Introduction
Aspirin, also known as acetylsalicylic acid, is a
>100-year-old drug widely used due to its analgesic, antipyretic
and anti-inflammatory properties (1).
Aspirin is capable of inhibiting the cyclooxygenase (COX) activity
of the enzyme prostaglandin G/H-synthase and blocking the
biosynthesis of prostaglandins (2).
The recognition that oral administration of aspirin may reduce the
risk of certain types of cancer makes aspirin a promising candidate
for tumor therapy. The high tumor-suppressive efficacy of aspirin
treatment observed in colorectal cancer (CRC) has increased the
number of studies investigating aspirin-induced CRC
suppression.
The pathogenesis of CRC is closely associated with
local inflammation in the intestine. Various inflammatory cells or
cytokines are responsible for the carcinogenesis of colorectal
cells (3). Among them, prostaglandin
is well-known for its long history of promoting survival,
proliferation and invasion in cancer cells (4). Therefore, the suppression of the
production of prostaglandin by inhibiting the activity of COX-2
with non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin
becomes a promising choice for CRC suppression. Compared with other
types of malignant tumors, CRC is relatively sensitive to aspirin
treatment, as revealed by several independent clinical trials
(5–7).
A number of studies have also demonstrated that COX-2 inhibition is
an important strategy for the chemopreventive treatment of
colon-associated disorders (8–11),
resulting in a lower risk of cancer. The inhibition of
COX-2-dependent signaling pathways is one mechanism involved in the
good tumor-suppressive efficacy of aspirin. COX-2-independent
pathways are also of importance; high concentrations of aspirin may
induce apoptosis via 15-lipoxygenase-1 in human HT-29 colonic
carcinoma cells (12).
Sex-determining region Y-box 7 (SOX7) is upregulated by aspirin and
is involved in the aspirin-mediated growth inhibition of human
SW480 colonic cancer cells (13). The
regulation of SOX7 by aspirin is implemented through the p38/MAPK
cascade (13). Although a number of
novel associations have been identified, the mechanism underlying
the inhibition of CRC by aspirin remains unclear.
Transforming growth factor-β1 (TGF-β1) is a
versatile cytokine involved in cell growth, differentiation and
immune modulation (14–16). TGF-β1 is a member of the TGF-β
superfamily and regulates proliferation and apoptosis in
epithelial, endothelial, neuronal and hematopoietic cells (17). TGF-β1 is also capable of inducing
apoptosis in a number of malignant tumor cells, contributing to
tumor suppression (18). In the
present study, the associations between aspirin, TGF-β1 expression
and CRC inhibitionwere examined, and a novel aspect of how aspirin
increases CRC inhibition and apoptosis was identified.
Materials and methods
Cell culture and treatment
The CT26 mouse coloncarcinoma cell line was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
maintained at 37°C in a humidified condition of 95% air and 5%
CO2. Cells were cultured in 75-cm2 flasks or
6-well plates with RPMI-1640 medium (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% heat-inactivated
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 U/ml streptomycin. Prior to the
addition of aspirin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
or LY364947 (Sigma-Aldrich; Merck KGaA), which is an inhibitor of
TGF-β receptor (R)-I, cells were allowed to adhere to 6- or 96-well
plates for 24 h. For the TGF-β1 treatment experiment, two groups
were used: i) 100 ng/ml TGF-β1 (BioLegend, Inc., San Diego, CA,
USA); ii) an equal amount of PBS for the control group. For the
aspirin treatment group, cells were divided into three groups: i)
3.5 µM aspirin; ii) combination of 3.5 µM aspirin and 1 µMLY364947
(LY364947 was added and after 2 h the culture medium was replaced
with an equal volume of RPMI-1640 medium containing 3.5 µM
aspirin); iii) an equal amount of dimethyl sulfoxide (DMSO) as the
control group. Each group was treated for 24, 36 and 48 h prior to
harvesting for additional study.
Cell viability assay
Cell viability was assessed using a MTT Cell
Proliferation and Cytotoxicity Assaykit (Beyotime Institute of
Biotechnology, Haimen, China). Cells (1×104) were seeded
on 96-well plates and cultured for 24, 36 and 48 h, followed by the
addition of MTT solution to the cells for 4 h. Subsequent to the
removal of the medium, the remaining MTT formazan crystals were
solubilized in DMSO and analyzed at 560 nm using a microplate
reader (Benchmark Electronics, Inc., Angleton, TX, USA).
ELISA
CT26 tumor cells were collected and then homogenized
in radioimmunoprecipitation assay buffer (0.1% SDS, 0.5%
deoxycholate, 1% Triton X-100, 150 mM NaCl and 50 mM Tris-HCl),
followed by centrifugation at 17,000 × g for 30 min at 4°C.
Adiethylaminobenzaldehyde assay was used to determine the protein
concentration of the samples. The prepared samples were stored at
−80°C until used. The levels of TGF-β1 in each sample were assessed
using mouse ELISA kits (R&D Systems, Inc., Minneapolis, MN,
USA), according to the manufacturer's protocol, and the
colorimetric reaction was measured at 450 nm using a microplate
reader (Benchmark Electronics, Inc.).
Flow cytometry analysis of
apoptosis
CT26 cells were treated, as aforementioned, then
harvested and washed in cold PBS prior to staining using an
Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA),
according to the manufacturer's protocol. Briefly, cells were
resuspended to a concentration of 1×105 cells/sample and
double-stained with fluorescein isothiocyanate-conjugated Annexin V
and propidium iodide. The samples were analyzed by flow cytometry,
as previously described (19)
(FACSAria™ SORP; BD Biosciences, Erembodegem, Belgium).
