Introduction
In total, 80–85% of all patients with lung cancer
are diagnosed with non-small cell lung cancer (NSCLC), resulting in
NSCLC being the most common type of lung cancer (1). In addition, NSCLC has remained a crucial
cause of mortality in patients with carcinoma, despite notable
advances in early diagnosis and treatment modalities. In the light
of the discovery of dendritic cells and current approaches in two
major areas, tumour-associated antigens (TAAs) and adoptive
cellular targeting, immunotherapy has resulted in an improved
prognosis for the treatment of patients with NSCLC at an advanced
stage of disease (2). Among the
numerous immunotherapies, dendritic cell (DC)-based adoptive
immunotherapy has emerged as the most viable option for such
patients. DCs are professional antigen-presenting cells that
efficiently activate T lymphocytes by presenting the antigens to
immature cluster of differentiation (CD)4+ cells via
major histocompatibility complex (MHC) class II and CD8+
cells through MHC class I (3,4). Studies have reported that DCs may be
generated from autologous monocytes (CD14+ cells
condensed by apheresis) by making use of a culture medium that is
complimented with interleukin (IL) 4, granulocyte-macrophage
colony-stimulating factor (GM-CSF) or IL-13 (1–4) and pulsed
ex vivo with TAAs to elicit a potent T cell-mediated immune
response and protect against additional tumour challenges (5–8).
However, collective data on the use of dendritic
cell-based immunotherapy for the treatment of NSCLC are limited,
and to the best of our knowledge, none of the previously reported
clinical trials have exclusively evaluated DC immunotherapies in
NSCLC (9,10). Studies have suggested that Survivin
and MAGE-3 are overexpressed in NSCLC and may play a vital role in
tumourigenesis (11,12). Therefore, the present study was
performed to identify the immunological response along with the
efficacy and harmlessness of the restorative vaccination using
autologous DCs pulsed with recombinant melanoma-associated antigen
(rMAGE-3) and recombinant Survivin (rSurvivin) peptide in patients
with NSCLC.
Materials and methods
Sample study and design
A total of 16 NSCLC patients were enrolled in the
present open-label non-randomised study. All patients had
histologically-confirmed diagnoses of stage I–IIIB disease.
Patients that had stable disease at the time of screening and had
completed definitive therapy (surgical, medical or multimodal) were
eligible to participate in the present study. The Ethics Committee
of the Central Hospital of Zibo (Zibo, China) approved the study
protocol; thus, prior to the start of the current study, written
informed consent was collected from all participating patients. The
present study followed all the required modifications under the
International Conference on Harmonisation and Good Clinical
Practice guidelines and was in agreement with the Declaration of
Helsinki, 1975. Between December 2013 and October 2014, patients
with disease duration of 6 weeks to 3 years (average, 8 months)
after definitive therapy were enrolled in the present study. A
heterogeneous group of patients was selected with respect to
medical history, stage of disease, risk of recurrence and treatment
of primary disease. Characteristics of the patients are summarised
in Table I.
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Patient code | Age, years | Histology | TNM stage | Prior treatment
received | Duration from last
treatment to DC immunotherapy, months |
|---|
| 1XU | 53 | Squamous | IIIA | Chemotherapy +
radiation therapy | 8 |
| 2LI | 63 | Squamous | IIIA | Chemotherapy +
radiation therapy | 5 |
| 3XB | 50 | Adenocarcinoma | IIIB | Neo/surgery/adjuvant
chemotherapy | 31 |
| 4YU | 65 | Bronchoalveolar | IB | Surgery | 6 |
| 5NC | 72 | Adenocarcinoma | IB | Surgery | 8 |
| 6QL | 59 | Adenocarcinoma | IA | Neo/surgery/adjuvant
chemotherapy | 5 |
| 7ZY | 66 | Adenocarcinoma | IIIA | Surgery/adjuvant
chemotherapy | 4 |
| 8XL | 57 | Squamous | IIIA | Chemotherapy +
radiation therapy | 4 |
| 9YP | 52 | Adenocarcinoma | IIB | Chemotherapy +
radiation therapy | 12 |
| 10LO | 65 | Adenocarcinoma | IB | Chemotherapy +
radiation therapy | 5 |
| 11XE | 61 | Squamous | IA | Chemotherapy +
radiation therapy | 7 |
| 12NZ | 58 | Adenocarcinoma | IIIA | Neo/surgery/adjuvant
chemotherapy | 3 |
| 13HE | 70 | Squamous | IIIA | Surgery/adjuvant
chemotherapy | 3 |
| 14PX | 56 | Squamous | IIIA |
Neo/surgery/adjuvant chemotherapy | 5 |
| 15SZ | 71 | Adenocarcinoma | IIIA | Chemotherapy +
radiation therapy | 7 |
| 16YU | 62 | Adenocarcinoma | IIB |
Surgery/chemotherapy | 11 |
Measurable immunological response to DC
immunotherapy was the primary endpoint, and obtaining comparative
immunological data from different NSCLC patients that had received
a definitive therapy was the secondary endpoint. To evaluate the
inhibitory effects of persistent tumour load and to assess the
impact of previous radiotherapy and chemotherapy on immunological
responses, the patients were primarily stratified according the
therapy they had received. Heterogeneity of the patients and small
sample size can prevent meaningful evaluation of therapeutic
effects. Therefore, it was important to incorporate the
immunotherapy into the therapeutic plan of the patient, with little
time commitment and risk. Routine safety laboratory measurements
were performed to evaluate clinical tolerability, and adverse
events were assessed according to the National Cancer Institute
Cancer Therapy Evaluation Program and Common Terminology Criteria
for Adverse Events (13).
