-resistant HCC4006 cells express high
levels of Axl, and siRNA-mediated Axl downregulation suppressed the
migratory capacity of these cells (55). Lay et al (56) established a series of cell lines with
different invasive abilities by the selection of increasingly
invasive cancer cell populations from a cell line of human lung
adenocarcinoma (CL1-0) using a Transwell invasion chamber assay. It
was revealed that Axl expression was highly associated with the
migratory ability of these cell lines. NF-κB signaling was
responsible for Axl-enhanced migration, with its suppression
blunting Axl-induced migration. Huang et al (57) additionally reported that Axl mediates
H2O2-induced migration by activating
PI3K/Akt/Ras-related C3 botulinum toxin substrate 1 signaling.
Furthermore, the first Ig-like domain and the intracellular domain
were vital for the function of Axl in these two models. These
studies indicated that Axl may not only activate
migration-promoting signals itself, but additionally mediate the
effect of other molecules to increase migration. Mechanistically,
Axl increases the expression of matrix metalloproteinase-9 (MMP9)
and MMP2, which promote tissue remodeling and cancer invasion
(58–61). Additionally, as discussed above, Axl
promotes the EMT of cancer cells (62,63), a
process associated with migratory and invasive properties (48). Axl has also been associated with
invasion through the modification of the cytoskeleton regulator Rac
(37,57,64),
leading to cytoskeletal reorganization and increased migration and
invasion.
Drug resistance is the main reason for the failure
of cancer treatments. Axl serves a notable function in the drug
resistance of a number of different cancer types (34,65–67); its
function in NSCLC drug resistance has been studied intensively
(68). Evidence is primarily derived
from in vitro cell line studies (Table II), with limited data from mouse
models (33,69) and the tumor tissues of patients with
cancer (69,70) (Table
III). Thus, these results may require further confirmation,
particularly in human tumors in vivo.
The majority of cell line-based studies use
drug-resistant cells, obtained by the long-term treatment of cancer
cells with increasing doses of drugs, and such studies have
revealed that Axl is upregulated in drug-resistant cancer cells
(Table II). One study additionally
revealed that suppression of Axl with genomic or pharmaceutical
methods may restore the sensitivity of cancer cells to drugs,
further informing on the function of Axl in these drug-resistant
models (69). Other studies
downregulated Axl expression in Axl-overexpressed NSCLC cancer
cells, including A549 and H462, to survey the change of the half
maximal inhibitory concentration of cancer cells to drugs, and it
was observed that the silencing of Axl enhanced the sensitivity of
cancer cells to drugs, including several chemotherapeutic drugs and
erlotinib (41,47). Zhang et al (69) used a combination of strategies,
including mouse models and matched NSCLC tumor tissues (tissues
from the same patient prior to treatment and following the
development of drug resistance), providing convincing evidence
supporting the involvement of Axl in the mechanisms underlying
NSCLC EGFR TKI resistance, and that targeting this molecule may
restore the sensitivity of resistant cells. Similarly, Brand et
al (33) used several differing
strategies, including the ectopic overexpression of Axl,
siRNA-mediated downregulation of Axl and mouse models, to
demonstrate that Axl serves a notable function in NSCLC resistance
to cetuximab, a chimeric monoclonal antibody targeting EGFR. In
addition to the work of Zhang et al (69), Ji et al (70) provides further evidence using the
tumor tissue of patients with NSCLC demonstrating that Axl
upregulation is an independent mechanism of NSCLC resistance to
EGFR-TKI (Table III).
A detailed mechanism for Axl-mediated drug
resistance is yet to be elucidated. However, a number of studies
have provided notable information. As previously discussed, the
overexpression of Axl is linked to EMT status, which was associated
with drug resistance (71,72). Additionally, Axl impedes
therapeutically induced apoptosis by exerting anti-apoptotic
effects by modulating the expression or activation of apoptosis
regulators, including B-cell lymphoma extra large, survivin
(41), B-cell lymphoma-2 (Bcl-2)
associated agonist of cell death (73), Bh3 interacting-domain death agonist
(74) and Bcl-2 (75). Additionally, a number of previous
studies have suggested that the ability of Axl to form complexes
with other RTKs may be one key aspect of this function.