Western blot analysis
The cells were harvested, lysed and the total
protein was quantified using a Micro Bicinchoninic Protein Assay
kit (Pierce; Thermo Fisher Scientific, Inc.) according to the
protocol of the manufacturer. Total protein (10 µg) from each
sample was separated by electrophoresis using 12% SDS-PAGE,
transferred onto polyvinylidene fluoride membranes, pre-blocked
with 5% skim milk (MerckKGaA) for 90 min at room temperature and
then incubated using the primary antibodies (1:1,000), including
mouse-anti-β-actin (cat. no., MAB8929), rabbit-anti-B-celllymphoma2
(Bcl-2; cat. no., AF810), rabbit-anti-Bcl-2-associated X protein
(Bax; cat. no., AF820), rabbit-anti-Caspase3 (cat. no., AF-605-NA;
R&D Systems, Inc.), rabbit-anti-Caspase8 (cat. no., 9429; Cell
Signaling Technology, Inc., Danvers, MA, USA) overnight at 4°C. The
corresponding secondary antibodies [anti-mouse Ig(H+L); cat. no.,
0216], [anti-rabbit Ig(H+L) cat. no., A0208; dilution, 1:10,000;
Beyotime Institute of Biotechnology, Haimen, China] were applied
for 1 h at room temperature β-catenin was used as a loading
control. Signals were developed on X-ray films following exposure
to electrochemiluminescence advanced luminescence and Image-Pro
Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA) was used to
test the iodine value of the blots.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analyses were performed using SPSS (version 16.0; SPSS
Inc., Chicago, IL, USA). Inter-group statistical significance was
determined using a Student's unpaired t-test. P<0.05 was
considered to indicate a statistically significant difference.
*P<0.05, **P<0.03, ***P<0.01, ****P<0.001.
Results
Aspirin induces the secretion of
TGF-β1 by CT26 cells
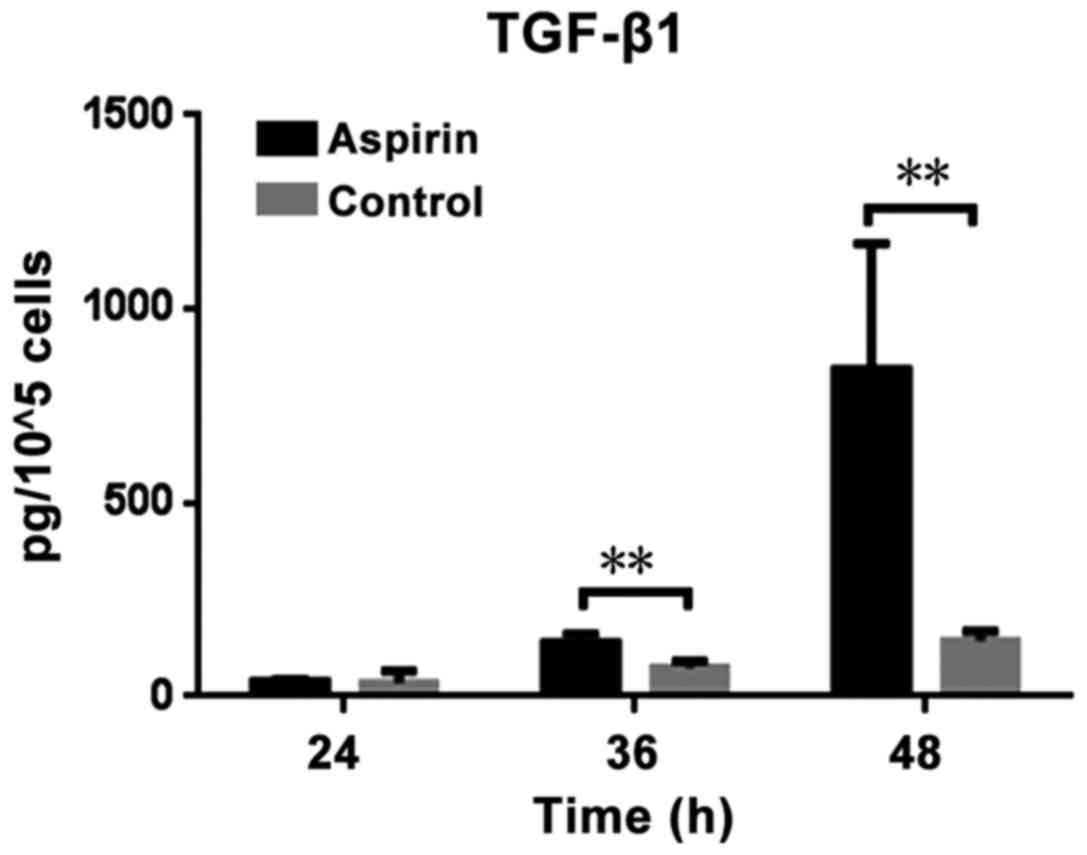
The levelsof TGF-β1 present in the supernatant fluid
of CT26 cells seeded in 6-well plates were determined using an
ELISA following aspirin treatment, and the amount of secreted
TGF-β1 by a certain number of CT26 cells was calculated: Firstly,
the amount of secreted TGF-β1 in 1×105 CT26 cells was
tested in each case. Secondly, the number of cells was counted.
Then, the average amount of TGF-β1 secreted by 1×105
CT26 cells in each case was calculated using the following formula:
Total amount of TGF-β1 in each case/the number of CT26 cells in
this case) ×105. The results demonstrated that the
levels of TGF-β1 vary based on the duration of treatment. Treatment
with aspirin significantly increased TGF-β1 secretion compared with
the control (P=0.0159 and P=0.0203 at 36 and 48 h, respectively;
Fig. 1). Treatment of CT26 cells with
aspirin for longer time points resulted in increased TGF-β1
secretion. While, one problem should be noted. The secretion of
TGF-β1 by CT26 cells is a continuous process and the present study
calculated the number of cells following collection of the
supernatant fluid of the CT26 cells. The number of cells in control
group increased subsequent to culturing for 24, 36 or 48 h, as
cellular proliferation had continued, while in the aspirin
treatment group, the number of cells increased by a small level, or
even decrease, as aspirin inhibits growth of cells (20). Despite this, a statistically
significant increase in secreted TGF-β1 in aspirin group was
observed.