Dosing schedule
The protocol followed, including the dose used and
the route and interval of administration, was selected on the basis
of the methods used previously (14).
The target dose selected was 108 DCs pulsed with rMAGE3
+ rSurvivin in a total 3 ml volume, and a prime immunotherapy
followed by one boost immunotherapy were intradermally administered
in the thigh 1 month apart. Overall, 16 prime and boost injections
containing 9.1×107 DCs and 8.2×107 DCs,
respectively, were administered. Following immunisation, patients
were monitored for 2 h in the outpatient clinic for immediate
unexpected adverse events.
Preparation of monocyte-derived DCs
(MODCs)
The DC immunotherapies were developed as described
by Hirschowitz et al (14).
Briefly, each patient was subjected to a 3-h leukapheresis
procedure and 1–3×1010 peripheral blood mononuclear
cells (PBMCs) were drawn. The cells were then placed in a tissue
culture flask at a density of 1×106 cells/cm2
in the presence of 1% human serum albumin (Baxter Healthcare,
Deerfield, IL, USA). Subsequent to incubating the cells in 5%
CO2 at 37°C for 2 h, the flask was washed with sterile
phosphate-buffered saline (PBS) to isolate non-adherent cells.
Adherent cells were then resuspended in a clinical grade CellGro DC
medium (CellGenix, Breisgau, Germany) containing 1,000 U/ml GM-CSF
(CellGenix), 50 ng/ml IL-4 (CellGenix) and were incubated for 5
days in 5% CO2 at 37°C. On the fifth day, DCs were split
into 2 aliquots, one for rMAGE3 and the other for rSurvivin. TAA
peptides at a concentration of10 µg/ml in 10 ml PBS were
individually added to every aliquot and then incubated at 37°C for
2 h. The aliquots were then transferred to a single vial. To induce
DC maturation, cytokine cocktail, IL-1β (Peprotech, Rocky Hill, NJ,
USA), IL-6 (Peprotech), tumour necrosis factor-α (TNF-α;
Peprotech), interferon-γ (IFN-γ; LG Life Sciences, Gurgaon,
Haryana, India), prostaglandin E2 (PGE2; Sigma Aldrich; Merck
Millipore, Darmstadt, Germany) and poly I:C (Sigma Aldrich; Merck
Millipore) were added to the culture between days 5 and 7. DCs were
later bathed twice and then resuspended in 1 ml PBS. Identification
of the morphology and immunophenotyping for CD14, CD83, CD86, CD1a
and human leukocyte antigen-antigen D related (HLA-DR) was
performed in MODCs and later detected for its sterility. The final
formulation contained rMAGE3-primed and rSurvivin-primed DCs in the
ratio of 1:1, and the total cell concentration was 5×106
DCs in each dose. All 5 doses were prepared at a time and were
frozen using automated cryopreservation.
Generation of recombinant
proteins
Procreation of the cDNAs encoding MAGE-3 or Survivin
into the pCTP vector was performed as previously described
(15). It was observed that
Escherichia coli BL21 (DE3) had both the antigens (MAGE-3
and Survivin) that were in the structure of 6×-His-attached fusion
proteins. Nickel-nitrilotriacetic acid column chromatography
(Qiagen, Hilden, Germany) was used to purify the antigens.
Endotoxin <1.0 EU/µg in limulus amoebocyte lysate test
(catalog no. 88282; Thermo Fischer Scientific, Waltham, MA, USA),
according to the manufacturer's instructions, and >95% purity in
SDS-PAGE analysis were performed for the quality control
authentication of all antigens. Working solutions were prepared by
dissolving 0.6 mg of each peptide in 30 ml dimethyl sulfoxide
(Wak-chemie Medical GmbH, Steinbach, Germany) and 270 ml sterile
water, resulting in a final concentration of 2 mg/ml.
In vitro characterisation
Each DC immunotherapy was subjected to sterility
testing and characterisation for the expression of CD14, CD86,
CD205 and HLA-DR. Mycoplasma contamination was checked with the use
of a MycoAlert Mycoplasma Detection kit (Lonza, Auckland, New
Zealand). A kinetic chromogenic limulus amoebocyte lysate test
(Lonza) was used in order to identify the endotoxin, according to
the manufacturer's instructions.
Evaluation of the phenotypes of
DCs
The phenotypes of mature DCs, immature DCs and
monocytes were determined using one- or two-colour fluorescence
analysis. In total, 3×105 cells were resuspended in 50
µl of buffer containing PBS, 2% foetal calf serum (FCS) and 1%
sodium azide. The cells were then incubated with 10 µl of
appropriate phycoerythrin-labelled monoclonal antibodies (mAbs) at
a dilution of 1:100 or fluorescein isothiocyanate (FITC) (HLA-DR:
catalog no. 130-098-176; clone, AC122; CD14: catalog no.