Cetuximab-resistant NSCLC cell line H226 cells exhibited
upregulation of Axl signaling, which was further elucidated to form
a physical complex with EGFR (33).
Furthermore, treatment with EGF or tumor necrosis factor resulted
in H226 cells resistant to cetuximab, with the formation of a
physical complex between Axl and EGFR, similar to that in resistant
cells. This indicates that the Axl-EGFR complex serves a role in
eliciting a drug-resistant phenotype of cancer cells. These studies
further demonstrated that Axl and EGFR were involved in maintaining
the growth of these resistant cells. Additionally, silencing the
expression of either Axl or EGFR with siRNA decreased the growth of
these cells. The Axl-EGFR complex was also observed in other drug
resistant cancer models. For example, as mentioned above, HNSCC and
ESCC cells induced to be resistant to PI3K inhibitor BYL719
additionally displayed this Axl-EGFR complex (34). In sensitive parental cells, EGFR
activated the PI3K/Akt pathway, which maintained mechanistic target
of rapamycin (mTOR) activity (76).
In resistant cells, the Axl-EGFR complex activated PLCγ-protein
kinase C (PKC) signaling, which in turn activated mTOR (76). Thus, tumor cells became less dependent
on PI3K/Akt signaling and resistant to PI3K inhibitor BYL719.
Combining BYL719 and R428 may reverse the resistant phenotype. This
complex additionally formed in breast cancer cells, and Axl in this
complex amplified and diversified EGFR signaling (32). Thus, it may be that this complex
increased the resistance of cancer cells to EGFR inhibitors and
that the suppression of Axl may enhance the efficacy of EGFR
inhibitors. Further studies revealed that the suppression of Axl
activation with R428 significantly increased the killing ability of
erlotinib (32). Furthermore, Wu
et al (54) revealed that Axl
and EGFR are co-expressed in a subset of NSCLC tumor tissues. This
may allow this complex to form readily, promoting drug resistance.
Other receptors, including human epidermal growth factor receptor 2
(HER2), HER3, MET proto-oncogene and platelet-derived growth factor
receptor-β have been reported to be associated with Axl (32). This association of Axl with other
receptors may have implications for targeted therapies, as these
complexes may change the response of RTKs to TKIs, or make cancer
cells less dependent on the signaling pathways that were targeted
(34), which may result in cancer
cells resistant to TKIs.
There are additional reports indicating a minimal
function of Axl in the resistance of NSCLCs to TKIs. For example,
the suppression of Axl with siRNA or chemical inhibitors in
erlotinib-resistant H358 (77) and
HCC827 (49) cells, which
demonstrated an increased expression of Axl, could not restore the
sensitivity of the cells to drug treatment.
Axl is a promising therapeutic target, considering
that it serves notable functions in NSCLC tumor growth, EMT,
invasion and drug resistance. A number of Axl inhibitors have been
developed and a number are in clinical trials, including foretinib
(XL880, GSK1363089), cabozantinib, crizotinib, ASLAN002 and BGB324
(R428) (3,78–81). It is
important to note that although a wide range of Axl kinase
inhibitors have been described, a majority of them are nonspecific
multi-kinase inhibitors. BGB324 (R428) was the first selective Axl
inhibitor to be developed (82). Oral
treatment with BGB324 (R428) in mouse xenograft models revealed
that it reduced breast cancer metastasis and prolonged survival. It
entered phase 1 clinical trials in 2013 (83). At present, clinical trials including
patients with melanoma, NSCLC and acute myeloid leukemia are
ongoing to determine the safety and efficiency of BGB324
(https://clinicaltrials.gov/ct2/results?cond=&term=BGB324&cntry1=&state1=&Search=Search).
The identification of appropriate biomarkers for the selection of
patients is another key issue for the development of Axl-targeted
therapies. Immunohistochemistry appears to be the most feasible
strategy for identifying appropriate biomarkers, with other
strategies including the detection of Axl expression in vivo
by single-photon emission computed tomography imaging using a
125I-labled Axl antibody (84). With the development of AXL inhibitors,
and an increase in the understanding of the underlying molecular
mechanisms responsible for NSCLC, patients who are suitable for
AXL-targeted therapies could be screened and treated with this form
of therapy.