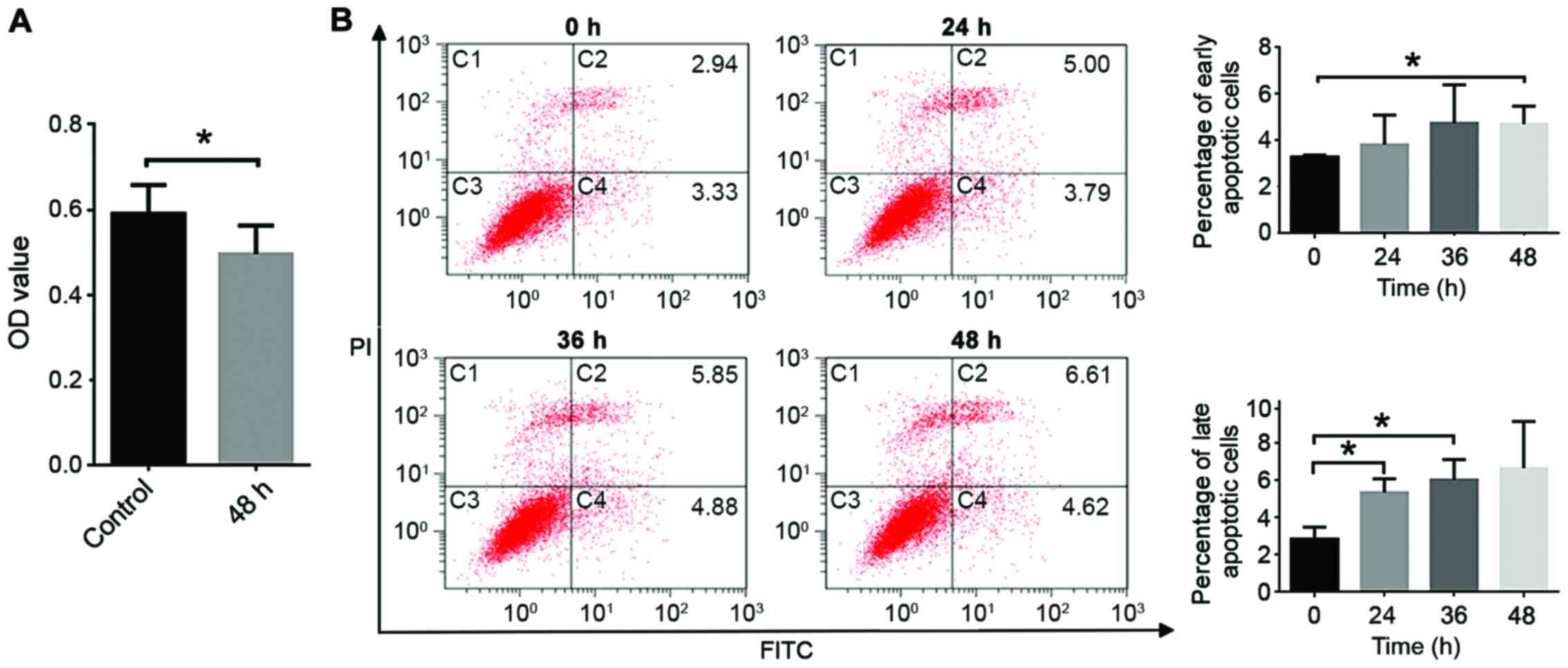
TGF-β1 induces the apoptosis of CT26
cells
An MTT assay demonstrated that TGF-β1 significantly
decreases the viability of CT26 cells (P<0.05; Fig. 2A), and induces early and late
apoptosis (Fig. 2B and C). Apoptosis
serves a key role in cellular development and is a stage-dependent
process, including early, intermediate and late-stage apoptotic
events. Cells may remain viable if stimulation by aspirin is
stopped during early apoptotic events, whereas for late apoptosis
defragmentation of DNA is typically observed and this will lead to
cell death even if stimulation is halted.
TGF-β1 mediates the effects of aspirin
on the viability of CT26 cells
In the present study, an MTT assay was used to
examine the effect of aspirin on CT26 cells. The concentration of
aspirin used was based on results from a previous study from our
group (Wang et al, unpublished). A concentration >10 µM
was excessive, while no apoptosis or proliferation of CT26 cells
was observed when <3 µM aspirin was used(data not shown). Data
from the present study demonstrated that treatment with 3 and 3.5
µM aspirin led to a significant decrease in cell viability
following 24, 48 and 72 h of treatment compared with the control
group (P<0.03, P<0.03 and P<0.01, respectively; Fig. 3A). However, co-treatment witha TGF-βR1
inhibitor, LY364947, resulted in a significant increase incell
viability following 48 and 72 h of treatment compared with cells
treated with aspirin alone (P<0.05), suggesting that the
inhibition caused by aspirin was mediated by TGF-β1. In addition,
co-treatment with aspirin and LY364947 significantly decreased the
percentage of early and late apoptotic events stimulated by aspirin
treatment alone following treatment for 36 h (late apoptotic
events) and 48 h (early and late apoptotic events) (P<0.05;
Fig. 3B).