130-110-576; clone, REA599; CD86: catalog no. 130-098-182; clone,
FM95; CD205: catalog no. 130-104-772; clone, HD30; Miltenyi Biotec,
Singapore) at 4°C for 30 min. Subsequent to incubation, the cells
were washed twice and resuspended in 500 µl of assay buffer. The
fluorescence was analysed by a flow cytometer (FACSCalibur; BD
Biosciences, Franklin Lakes, NJ, USA). There was a build up of
15,000 events for every sample, in addition to delineation of the
number of positive cells. DCs were characterised using human
HLA-DR-, CD14-, CD86- and CD205-specific mAbs (Miltenyi Biotec) and
control immunoglobulins G1 and G2a (IgG1 and IgG2a; BD
Biosciences).
Intracytoplasmic IFN-γ detection
assay
The procedure used was determined by Kern et
al (16) for the intracellular
staining of IFN-γ released by lymphocytes. Briefly,
5×106 CD14-peripheral mononuclear cells were obtained
prior to the first injection (T0) and subsequent to the fourth
injection (T4). Co-culturing of the cells was then performed for 18
h with 1×106 mature MODCs pulsed with rMAGE3 +
rSurvivin. Protein secretion was blocked during the last 3 h using
10 µmol of monensin (Sigma Aldrich; Merck Millipore). T0 and T4
cells that were not exposed to rMAGE3 + rSurvivin were used as
controls. Intermingling of ionomycin (500 ng/ml; Sigma Aldrich;
Merck Millipore) and phorbol myristate acetate (PMA; 50 ng/ml;
Sigma Aldrich; Merck Millipore) was performed with the cell
suspensions in a correspondent experimental lay down. Subsequent to
harvesting, washing and permeabilising the cells with a
permeabilisation agent (Immunotech Laboratories, Inc.), according
to the manufacturer's protocol, the cells were double-stained with
IFN-γ-specific or CD69-specific antibody labelled with
phycoerythrin (catalog no. 130-098-901; clone, FN50; Miltenyi
Biotec) and CD3-specific antibody (catalog no., 130-098-162; clone,
BW264/56; dilution, 1:15; Miltenyi Biotec) labelled with FITC. IgG1
antibodies were utilized as isotype controls. The samples were
examined using a flow cytometer (FACSCalibur; BD Biosciences).
Ratio of CD4+ and
CD8+ cells
The CD4 and CD8 lymphocyte count was analysed in
accordance with the technique described by Bapsy et al
(17). Briefly, 2–3 ml of peripheral
blood was incubated with anti-human CD3-PC5, CD4-FITC, CD8-PE and
CD16-FITC mAbs (BD Biosciences). Subsequent to staining the cells,
they were fixed with 1% paraformaldehyde and examination was
performed using FACSCalibur flow cytometer and CellQuest Pro
software (BD Biosciences). Lymphocytes are characterised by their
side and forward light scattering properties; therefore, the
analysis and acquisition gates were limited to the lymphocyte gate.
Cells that expressed CD markers were acquired and analysed in the
FL1 or FL2 logarithmic scale by using the set gates.
Delayed-type hypersensitivity
test
rMAGE3 + rSurvivin-pulsed DCs and unpulsed DCs were
administered intra-dermally into the forearm, at the time of T0 and
T4. Erythema >1.5 cm and skin induration 48 h after intradermal
injection were considered as positive delayed-type
hypersensitivity.
Response evaluation
Patients were followed-up by the primary physicians.
The follow-up included physical examination and routine history.
Chest X-rays or computed tomography (CT) scans were also obtained
for assessment depending on the signs and symptoms of tumour
recurrence or at regular intervals. Toxicity was graded according
to World Health Organisation criteria.
Statistical evaluation
Paired Student's t-test was used to examine the
data. P<0.05 was considered to indicate a statistically
significant difference. The correlation between two interval-scaled
variables was tested using Pearson's correlation, whereas the
correlation between two ordinal-scaled variables was tested using
Spearman's rank correlation. Non-parametric Wilcoxon rank sum test
was used to compare the immunological outcomes (e.g. ratio of
CD4/CD8 count in blood and IFN-γ release from macrophages) between
clinical responders (CD4/CD8 >1) and non-responders (IFN-γ
>1%). Kaplan-Meier survival analysis was performed using
GraphPad Prism software (GraphPad Software, San Diego, CA,
USA).
Results
As aforementioned, 16 patients were enrolled in the
present study between December 2013 and October 2014. A total of 19
patients were screened, of which 16 patients met the inclusion
criteria. Patient characteristics and clinical outcomes are
presented in Table II.
| Table II.Treatment characteristics and DC
vaccine-associated clinical events. |
Table II.