The present review was supported by the Project of
Shandong Province Higher Educational Science and Technology Program
(grant no. J13LK14).
|
1
|
Linger RM, Keating AK, Earp HS and Graham
DK: TAM receptor tyrosine kinases: Biologic functions, signaling,
and potential therapeutic targeting in human cancer. Adv Cancer
Res. 100:35–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Graham DK, DeRyckere D, Davies KD and Earp
HS: The TAM family: Phosphatidylserine sensing receptor tyrosine
kinases gone awry in cancer. Nat Rev Cancer. 14:769–785. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rothlin CV, Carrera-Silva EA, Bosurgi L
and Ghosh S: TAM receptor signaling in immune homeostasis. Annu Rev
Immunol. 33:355–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lemke G and Rothlin CV: Immunobiology of
the TAM receptors. Nat Rev Immunol. 8:327–336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stitt TN, Conn G, Gore M, Lai C, Bruno J,
Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al: The
anticoagulation factor protein S and its relative, Gas6, are
ligands for the Tyro 3/Axl family of receptor tyrosine kinases.
Cell. 80:661–670. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Varnum BC, Young C, Elliott G, Garcia A,
Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, et
al: Axl receptor tyrosine kinase stimulated by the vitamin
K-dependent protein encoded by growth-arrest-specific gene 6.
Nature. 373:623–626. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Manfioletti G, Brancolini C, Avanzi G and
Schneider C: The protein encoded by a growth arrest-specific gene
(gas6) is a new member of the vitamin K-dependent proteins related
to protein S, a negative coregulator in the blood coagulation
cascade. Mol Cell Biol. 13:4976–4985. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caberoy NB, Alvarado G, Bigcas JL and Li
W: Galectin-3 is a new MerTK-specific eat-me signal. J Cell
Physiol. 227:401–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caberoy NB, Zhou Y and Li W: Tubby and
tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO
J. 29:3898–3910. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verma A, Warner SL, Vankayalapati H,
Bearss DJ and Sharma S: Targeting Axl and Mer kinases in cancer.
Mol Cancer Ther. 10:1763–1773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu E, Hjelle B and Bishop JM:
Transforming genes in chronic myelogenous leukemia. Proc Natl Acad
Sci USA. 85:pp. 1952–1956. 1988; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Bryan JP, Frye RA, Cogswell PC, Neubauer
A, Kitch B, Prokop C, Espinosa R III, Le Beau MM, Earp HS and Liu
ET: axl, a transforming gene isolated from primary human myeloid
leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell
Biol. 11:5016–5031. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Janssen JW, Schulz AS, Steenvoorden AC,
Schmidberger M, Strehl S, Ambros PF and Bartram CR: A novel
putative tyrosine kinase receptor with oncogenic potential.
Oncogene. 6:2113–2120. 1991.PubMed/NCBI
|
|
15
|
O'Bryan JP, Fridell YW, Koski R, Varnum B
and Liu ET: The transforming receptor tyrosine kinase, Axl, is
post-translationally regulated by proteolytic cleavage. J Biol
Chem. 270:551–557. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reichl P, Fang M, Starlinger P, Staufer K,
Nenutil R, Muller P, Greplova K, Valik D, Dooley S, Brostjan C, et
al: Multicenter analysis of soluble Axl reveals diagnostic value
for very early stage hepatocellular carcinoma. Int J Cancer.
137:385–394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hafizi S and Dahlbäck B: Signalling and
functional diversity within the Axl subfamily of receptor tyrosine
kinases. Cytokine Growth Factor Rev. 17:295–304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung BI, Malkowicz SB, Nguyen TB,
Libertino JA and McGarvey TW: Expression of the proto-oncogene Axl
in renal cell carcinoma. DNA Cell Biol. 22:533–540. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prieto AL, Weber JL and Lai C: Expression
of the receptor protein-tyrosine kinases Tyro-3, Axl, and mer in
the developing rat central nervous system. J Comp Neurol.