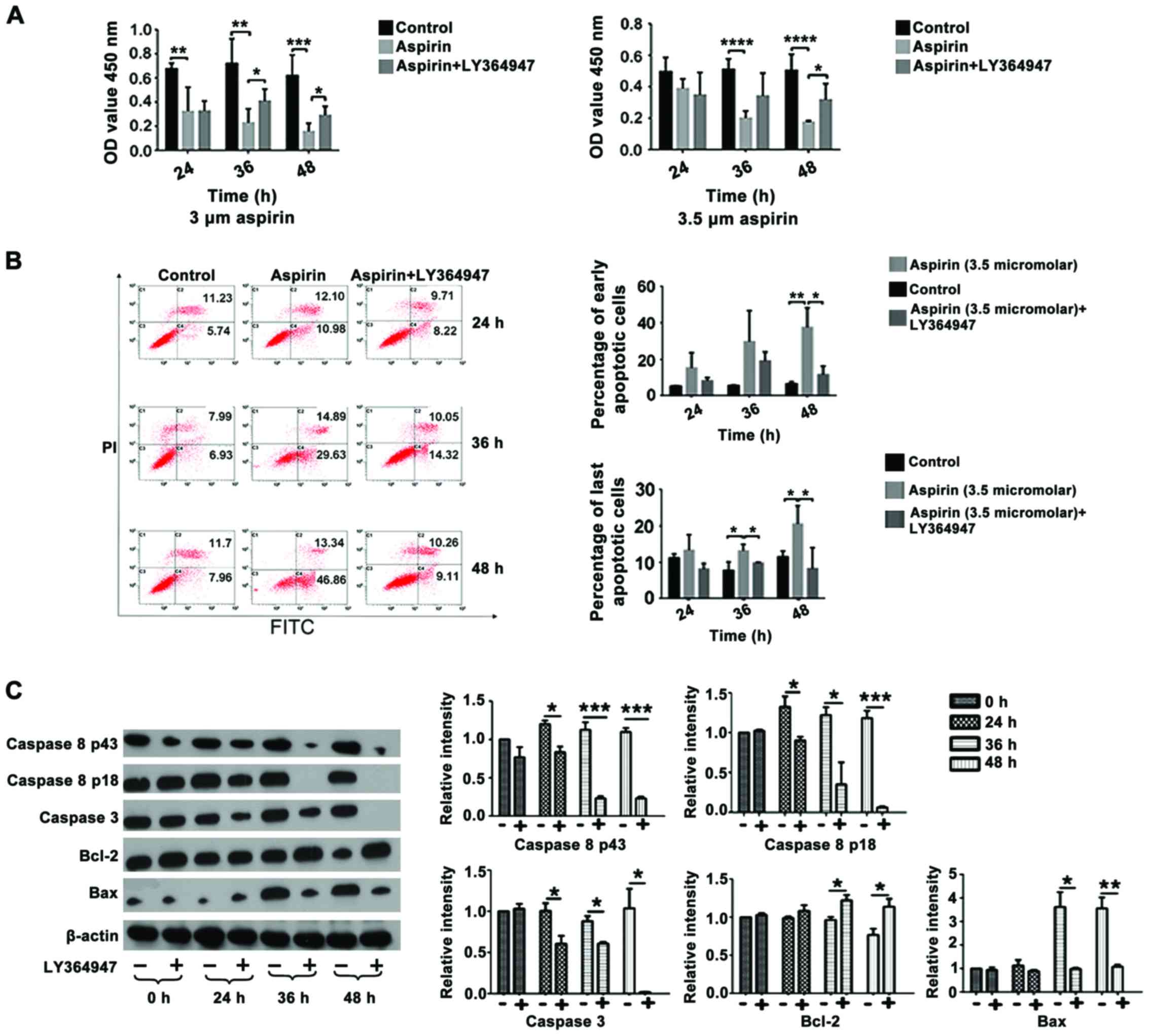 | Figure 3.LY364947 partially rescues the effect
of aspirin on CT26 cell viability. (A) Viability of CT26 cells
following treatment with 3 or 3.5 µM aspirin and/or 1 µM LY364947.
(B) Flow cytometry results and percentage of early and late
apoptotic cells following treatment with 3.5 µM aspirin and/or 1 µM
LY364947 for the indicated time points. (C) Representative western
blot analysis and quantification of protein levels of caspase 8,
caspase 3, Bcl-2 and Bax following treatment with 3.5 µM aspirin
and/or 1 µM LY364947 for the indicated time points. The control
group was 0 h, which received no treatment. *P<0.05,
**P<0.03, ***P<0.01, ****P<0.001. FITC, fluorescein
isothiocyanate; PI, propidium iodide; OD, optical density; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-associated X protein. |
The expression of a number of apoptosis-associated
proteins was analyzed by western blot analysis following treatment
with 3.5 µM aspirin and/or 1 µM LY364947. Changes in the expression
of caspase 8 P43, caspase 8 P18, caspase 3, Bcl-2 and Bax following
treatment with aspirin (data not shown) were significantly rescued
when cells were co-treated with aspirin and LY364947 at 24, 36 and
48 h following treatment (Fig.
3C).
Discussion
In the present study, it was demonstrated that
aspirin induced the secretion of TGF-β1 by CT26 cells, and that
TGF-β1 in turn led to a decrease in viability and increase in the
apoptotic rate CT26 cells. To the best of our knowledge, this is a
novel mechanism by which aspirin affects CRC suppression. TGF-β1 is
one of the mediators of aspirin-induced apoptosis and, through
TGF-β1, aspirin treatment resulted in the downregulation of certain
proteins known to be involved in apoptosis, including Bcl-2,
caspase3 and caspase8 fragments. It has previously been implicated
that the downregulation of Bcl-2 family members (21,22),
upregulation of the pro-apoptotic factor Bax (23) and the activation of caspase proteases
(24) are involved in TGF-β1-induced
apoptosis. Bcl-2 and Bax are well known for their roles in
apoptosis. Apoptotic caspase proteases are classified into
initiators and effectors, according to their point of entry into
the apoptotic cascade. Initiator caspases, such as caspase-8, are
the first to be activated in a particular cell-death pathway, and
they activate effector caspases, such as caspase-3, via the
cleavage of linker segments (25,26). Once
activated, caspases cleave a variety of important structural
proteins, enzymes and regulatory molecules (27), which leads to DNA fragmentation
(28–30) and subsequentcell apoptosis. The
present study also demonstrated an increase in the levels of
caspase-8. Aspirin suppresses tumor cell growth indirectly by
inducing the secretion of TGF-β1. This is similar to the results of
a previous study that suggested that ethanol exposure increased
TGF-β1 signaling through the suppression of Bcl-2 and
retinoblastoma proteins, which may have led to apoptotic cell
death, including β-endorphin neurons in the arcuate nucleus of the
hypothalamus (31).