Treatment characteristics and DC
vaccine-associated clinical events.
| Patient code | Recurrence | Time to recurrence
from treatment, months | Time to recurrence
from DC immunotherapy, months | Survival from
treatment, months | Survival from DC
immunotherapy, months |
|---|
| 1XU | No | 22 | – | – | NA |
| 2LI | No | 12 | – | – | NA |
| 3XB | Yes | – | 7 | 12 | 7 |
| 4YU | No | 17 | – | – | NA |
| 5NC | No | 25 | – | 15 | 6 |
| 6QL | No | 5 | 3 | 16 | 12 |
| 7ZY | Yes | 15 | 3 | 12 | NA |
| 8XL | No | 20 | – | – | NA |
| 9YP | Yes | 14 | – | – | NA |
| 10LO | No | 17 | – | – | NA |
| 11XE | No | 13 | – | – | NA |
| 12NZ | No | 25 | – | – | NA |
| 13HE | No | 16 | – | – | NA |
| 14PX | No | – | – | – | NA |
| 15SZ | Yes | 11 | 16 | NA | NA |
| 16YU | Yes | – | 8 | 11 | NA |
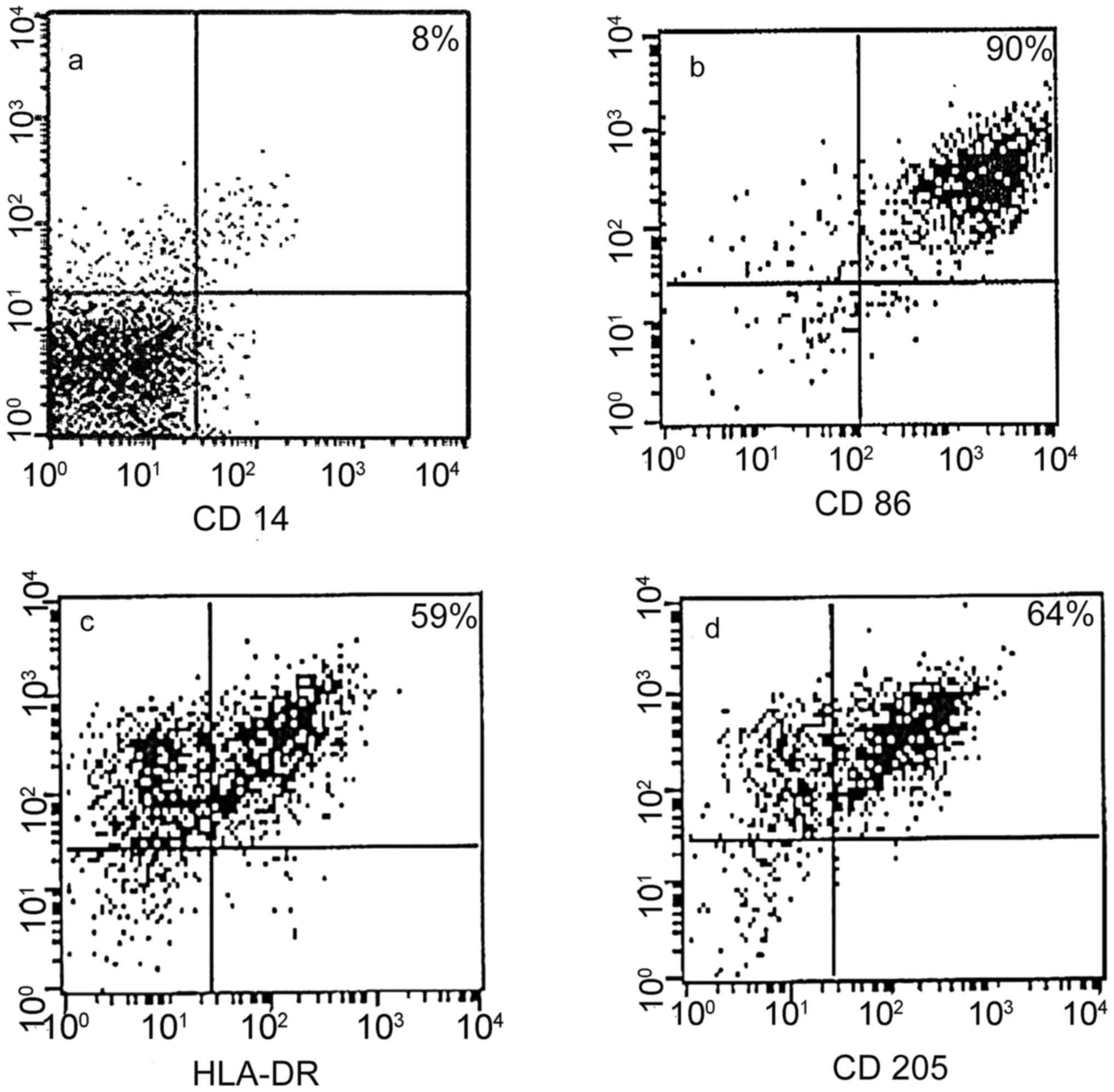
Phenotype of DCs
The final immunotherapy products did not express
CD14, and the majority of the cells expressed CD86 (90%), CD205
(60–75%) and HLA-DR (55–62%) (Fig.