425:295–314. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lafdil F, Chobert MN, Couchie D, Brouillet
A, Zafrani ES, Mavier P and Laperche Y: Induction of Gas6 protein
in CCl4-induced rat liver injury and anti-apoptotic effect on
hepatic stellate cells. Hepatology. 44:228–239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Melaragno MG, Fridell YW and Berk BC: The
Gas6/Axl system: A novel regulator of vascular cell function.
Trends Cardiovasc Med. 9:250–253. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruan GX and Kazlauskas A: Axl is essential
for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 31:1692–1703.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neubauer A, Fiebeler A, Graham DK, O'Bryan
JP, Schmidt CA, Barckow P, Serke S, Siegert W, Snodgrass HR, Huhn
D, et al: Expression of axl, a transforming receptor tyrosine
kinase, in normal and malignant hematopoiesis. Blood. 84:1931–1941.
1994.PubMed/NCBI
|
|
24
|
Angelillo-Scherrer A, de Frutos P,
Aparicio C, Melis E, Savi P, Lupu F, Arnout J, Dewerchin M,
Hoylaerts M, Herbert J, et al: Deficiency or inhibition of Gas6
causes platelet dysfunction and protects mice against thrombosis.
Nat Med. 7:215–221. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagata K, Ohashi K, Nakano T, Arita H,
Zong C, Hanafusa H and Mizuno K: Identification of the product of
growth arrest-specific gene 6 as a common ligand for Axl, Sky, and
Mer receptor tyrosine kinases. J Biol Chem. 271:30022–30027. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schneider C, King RM and Philipson L:
Genes specifically expressed at growth arrest of mammalian cells.
Cell. 54:787–793. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laurance S, Lemarié CA and Blostein MD:
Growth Arrest-specific gene 6 (gas6) and vascular hemostasis. Adv
Nutr. 3:196–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hafizi S and Dahlbäck B: Gas6 and protein
S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase
subfamily. FEBS J. 273:5231–5244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sasaki T, Knyazev PG, Clout NJ, Cheburkin
Y, Göhring W, Ullrich A, Timpl R and Hohenester E: Structural basis
for Gas6-Axl signalling. EMBO J. 25:80–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsou WI, Nguyen KQ, Calarese DA, Garforth
SJ, Antes AL, Smirnov SV, Almo SC, Birge RB and Kotenko SV:
Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate
distinct patterns and complex regulation of ligand-induced
activation. J Biol Chem. 289:25750–25763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lew ED, Oh J, Burrola PG, Lax I, Zagórska
A, Través PG, Schlessinger J and Lemke G: Differential TAM
receptor-ligand-phospholipid interactions delimit differential TAM
bioactivities. Elife. 3:2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meyer AS, Miller MA, Gertler FB and
Lauffenburger DA: The receptor AXL diversifies EGFR signaling and
limits the response to EGFR-targeted inhibitors in triple-negative
breast cancer cells. Sci Signal. 6:ra662013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brand TM, Iida M, Stein AP, Corrigan KL,
Braverman CM, Luthar N, Toulany M, Gill PS, Salgia R, Kimple RJ and
Wheeler DL: AXL mediates resistance to cetuximab therapy. Cancer
Res. 74:5152–5164. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Elkabets M, Pazarentzos E, Juric D, Sheng
Q, Pelossof RA, Brook S, Benzaken AO, Rodon J, Morse N, Yan JJ, et
al: AXL mediates resistance to PI3Kα inhibition by activating the
EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell
carcinomas. Cancer Cell. 27:533–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fridell YW, Jin Y, Quilliam LA, Burchert
A, McCloskey P, Spizz G, Varnum B, Der C and Liu ET: Differential
activation of the Ras/extracellular-signal-regulated protein kinase
pathway is responsible for the biological consequences induced by
the Axl receptor tyrosine kinase. Mol Cell Biol. 16:135–145. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Braunger J, Schleithoff L, Schulz AS,
Kessler H, Lammers R, Ullrich A, Bartram CR and Janssen JW:
Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is
mediated mainly by a multi-substrate docking-site. Oncogene.
14:2619–2631. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Abu-Thuraia A, Gauthier R, Chidiac R,
Fukui Y, Screaton RA, Gratton JP and Côté JF: Axl phosphorylates
elmo scaffold proteins to promote Rac activation and cell invasion.