Previously, aspirin has attracted attention due to
its potential benefit in the chemoprevention of CRC (32). Several clinical studies also reported
that aspirin may prolong the overall survival of patients with CRC
as a fore mentioned (5–7). A systematic review concluded that the
regular use of aspirin reduced the risk of CRC and the 20-year risk
of mortality due to CRC (33).
Another study which combined the analyses of four randomized trials
designed for the primary or secondary prevention of cardiovascular
events, identified that the use of aspirin at dosages of ≥75 mg
daily was associated with a 24% reduction in incidence and a 35%
reduction in mortality from colon cancer (34). However, the molecular mechanisms by
which aspirin inhibits CRC formation and growth have remained
unclear; therefore, the utilization of aspirin in CRC therapy
remains a disputed issue.
The best known molecular target of aspirin is COX.
Aspirin, but not other NSAIDs, may cause irreversible inactivation
of COX isozymes through the acetylation of a specific serine moiety
(35); yet there is evidence that, in
COX negative SW480 colonic cancer cells, aspirin is also able to
inhibit the growth of the cells (36). Another study also identified that
aspirin may act through COX-independent mechanisms that result in
an increased expression of DNA mismatch repair proteins and the
subsequent apoptosis in SW480 cells (37). COX-dependent and COX-independent
pathways used by aspirin may work cooperatively. For example, it
has been suggested that aspirin may sensitize tumor cells to tumor
necrosis factor-related apoptosis-inducing ligand, and may act
synergistically with the inhibition of COX-2-dependent
prostaglandin formation (38,39). The two pathways work together and
result in the apoptosis of tumor cells.
The life span of cancer cells within a living system
is significantly affected by the rate of apoptosis (40), which has been recognized as an
important physiological event in the pharmacology of anticancer
agents (41,42). The regulation of apoptosis has become
an area of extensive analysis in cancer studies (43). Aspirin may affect apoptosis through
prostaglandin-dependent or prostaglandin-independent pathways
(44). In prostaglandin-dependent
pathways, the inhibition of COX, particularly COX-2, is the primary
mechan is minvolved in CRC suppression by aspirin (8–11). COX-2
is undetectable in the normal epithelium, but is detectable in
>80% of patients with CRC and is associated with larger tumor
size and deeper tissue invasion (45). However, not all human colonic cancer
cells express COX-2 and produce prostaglandins (46,47).
Several studies have demonstrated that high aspirin dosages may
also affect apoptosis and cell proliferation in CRC via COX-2
independent pathways (37,48,49). These
mechanisms include: i) Wnt/β-catenin pathway inhibition (50); ii) interleukin-6/signal transducer and
activator of transcription 3 signaling pathway (51); iii) the downregulation of special
protein(Sp)1, Sp3, Sp4 and numerous Sp-regulated gene products
(52); iv) the regulation of various
targets, including 15-lipoxygenase-1 (53) or the
pro-apoptoticproteinprotease-activated receptor 4 (54); v) activation of p38/mitogen-activated
protein kinase (MAPK) (55), caspases
(56) or the ceramide pathway
(57); vi) the release of
mitochondrial cytochrome c (58); vii) the induction of autophagy in
colorectal cancer cells (59); viii)
inhibiting the activation of nuclear factor-κB (60), which has been implicated in cellular
adhesion (61), and the promotion of
metastasis (62).
In addition, aspirin may be administered for cancer
treatment due to its indirect function in reducing drug resistance
or enhancing the antitumor effects of other drugs (63). Furthermore, aspirin may suppress the
pro-invasion and pro-metastasis effects of sorafenib in
hepatocellular carcinoma (HCC) through the upregulation of
oxidoreductase HTATIP2, which is potentially mediated by the
inhibition of COX2 expression, and it may improve the efficacy of
sorafenib (63). An additional study
revealed that aspirin enhanced doxorubicin-induced apoptosis and
reduced tumor growth in human HCC cells in vitro and in
vivo (64).
TGF-β1is a versatile cytokine that is involved in
cell-cycle control, the regulation of early development,
differentiation, extracellular matrix formation, hematopoiesis,
angiogenesis, chemotaxis and immune functions (15). Proliferation of a number of epithelial
cell types may be inhibited by TGF-β1, including intestinal
epithelial cells, whereas the growth of mesenchymal cells is
stimulated (65). TGF-β1 is also an
inhibitor of tumor growth that is capable of inducing apoptosis in
various types of cancer cells; however, the reaction of diverse
malignant cells to TGF-β1 is different: During the process of
cancer development, certain transformed cells become partly or
completely resistant to TGF-β1 growth inhibition (12), resulting in the uncontrolled
proliferation of the cells (52).
However, specific cancer cells remain sensitive to TGF-β1, which
may lead to proliferation inhibition and the induction of apoptosis
in these cells (16). A number of
transgenic mouse studies have also provided evidence for the
hypothesis that one of the roles of the TGF-β1 signaling pathway is
to provide protection against malignant transformation (17,18).
Transgenic mice that produce a constitutively active form of TGF-β1
are resistant to DMBA-induced mammary tumor formation (17). Patients with breast cancer and high
expression levels of TGF-β1 appear to exhibit longer disease-free
intervals and a significantly improved probability of survival
(66).
In summary, the present study has suggested a novel
pathway for aspirin-induced CRC cell apoptosis. More specifically,
aspirin induces CRC cell apoptosis by elevating the secretion of
TGF-β1 by those cells, and the increased levels of TGF-β1 in turn
lead to apoptosis and proliferation inhibition in the CRC cells.
There are certain limitations associated with the present study.