1). However, with respect to cytokine secretion, antigen-pulsed
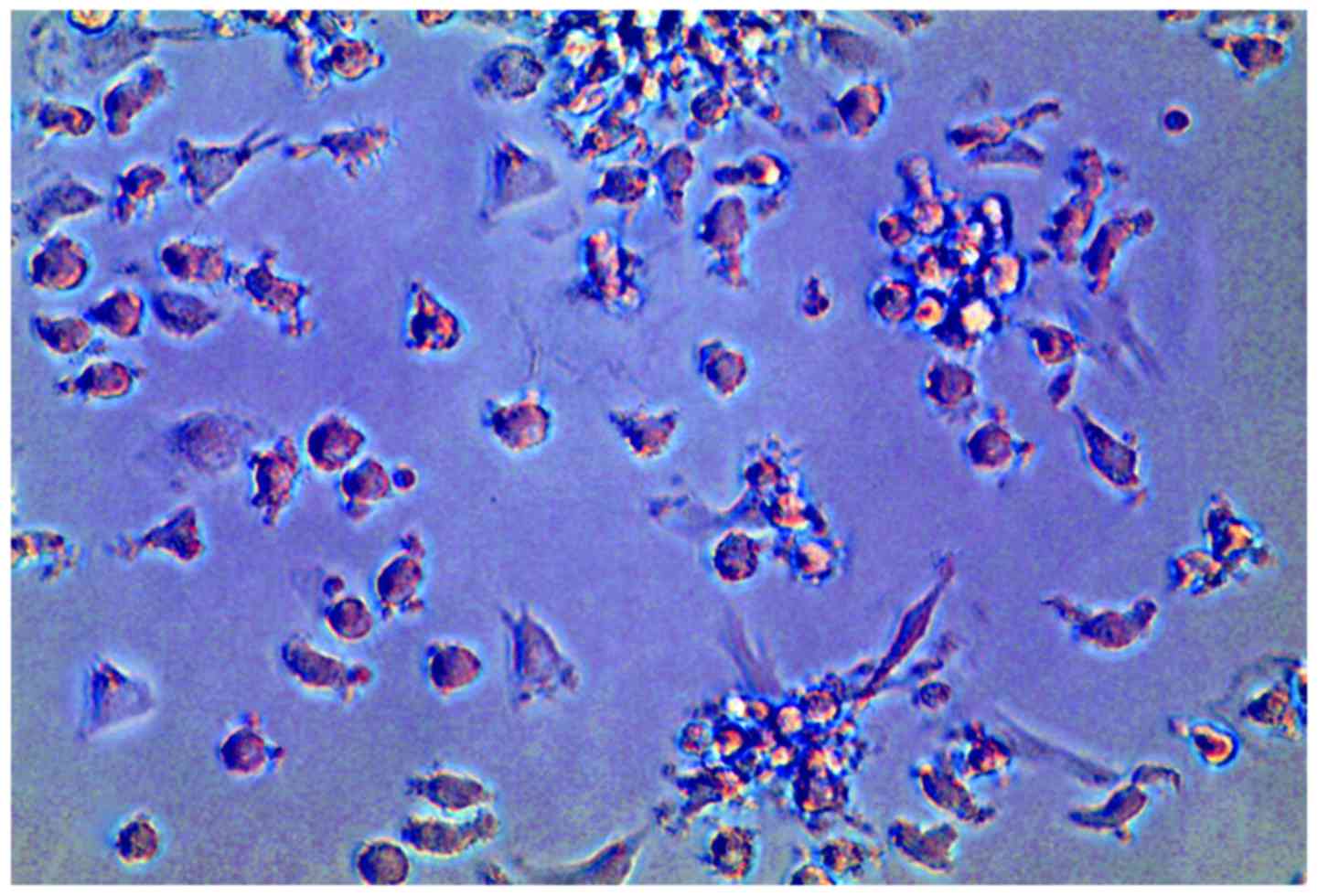
DC/T cell maturation factor-treated DCs appeared to be more mature
compared with naive DCs. Light microscopy of cells cultured for 8
days revealed predominantly mature DCs (Fig. 2).
Delayed-type hypersensitivity
test
DCs pulsed with rMAGE3 + rSurvivin were introduced
into the forearm intra-dermally to determine DTH reactivity. An
induration >1.5 cm in diameter was considered as a non-negative
DTH reaction. Subsequent to the first vaccination, every patient
showed a truly positive DTH reaction.