Mol Cell Biol. 35:76–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Linger RM, Keating AK, Earp HS and Graham
DK: Taking aim at Mer and Axl receptor tyrosine kinases as novel
therapeutic targets in solid tumors. Expert Opin Ther Targets.
14:1073–1090. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yanagita M, Arai H, Nakano T, Ohashi K,
Mizuno K, Fukatsu A, Doi T and Kita T: Gas6 induces mesangial cell
proliferation via latent transcription factor STAT3. J Biol Chem.
276:42364–42369. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brandão L, Migdall-Wilson J, Eisenman K
and Graham DK: TAM receptors in leukemia: Expression, signaling,
and therapeutic implications. Crit Rev Oncog. 16:47–63. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Linger RM, Cohen RA, Cummings CT, Sather
S, Migdall-Wilson J, Middleton DH, Lu X, Barón AE, Franklin WA,
Merrick DT, et al: Mer or Axl receptor tyrosine kinase inhibition
promotes apoptosis, blocks growth and enhances chemosensitivity of
human non-small cell lung cancer. Oncogene. 32:3420–3431. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iida S, Miki Y, Suzuki T, Mori K, Saito M,
Niikawa H, Kondo T, Yamada-Okabe H and Sasano H: Activation of AXL
and antitumor effects of a monoclonal antibody to AXL in lung
adenocarcinoma. Anticancer Res. 34:1821–1827. 2014.PubMed/NCBI
|
|
43
|
Fujimori T, Grabiec AM, Kaur M, Bell TJ,
Fujino N, Cook PC, Svedberg FR, MacDonald AS, Maciewicz RA, Singh D
and Hussell T: The Axl receptor tyrosine kinase is a discriminator
of macrophage function in the inflamed lung. Mucosal Immunol.
8:1021–1030. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schmid ET, Pang IK, Carrera Silva EA,
Bosurgi L, Miner JJ, Diamond MS, Iwasaki A and Rothlin CV: AXL
receptor tyrosine kinase is required for T cell priming and
antiviral immunity. Elife. 5:pii: e124142016. View Article : Google Scholar
|
|
45
|
Healy AM, Schwartz JJ, Zhu X, Herrick BE,
Varnum B and Farber HW: Gas 6 promotes Axl-mediated survival in
pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol.
280:L1273–L1281. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang Y, Xia H, Zhuang Z, Miao L, Chen X
and Cai H: Axl-altered microRNAs regulate tumorigenicity and
gefitinib resistance in lung cancer. Cell Death Dis. 5:e12272014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu F, Li J, Jang C, Wang J and Xiong J:
The role of Axl in drug resistance and epithelial-to-mesenchymal
transition of non-small cell lung carcinoma. Int J Clin Exp Pathol.
7:6653–6661. 2014.PubMed/NCBI
|
|
48
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wilson C, Ye X, Pham T, Lin E, Chan S,
McNamara E, Neve RM, Belmont L, Koeppen H, Yauch RL, et al: AXL
inhibition sensitizes mesenchymal cancer cells to antimitotic
drugs. Cancer Res. 74:5878–5890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Byers LA, Diao L, Wang J, Saintigny P,
Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al: An
epithelial-mesenchymal transition gene signature predicts
resistance to EGFR and PI3K inhibitors and identifies Axl as a
therapeutic target for overcoming EGFR inhibitor resistance. Clin
Cancer Res. 19:279–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ishikawa M, Sonobe M, Nakayama E,
Kobayashi M, Kikuchi R, Kitamura J, Imamura N and Date H: Higher
expression of receptor tyrosine kinase Axl, and differential
expression of its ligand, Gas6, predict poor survival in lung
adenocarcinoma patients. Ann Surg Oncol. 20 Suppl 3:S467–S476.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shieh YS, Lai CY, Kao YR, Shiah SG, Chu
YW, Lee HS and Wu CW: Expression of axl in lung adenocarcinoma and
correlation with tumor progression. Neoplasia. 7:1058–1064. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu Z, Bai F, Fan L, Pang W, Han R, Wang J,
Liu Y, Yan X, Duan H and Xing L: Coexpression of receptor tyrosine