Firstly, concentrations of 3 and 3.5 µM aspirin were used to treat
the CT26 cells, as lower doses of aspirin were unable to induce
apoptosis and proliferation inhibition following treatment for 48
h; this concentration of aspirin induced a secretion of ~1,000 pg
TGF-β1 by 1×105CT26 cells. However, 100 ng/ml TGF-β1,
a20-fold lower concentration compared with the amount induced by
aspirin, was used to examine whether TGF-β1 is capable of inducing
apoptosis and proliferation inhibition in CT26 cells. It was not
possible to test higher concentrations of TGF-β1 induced by aspirin
due to the high cost of commercial TGF-β1 protein. However, if a
significantly lower dose of TGF-β1 was able to induce apoptosis and
proliferation inhibition in CT26 cells, it is reasonable to predict
that higher concentrations would elicita similar effect.
Alterations in the expression levels of various apoptotic proteins
in CT26 cells following treatment with 3.5 µM aspirin were
observed, but these changes did not appear in the cells following
treatment with 100 ng/ml TGF-β1. Whether 100 ng/ml TGF-β1 is able
to induce apoptosis in CT26 cells without affecting the caspase
proteins remains to be determined. Furthermore, caspase-independent
pathways, such as the AIF-endonuclease G-Omi/HtrA pathway, may also
induce apoptosis in these cells (67). Whether this pathway was involved at
the relatively low concentrations of TGF-β1-induced apoptosis in
the present study, and whether higher and lower concentrations of
TGF-β1may induce apoptosis via alternate pathways remains to be
elucidated, and additional studies are required. Secondly, the
present study only focused on a small number of apoptotic proteins,
including the caspase 8 fragments p43 and p18, caspase3, Bcl-2 and
Bax. An association has previously been demonstrated between TGF-β1
and the well-known extracellular signal-regulated kinase
(ERK)1/2-MAPK signaling pathway (68). Whether aspirin exhibits any
significant effects on this pathway has yet to be determined and
additional studies are required.
Acknowledgements
The present study was funded by The National Natural
Science Foundation of China (grant no. 81201787).
References
|
1
|
Ugurlucan M, Caglar IM, Caglar FN, Ziyade
S, Karatepe O, Yildiz Y, Zencirci E, Ugurlucan FG, Arslan AH,
Korkmaz S, et al: Aspirin: From a historical perspective. Recent
Pat Cardiovasc Drug Discov. 7:71–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vane JR, Flower RJ and Botting RM: History
of aspirin and its mechanism of action. Stroke. 21 12
Suppl:IV12–IV23. 1990.PubMed/NCBI
|
|
3
|
Wang S, Liu Z, Wang L and Zhang X:
NF-kappaB signaling pathway, inflammation and colorectal cancer.
Cell Mol Immunol. 6:327–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Menter DG and Dubois RN: Prostaglandins in
cancer cell adhesion, migration, and invasion. Int J Cell Biol.
2012:7234192012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kanaoka S, Yoshida K, Miura N, Sugimura H
and Kajimura M: Potential usefulness of detecting cyclooxygenase 2
messenger RNA in feces for colorectal cancer screening.
Gastroenterology. 127:422–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fenwick SW, Toogood GJ, Lodge JP and Hull
MA: The effect of the selective cyclooxygenase-2 inhibitor
rofecoxib on human colorectal cancerliver metastases.
Gastroenterology. 125:716–729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Krishnan K, Ruffin MT, Normolle D,
Shureiqi I, Burney K, Bailey J, Peters-Golden M, Rock CL, Boland CR
and Brenner DE: Colonic mucosal prostaglandin E2 and cyclooxygenase
expression before and after low aspirin doses in subjects at high
risk or at normal risk for colorectal cancer. Cancer Epidemiol
Biomarkers Prev. 10:447–453. 2001.PubMed/NCBI
|
|
8
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signaling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moon CM, Kwon JH, Kim JS, Oh SH, Jin Lee
K, Park JJ, Pil Hong S, Cheon JH, Kim TI and Kim WH: Nonsteroidal
anti-inflammatory drugs suppress cancer stem cells via inhibiting
PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in
colorectal cancer. Int J Cancer. 134:519–529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vaish V, Tanwar L and Sanyal SN: The role
of NF-κB and PPARγ in experimentally induced colorectal cancer and
chemoprevention by cyclooxygenase-2 inhibitors. Tumour Biol.
31:427–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishizaki T, Katsumata K, Tsuchida A, Wada
T, Mori Y, Hisada M, Kawakita H and Aoki T: Etodolac, a selective
cyclooxygenase-2 inhibitor, inhibits liver metastasis of colorectal
cancercells via the suppression of MMP-9 activity. Int J Mol Med.
17:357–362. 2006.PubMed/NCBI
|
|
12
|
Shureiqi I, Chen D, Lotan R, Yang P,
Newman RA, Fischer SM and Lippman SM: 15-Lipoxygenase-1 mediates
nonsteroidalanti-inflammatory drug-induced apoptosis independently
of cyclooxygenase-2 in colon cancer cells. Cancer Res.