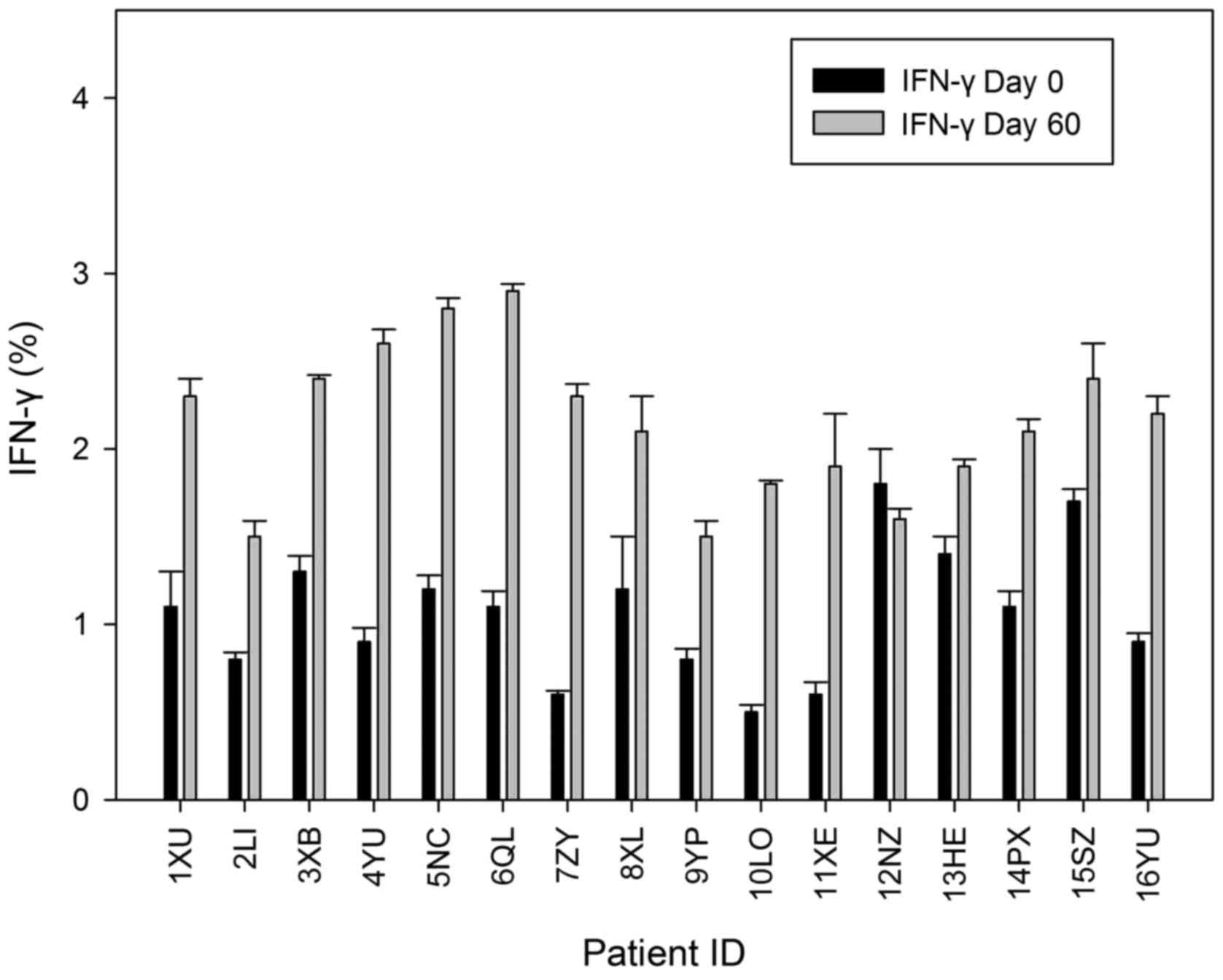
In vitro IFN-γ assay
The assay that was elaborated in order to identify
the intracellular IFN-γ production in the peripheral T cells was
utilized to identify the capability of the DC immunotherapy for the
progression of an immune response, specifically against tumour
cells. The present study used flow cytometry to assess the
production of IFN-γ in CD3+ lymphocytes in patients on
days 0 and 60. In CD3+ cells that were generated
subsequent to T4 (DC immunotherapy arm) and could not be
invigorated with ionomycin or PMA, it was observed that there was a
significantly increased level of IFN-γ expression compared with
cells obtained on day 0 (P=0.044). IFN-γ expression on day 60 was
significantly increased compared with day 0 (P=0.48) (Fig. 2).
CD4:CD8 levels
There was an increasing trend in the mean CD4:CD8
values between day 30 and day 60 (Fig.
3); however, the increase was not statistically significant
(P=0.150). In the majority of the patients, the basal values were
found to be <2.
Toxicity
In total, 32 DC injections were subcutaneously
administered to the thigh. All injections were well tolerated;
however, one incident of temporary exanthema was observed in one
patient (patient ID, 5NC). It was observed that the exanthema
vanished without any supplementary treatment. At the DC injection
region, 18.75% (3/16) of patients reported a small itching
induration. No patients showed any serious adverse events. Overall,
DC immunotherapy was found to be safe and well tolerated and only
incidence of grade 1 chills, fever and fatigue was observed.
Response evaluation
At least 12 months of follow up was performed for
all patients subsequent to primary immunisation, and the clinical
follow-up data are shown in Table
II. The disease recurred or progressed in 5 patients, 3 of
which succumbed to NSCLC 4–9 months after detection due to disease
progression to an advanced stage (stage IV). One NSCLC patient
(patient ID, DC10), who had stage I disease and had received
radiotherapy and chemotherapy, developed solitary brain metastasis
2 months subsequent to T0. Subsequently, 15 months after local
resection of stage IV disease, the patient showed no evidence of
NSCLC. One patient with stage IIIB unresectable disease developed
local progression 16 months subsequent to T0 and 19 months
subsequent to chemoradiation. A sixth patient (patient ID, DC16)
with stage IIIA disease that could be resected surgically and had
received multimodality therapy developed radiographically
persistent nodule 12 months subsequent to T0 and 21 months
subsequent to completion of treatment and was currently receiving
chemotherapy.