kinase AXL and EGFR in human primary lung adenocarcinomas. Hum
Pathol. 46:1935–1944. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kurokawa M, Ise N, Omi K, Goishi K and
Higashiyama S: Cisplatin influences acquisition of resistance to
molecular-targeted agents through epithelial-mesenchymal
transition-like changes. Cancer Sci. 104:904–911. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lay JD, Hong CC, Huang JS, Yang YY, Pao
CY, Liu CH, Lai YP, Lai GM, Cheng AL, Su IJ and Chuang SE:
Sulfasalazine suppresses drug resistance and invasiveness of lung
adenocarcinoma cells expressing AXL. Cancer Res. 67:3878–3887.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huang JS, Cho CY, Hong CC, Yan MD, Hsieh
MC, Lay JD, Lai GM, Cheng AL and Chuang SE: Oxidative stress
enhances AXL-mediated cell migration through AKT1/Rac1-dependent
mechanism. Free Radic Biol Med. 65:1246–1256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tai KY, Shieh YS, Lee CS, Shiah SG and Wu
CW: Axl promotes cell invasion by inducing MMP-9 activity through
activation of NF-kappaB and Brg-1. Oncogene. 27:4044–4055. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Reichl P, Dengler M, van Zijl F, Huber H,
Führlinger G, Reichel C, Sieghart W, Peck-Radosavljevic M,
Grubinger M and Mikulits W: Axl activates autocrine transforming
growth factor-β signaling in hepatocellular carcinoma. Hepatology.
61:930–941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chiu KC, Lee CH, Liu SY, Yeh CT, Huang RY,
Yuh DY, Cheng JC, Chou YT and Shieh YS: Protumoral effect of
macrophage through Axl activation on mucoepidermoid carcinoma. J
Oral Pathol Med. 43:538–544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Han J, Tian R, Yong B, Luo C, Tan P, Shen
J and Peng T: Gas6/Axl mediates tumor cell apoptosis, migration and
invasion and predicts the clinical outcome of osteosarcoma
patients. Biochem Biophys Res Commun. 435:493–500. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Asiedu MK, Beauchamp-Perez FD, Ingle JN,
Behrens MD, Radisky DC and Knutson KL: AXL induces
epithelial-to-mesenchymal transition and regulates the function of
breast cancer stem cells. Oncogene. 33:1316–1324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cheng P, Phillips E, Kim SH, Taylor D,
Hielscher T, Puccio L, Hjelmeland AB, Lichter P, Nakano I and
Goidts V: Kinome-wide shRNA screen identifies the receptor tyrosine
kinase AXL as a key regulator for mesenchymal glioblastoma
stem-like cells. Stem Cell Reports. 4:899–913. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Allen MP, Linseman DA, Udo H, Xu M,
Schaack JB, Varnum B, Kandel ER, Heidenreich KA and Wierman ME:
Novel mechanism for gonadotropin-releasing hormone neuronal
migration involving Gas6/Ark signaling to p38 mitogen-activated
protein kinase. Mol Cell Biol. 22:599–613. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Mahadevan D, Cooke L, Riley C, Swart R,
Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H,
et al: A novel tyrosine kinase switch is a mechanism of imatinib
resistance in gastrointestinal stromal tumors. Oncogene.
26:3909–3919. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Giles KM, Kalinowski FC, Candy PA, Epis
MR, Zhang PM, Redfern AD, Stuart LM, Goodall GJ and Leedman PJ: Axl
mediates acquired resistance of head and neck cancer cells to the
epidermal growth factor receptor inhibitor erlotinib. Mol Cancer
Ther. 12:2541–2558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu L, Greger J, Shi H, Liu Y, Greshock J,
Annan R, Halsey W, Sathe GM, Martin AM and Gilmer TM: Novel
mechanism of lapatinib resistance in HER2-positive breast tumor
cells: Activation of AXL. Cancer Res. 69:6871–6878. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Paccez JD, Vogelsang M, Parker MI and
Zerbini LF: The receptor tyrosine kinase Axl in cancer: Biological
functions and therapeutic implications. Int J Cancer.