60:6846–6850. 2000.PubMed/NCBI
|
|
13
|
Zhou X, Huang SY, Feng JX, Gao YY, Zhao L,
Lu J, Huang BQ and Zhang Y: SOX7 is involved in aspirin-mediated
growth inhibition of human colorectal cancer cells. World J
Gastroenterol. 17:4922–4927. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Govinden R and Bhoola KD: Genealogy,
expression, and cellular function of transforming growth
factor-beta. Pharmacol Ther. 98:257–265. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sánchez-Capelo A: Dual role for TGF-beta1
in apoptosis. Cytokine Growth Factor Rev. 16:15–34. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pierce DF Jr, Gorska AE, Chytil A, Meise
KS, Page DL, Coffey RJ Jr and Moses HL: Mammary tumor suppression
by transforming growth factor beta 1 transgene expression. Proc
Natl Acad Sci USA. 92:pp. 4254–4258. 1995; View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen L, Lu X, Zeng T, Chen Y, Chen Q, Wu
W, Yan X, Cai H, Zhang Z, Shao Q and Qin W: Enhancement of
DEN-induced liver tumourigenesis in hepatocyte-specific
Lass2-knockout mice coincident with upregulation of the
TGF-β1-Smad4-PAI-1 axis. Oncol Rep. 31:885–893. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Elmore S: Apoptosis: A review of
programmed death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Jiang M, Li Z, Wang J, Du C,
Yanyang L, Yu Y, Wang X, Zhang N, Zhao M, et al: Hypoxia and TGF-β1
lead to endostatin resistance by cooperatively increasing cancer
stem cellsin A549 transplantation tumors. Cell Biosci. 5:722015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yue W, Zheng X, Lin Y, Yang CS, Xu Q,
Carpizo D, Huang H, DiPaola RS and Tan XL: Metformin combined with
aspirin significantly inhibit pancreatic cancer cell growth in
vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and
Bcl-2. Oncotarget. 6:21208–21224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koseki T, Yamato K, Krajewski S, Reed JC,
Tsujimoto Y and Nishihara T: Activin A-induced apoptosis is
suppressed by BCL-2. FEBS Lett. 376:247–250. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saltzman A, Munro R, Searfoss G, Franks C,
Jaye M and Ivashchenko Y: Transforming growth factor-beta-mediated
apoptosis in the Ramos B-lymphoma cell line is accompanied by
caspase activation and Bcl-XL downregulation. Exp Cell Res.
242:244–254. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fukuchi Y, Kizaki M, Yamato K, Kawamura C,
Umezawa A, Hata Ji, Nishihara T and Ikeda Y: Mcl-1, an
early-induction molecule, modulates activin A-induced apoptosis and
differentiation of CML cells. Oncogene. 20:704–713. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen RH and Chang TY: Involvement of
caspase family proteases in transforming growth factor-beta-induced
apoptosis. Cell Growth Differ. 8:821–827. 1997.PubMed/NCBI
|
|
26
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alnemri ES: Mammalian cell death
proteases: A family of highly conserved aspartate specific cysteine
proteases. J Cell Biochem. 64:33–42. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han BH, DeMattos RB, Dugan LL, Kim-Han JS,
Brendza RP, Fryer JD, Kierson M, Cirrito J, Quick K, Harmony JA, et
al: Clusterin contributes to caspase-3-independent brain injury
following neonatal hypoxia-ischemia. Nat Med. 7:338–343. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuhn P and Sarkar DK: Ethanol induces
apoptotic death of b-Endorphin neurons in the rat Hypothalamus by a
TGF-beta 1-dependent mechanism. Alcohol Clin Exp Res. 32:706–714.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jänne PA and Mayer RJ: Chemoprevention of
colorectal cancer. N Engl J Med. 342:1960–1968. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Algra AM and Rothwell PM: Effects of
regular aspirin on long-term cancer incidence and metastasis: A
systematic comparison of evidence from observational studies versus
randomised trials. Lancet Oncol. 13:518–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rothwell PM, Wilson CE, Elwin M, Norrving
B, Algra A, Warlow CP and Meade TW: Long-term effect of aspirin on
colorectal cancer incidence and mortality: 20-year follow-up of
five randomized trials. Lancet. 376:1741–1750. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Patrono C and Rocca B: Aspirin: promise
and resistance in the new millennium. Arterioscler Thromb Vasc
Biol. 28 Suppl:s25–s32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lai MY, Huang JA, Liang ZH, Jiang HX and
Tang GD: Mechanisms underlying aspirin-mediated growth inhibition
and apoptosis induction of cyclooxygenase-2 negative colon cancer
cell line SW480. World J Gastroenterol. 14:4227–4233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bos CL, Kodach LL, van den Brink GR, Diks
SH, van Santen MM, Richel DJ, Peppelenbosch MP and Hardwick JC:
Effect of aspirin on the Wnt/beta-catenin pathway is mediated via
protein phosphatase 2A. Oncogene. 25:6447–6456. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martin S, Phillips DC, Szekely-Szucs K,
Elghazi L, Desmots F and Houghton JA: Cyclooxygenase-2 inhibition
sensitizes human colon carcinoma cells to TRAIL-induced apoptosis
through clustering of DR5 and concentrating death-inducing
signaling complex components into ceramide-enriched caveolae.
Cancer Res. 65:11447–11458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim KM, Song JJ, An JY, Kwon YT and Lee
YJ: Pretreatment of acetylsalicylic acid promotes tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis by
down-regulating BCL-2 gene expression. J Biol Chem.
280:41047–41056. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chinnaiyan AM and Dixit VM: The cell-death
machine. Curr Biol. 6:555–562. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kasibhatla S and Tseng B: Why target
apoptosis in cancer treatment? Mol Cancer Ther. 2:573–580.
2003.PubMed/NCBI
|
|
44
|
Makin G: Targeting apoptosis in cancer
chemotherapy. Expert Opin Ther Targets. 6:73–84. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marx J: Cancer research.
Anti-inflammatories inhibit cancer growth-but how? Science.
291:581–582. 2001.PubMed/NCBI
|
|
46
|
Fujita T, Matsui M, Takaku K, Uetake H,
Ichikawa W, Taketo MM and Sugihara K: Size- and invasion-dependent
increase in cyclooxygenase 2 levels in human colorectal carcinomas.
Cancer Res. 58:4823–4826. 1998.PubMed/NCBI
|
|
47
|
Marnett LJ: Aspirin and the potential role
of prostaglandins in colon cancer. Cancer Res. 52:5575–5589.
1992.PubMed/NCBI
|
|
48
|
Hanif R, Pittas A, Feng Y, Koutsos MI,
Qiao L, Staiano-Coico L, Shiff SI and Rigas B: Effects of
nonsteroidal anti-inflammatory drugs onproliferation and on
induction of apoptosis in colon cancer cells by a
prostaglandin-independent pathway. Biochem Pharmacol. 52:237–245.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Thun MJ, Jacobs EJ and Patrono C: The role
of aspirin in cancer prevention. Nat Rev Clin Oncol. 9:259–267.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun Y, Chen J and Rigas B: Chemopreventive
agents induce oxidative stress in cancer cells leading to COX-2
overexpression and COX-2-independent cell death. Carcinogenesis.