Discussion
The current study has validated the concept of
cellular immunotherapy using MODCs as a viable treatment option in
treating NSCLC. No patients demonstrated treatment-associated
haematological, hepatic, renal or neurological toxicity, or
autoimmune disease, indicating that the autologous DC immunotherapy
was safe. DCs also met the specifications for quality control
described by Sabado et al (18). Aggressive treatment of NSCLC has lead
to improved outcomes (19,20), and survival can further be increased
by expanding the scope of available therapeutic options for NSCLC
(21,22). Immunotherapy specifically targets
malignant cells and is an attractive systemic approach. Evidence
that autologous tumour immunotherapy expressing GM-CSF (GVAX)
elicits a durable clinical response in patients with NSCLC
indicates that it is possible to modulate the immune system to
benefit NSCLC patients (14).
Although it is questionable whether immunotherapy can adequately
and consistently treat such considerable diseases, efficient
immunotherapy can act as an adjuvant therapy for surgical
multimodality or medical therapy that shows definitive clinical
responses. The ultimate objective of the present study was to
identify the role of immunotherapy as an adjuvant therapy in the
treatment of stage I–IIIB NSCLC. Therefore, the main aim of the
current study was to identify the immunological response generated
by autologous DC immunotherapy in 16 patients with NSCLC. In total,
5 patients experienced disease recurrence or progression, of which
3 patients succumbed to disease progression. In addition, 3
patients experienced therapeutic efficacy. One patient, who had
stage IB disease, developed solitary brain metastasis 2 months
subsequent to vaccination (DC immunotherapy); however, following
surgical resection of stage IV disease, the patient survived for 15
months. Additionally, no disease progression was observed in 2
patients with stage III unresectable disease 23 months and 35
months after chemoradiation, respectively. One patient with
bronchoalveolar carcinoma, who had resected stage IIIB disease,
also remained tumour-free 19 months subsequent to vaccination and
28 months subsequent to surgical resection.
A positive DTH response against the TAAs used for
priming the DC was observed in all patients. Previous studies have
not shown a statistically significant increase in the release of
IFN-γ (17,18). However, the levels of IFN-γ released
by CD3+ cells in the present study support the
activation of the immune response by DC immunotherapy. In the
present study, MAGE3 and Survivin were used as TAAs for DC
immunotherapy and for generating a Th1 immune response; the use of
purified and defined MAGE3 and Survivin peptides to prime DCs has
already been demonstrated (23,24).
Therapeutic cancer immunotherapies have become a
reality (17). Initial failures have
increased knowledge of the immune response against tumours and
prompted the development of immunotherapies and immunotherapeutic
agents that are more potent and considerably less toxic than
chemotherapies or targeted therapies (6,10). Trials
and approval of the first DC immunotherapy in the US have shown
that activating the immune system with a therapeutic cancer
immunotherapy can provide clinical benefit to cancer patients for a
prolonged period (25).
Immunotherapies have been more successful in prostate cancer due to
the generally indolent progression of prostate cancer (25). In the present study, only patients
with advanced tumour stage were enrolled; however, the optimal
setting to apply DC immunotherapy may be minimal residual disease.
The foci of on-going and forthcoming studies are various aspects of
immunotherapy optimisation, antigen preparation and methods of
application (26). The current study
showed that DCs can be used in adoptive immunotherapy for the
treatment of NSCLC. However, if these promising results can be
confirmed in a larger patient population, then DC immunotherapy
based on the combination of rMAGE3 and rSurvivin may become a
sought after option for treating NSCLC. Several questions
associated with the manufacturing and quality of immunotherapy,
immune monitoring, patient selection and immunotherapy delivery
strategies need to be addressed. Future studies should address
prominent issues, including secretion of immunosuppressive
cytokines and mechanisms of tumour escape from immune surveillance,
through down-regulation of antigen and MHC expression.
References
|
1
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clinic Proc. 83:584–594.
2008. View Article : Google Scholar
|
|
2
|
Reck M: What future opportunities may
immuno-oncology provide for improving the treatment of patients
with lung cancer? Ann Oncol. 23 suppl 8:viii28–viii34. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Romani N, Reider D, Heuer M, Ebner S,
Kämpgen E, Eibl B, Niederwieser D and Schuler G: Generation of
mature dendritic cells from human blood. An improved method with
special regard to clinical applicability. J Immunol Methods.
196:137–151. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sallusto F and Lanzavecchia A: Efficient
presentation of soluble antigen by cultured human dendritic cells
is maintained by granulocyte/macrophage colony-stimulating factor
plus interleukin 4 and downregulated by tumor necrosis factor
alpha. J Exp Med. 179:1109–1118. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He L, Feng H, Raymond A, Kreeger M, Zeng
Y, Graner M, Whitesell L and Katsanis E: Dendritic-cell-peptide
immunization provides immunoprotection against bcr-abl-positive
leukemia in mice. Cancer Immunol Immunother. 50:31–40. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mayordomo JI, Zorina T, Storkus WJ,
Zitvogel L, Celluzzi C, Falo LD, Melief CJ, Ildstad ST, Kast WM,
Deleo AB, et al: Bone marrow-derived dendritic cells pulsed with
synthetic tumour peptides elicit protective and therapeutic
antitumour immunity. Nat Med. 1:1297–1302. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schuler G and Steinman RM: Dendritic cells
as adjuvants for immune-mediated resistance to tumors. J Exp Med.