134:1024–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ji W, Choi CM, Rho JK, Jang SJ, Park YS,
Chun SM, Kim WS, Lee JS, Kim SW, Lee DH and Lee JC: Mechanisms of
acquired resistance to EGFR-tyrosine kinase inhibitor in Korean
patients with lung cancer. BMC Cancer. 13:6062013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lee WP, Wen Y, Varnum B and Hung MC: Akt
is required for Axl-Gas6 signaling to protect cells from
E1A-mediated apoptosis. Oncogene. 21:329–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Papadakis ES, Cichoń MA, Vyas JJ, Patel N,
Ghali L, Cerio R, Storey A and O'Toole EA: Axl promotes cutaneous
squamous cell carcinoma survival through negative regulation of
pro-apoptotic Bcl-2 family members. J Invest Dermatol. 131:509–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hong CC, Lay JD, Huang JS, Cheng AL, Tang
JL, Lin MT, Lai GM and Chuang SE: Receptor tyrosine kinase AXL is
induced by chemotherapy drugs and overexpression of AXL confers
drug resistance in acute myeloid leukemia. Cancer Lett.
268:314–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Meric-Bernstam F and Gonzalez-Angulo AM:
Targeting the mTOR signaling network for cancer therapy. J Clin
Oncol. 27:2278–2287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Suda K, Mizuuchi H, Sato K, Takemoto T,
Iwasaki T and Mitsudomi T: The insulin-like growth factor 1
receptor causes acquired resistance to erlotinib in lung cancer
cells with the wild-type epidermal growth factor receptor. Int J
Cancer. 135:1002–1006. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Myers SH, Brunton VG and Unciti-Broceta A:
AXL inhibitors in cancer: A medicinal chemistry perspective. J Med
Chem. 59:3593–3608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Feneyrolles C, Spenlinhauer A, Guiet L,
Fauvel B, Daydé-Cazals B, Warnault P, Chevé G and Yasri A: Axl
kinase as a key target for oncology: Focus on small molecule
inhibitors. Mol Cancer Ther. 13:2141–2148. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Levin PA, Brekken RA, Byers LA, Heymach JV
and Gerber DE: Axl receptor axis: A new therapeutic target in lung
cancer. J Thorac Oncol. 11:1357–1362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu X, Liu X, Koul S, Lee CY, Zhang Z and
Halmos B: AXL kinase as a novel target for cancer therapy.
Oncotarget. 5:9546–9563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sheridan C: First Axl inhibitor enters
clinical trials. Nat Biotechnol. 31:775–776. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Nimmagadda S, Pullambhatla M, Lisok A, Hu
C, Maitra A and Pomper MG: Imaging Axl expression in pancreatic and
prostate cancer xenografts. Biochem Biophys Res Commun.
443:635–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Qu XH, Liu JL, Zhong XW, Li XI and Zhang
QG: Insights into the roles of hnRNP A2/B1 and AXL in non-small
cell lung cancer. Oncol Lett. 10:1677–1685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Choi YJ, Kim SY, So KS, Baek IJ, Kim WS,
Choi SH, Lee JC, Bivona TG, Rho JK and Choi CM: AUY922 effectively
overcomes MET- and AXL-mediated resistance to EGFR-TKI in lung
cancer cells. PLoS One. 10:e01198322015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bae SY, Hong JY, Lee HJ, Park HJ and Lee
SK: Targeting the degradation of AXL receptor tyrosine kinase to
overcome resistance in gefitinib-resistant non-small cell lung
cancer. Oncotarget. 6:10146–10160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yoshida T, Zhang G, Smith MA, Lopez AS,
Bai Y, Li J, Fang B, Koomen J, Rawal B, Fisher KJ, et al: Tyrosine
phosphoproteomics identifies both codrivers and cotargeting
strategies for T790M-related EGFR-TKI resistance in non-small cell
lung cancer. Clin Cancer Res. 20:4059–4074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kim HR, Kim WS, Choi YJ, Choi CM, Rho JK
and Lee JC: Epithelial-mesenchymal transition leads to crizotinib
resistance in H2228 lung cancer cells with EML4-ALK translocation.
Mol Oncol. 7:1093–1102. 2013. View Article : Google Scholar : PubMed/NCBI
|