30:93–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shtivelband MI, Juneja HS, Lee S and Wu
KK: Aspirin and salicylate inhibit colon cancer medium- and
VEGF-induced endothelial tube formation: Correlation with
suppression of cyclooxygenase-2 expression. J ThrombHaemost.
1:2225–2233. 2003. View Article : Google Scholar
|
|
52
|
Tian Y, Ye Y, Gao W, Chen H, Song T, Wang
D, Mao X and Ren C: Aspirin promotes apoptosis in a murine model of
colorectal cancer by mechanisms involving downregulation of
IL-6-STAT3 signaling pathway. Int J Colorectal Dis. 26:13–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pathi S, Jutooru I, Chadalapaka G, Nair V,
Lee SO and Safe S: Aspirin inhibits colon cancer cell and tumor
growth and downregulates specificity protein (Sp) transcription
factors. PLoS One. 7:e482082012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shureiqi I, Xu X, Chen D, Lotan R, Morris
JS, Fischer SM and Lippman SM: Nonsteroidal anti-inflammatory drugs
induce apoptosis in esophageal cancer cells by restoring
15-lipoxygenase-1 expression. Cancer Res. 61:4879–4884.
2001.PubMed/NCBI
|
|
55
|
Zhang Z and DuBois RN: Par-4, a
proapoptoticgene, is regulated by NSAIDs in human colon carcinoma
cells. Gastroenterol. 118:1012–1017. 2000. View Article : Google Scholar
|
|
56
|
Bellosillo B, Piqué M, Barragán M, Castaño
E, Villamor N, Colomer D, Montserrat E, Pons G and Gil J: Aspirin
and salicylate induce apoptosis and activation of caspases in
B-cell chronic lymphocytic leukaemia cells. Blood. 92:1406–1414.
1998.PubMed/NCBI
|
|
57
|
Schwenger P, Bellosta P, Vietor I,
Basilico C, Skolnik Y and Vilcek J: Sodium salicylate induces
apoptosis via p38 mitogen activated protein kinase but inhibits
tumour necrosis factor-induced c-Jun N-terminal
kinase/stress-activated protein kinase activation. Proc Natl Acad
Sci USA. 94:pp. 2869–2873. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chan TA, Morin PJ, Vogelstein B and
Kinzler KW: Mechanisms underlying non-steroidal anti-inflammatory
drug-mediated apoptosis. Proc Natl Acad Sci USA. 95:pp. 681–686.
1998; View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zimmermann KC, Waterhouse NJ, Goldstein
JC, Schuler M and Green DR: Aspirin induces apoptosis through
release of cytochrome c from mitochondria. Neoplasia. 2:505–513.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Din FV, Valanciute A, Houde VP, Zibrova D,
Green KA, Sakamoto K, Alessi DR and Dunlop G: Aspirin inhibits mTOR
signaling, activates AMP-activated protein kinase, and induces
autophagy in colorectal cancer cells. Gastroenterology.
142:1504–1515.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Takada Y, Bhardwaj A, Potdar P and
Aggarwal BB: Nonsteroidal anti-inflammatory agents differ in their
ability to suppress NF-kappaB activation, inhibition of expression
of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell
proliferation. Oncogene. 23:9247–9258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tozawa K, Sakurada S, Kohri K and Okamoto
T: Effects of anti-nuclear factor kappa B reagents in blocking
adhesion of human cancer cells to vascular endothelial cells.
Cancer Res. 55:4162–4167. 1995.PubMed/NCBI
|
|
63
|
Fujioka S, Sclabas GM, Schmidt C,
Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C and Chiao
PJ: Function of nuclear factor kappaB in pancreatic cancer
metastasis. Clin Cancer Res. 9:346–354. 2003.PubMed/NCBI
|
|
64
|
Lu L, Sun HC, Zhang W, Chai ZT, Zhu XD,
Kong LQ, Wang WQ, Zhang KZ, Zhang YY, Zhang QB, et al: Aspirin
minimized the pro-metastasis effect of sorafenib and improved
survival by up-regulating HTATIP2 in hepatocellular carcinoma. PLoS
One. 8:e650232013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hossain MA, Kim DH, Jang JY, Kang YJ, Yoon
JH, Moon JO, Chung HY, Kim GY, Choi YH, Copple BL and Kim ND:
Aspirin enhances doxorubicin-induced apoptosis and reduces tumor
growth in human hepatocellular carcinoma cells in vitro and in
vivo. Int J Oncol. 40:1636–1642. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Smith RD: Evidence for autocrine growth
inhibition of rat intestinal epithelial (RIE-1) cells by
transforming growth factor type-beta. Biochem Mol Biol Int.
35:1315–1321. 1995.PubMed/NCBI
|
|
67
|
Murray PA, Barrett-Lee P, Travers M,
Luqmani Y, Powles T and Coombes RC: The prognostic significance of
transforming growth factors in human breast cancer. Br J Cancer.
67:1408–1412. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sun Y, Gao W, Zhao Y, Cao W, Liu Z, Cui G,
Tong L, Lei F and Tang B: Visualization and inhibition of
mitochondria-nuclear translocation of apoptosis inducing factor by
a graphene oxide-DNA nanosensor. Anal Chem. 89:4642–4647. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Oussaief L, Hippocrate A, Ramirez V,
Rampanou A, Zhang W, Meyers D, Cole P, Khelifa R and Joab I:
Phosphatidylinositol 3-kinase/Akt pathway targets acetylation of
Smad3 through Smad3/CREB-binding protein interaction: Contribution
to transforming growth factor beta1-induced Epstein-Barr virus
reactivation. J Biol Chem. 284:23912–23924. 2009. View Article : Google Scholar : PubMed/NCBI
|