186:1183–1187. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Banchereau J and Steinman RM: Dendritic
cells and the control of immunity. Nature. 392:245–252. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fong L, Hou Y, Rivas A, Benike C, Yuen A,
Fisher GA, Davis MM and Engleman EG: Altered peptide ligand
vaccination with Flt3 ligand expanded dendritic cells for tumor
immunotherapy. Proc Natl Acad Sci USA. 98:8809–8814. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nair SK, Hull S, Coleman D, Gilboa E,
Lyerly HK and Morse MA: Induction of carcinoembryonic antigen
(CEA)-specific cytotoxic T-lymphocyte responses in vitro using
autologous dendritic cells loaded with CEA peptide or CEA RNA in
patients with metastatic malignancies expressing CEA. Int J Cancer.
82:121–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Falleni M, Pellegrini C, Marchetti A,
Oprandi B, Buttitta F, Barassi F, Santambrogio L, Coggi G and
Bosari S: Survivin gene expression in early-stage non-small cell
lung cancer. J Pathol. 200:620–626. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weiser TS, Ohnmacht GA, Guo ZS, Fischette
MR, Chen GA, Hong JA, Nguyen DM and Schrump DS: Induction of MAGE-3
expression in lung and esophageal cancer cells. Ann Thorac Surg.
71:295–302. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
US Department of Health and Human
Services, National Institutes of Health, National Cancer Institute:
Common terminology criteria for adverse events. version, 4.0.
https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdfOctober.
2014
|
|
14
|
Hirschowitz EA, Foody T, Kryscio R,
Dickson L, Sturgill J and Yannelli J: Autologous dendritic cell
vaccines for non-small-cell lung cancer. J Clin Oncol.
22:2808–2815. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim D, Jeon C, Kim JH, Kim MS, Yoon CH,
Choi IS, Kim SH and Bae YS: Cytoplasmic transduction peptide (CTP):
New approach for the delivery of biomolecules into cytoplasm in
vitro and in vivo. Exp Cell Res. 312:1277–1288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kern F, Surel IP, Brock C, Freistedt B,
Radtke H, Scheffold A, Blasczyk R, Reinke P, Schneider-Mergener J,
Radbruch A, et al: T-cell epitope mapping by flow cytometry. Nat
Med. 4:975–978. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bapsy PP, Sharan B, Kumar C, Das RP,
Rangarajan B, Jain M, Attili Suresh VS, Subramanian S, Aggarwal S,
Srivastava M and Vaid A: Open-label, multi-center, non-randomized,
single-arm study to evaluate the safety and efficacy of dendritic
cell immunotherapy in patients with refractory solid malignancies,
on supportive care. Cytotherapy. 16:234–244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sabado RL, Miller E, Spadaccia M, Vengco
I, Hasan F and Bhardwaj N: Preparation of tumor antigen-loaded
mature dendritic cells for immunotherapy. J Vis Exp. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Spira A and Ettinger DS: Multidisciplinary
management of lung cancer. N Engl J Med. 350:379–392. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arriagada R, Bergman B, Dunant A, Le
Chevalier T, Pignon JP and Vansteenkiste J: International Adjuvant
Lung Cancer Trial Collaborative Group: Cisplatin-based adjuvant
chemotherapy in patients with completely resected non-small-cell
lung cancer. N Engl J Med. 350:351–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dy GK and Adjei AA: Novel targets for lung
cancer therapy: Part II. J Clin Oncol. 20:3016–3028. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dy GK and Adjei AA: Novel targets for lung
cancer therapy: Part I. J Clin Oncol. 20:2881–2894. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu X, Sun N, Dong Y, Li J, Liu Y, Ren Y,
Yang C, Zhang L, Zhou Y, Tong Z, et al: Anticancer effects of
adenovirus-mediated calreticulin and melanoma-associated antigen 3
expression on non-small cell lung cancer cells. Int
Immunopharmacol. 25:416–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hobo W, Strobbe L, Maas F, Fredrix H,
Greupink-Draaisma A, Esendam B, de Witte T, Preijers F, Levenga H,
van Rees B, et al: Immunogenicity of dendritic cells pulsed with
MAGE3, Survivin and B-cell maturation antigen mRNA for vaccination
of multiple myeloma patients. Cancer Immunol Immunother.
62:1381–1392. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheever MA and Higano CS: PROVENGE
(Sipuleucel-T) in prostate cancer: The first FDA-approved
therapeutic cancer vaccine. Clin Cancer Res. 17:3520–3526. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tuyaerts S, Aerts JL, Corthals J, Neyns B,
Heirman C, Breckpot K, Thielemans K and Bonehill A: Current
approaches in dendritic cell generation and future implications for
cancer immunotherapy. Cancer Immunol Immunother. 56:1513–1537.
2007. View Article : Google Scholar : PubMed/NCBI
|