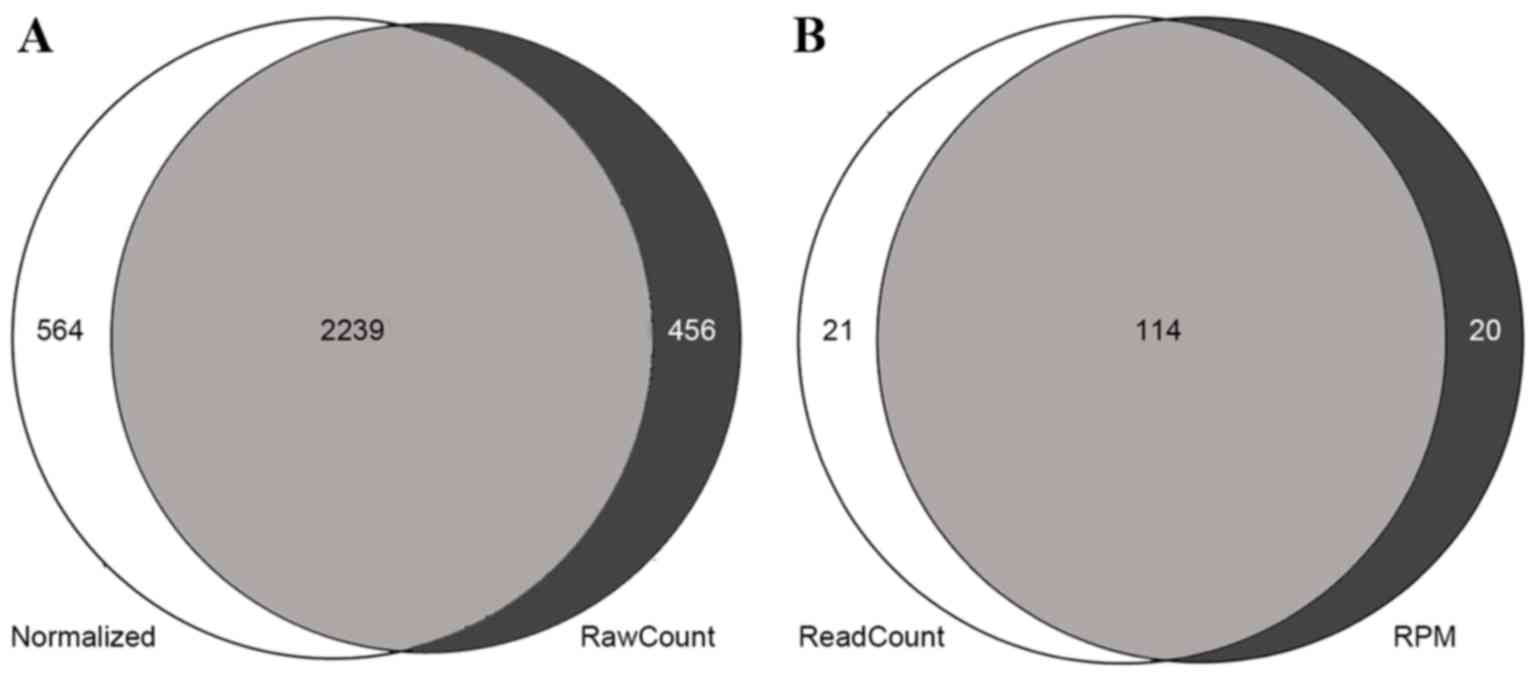
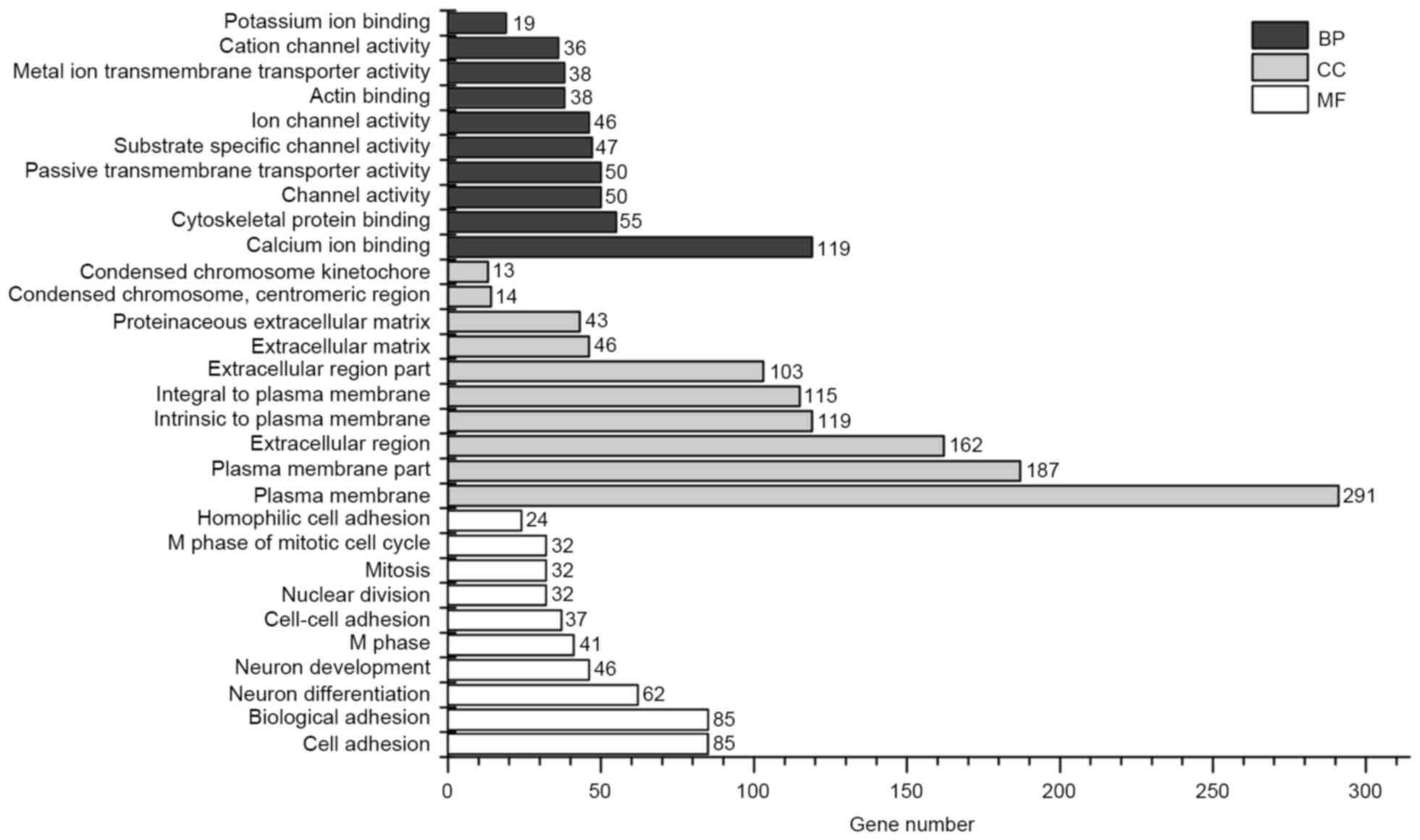
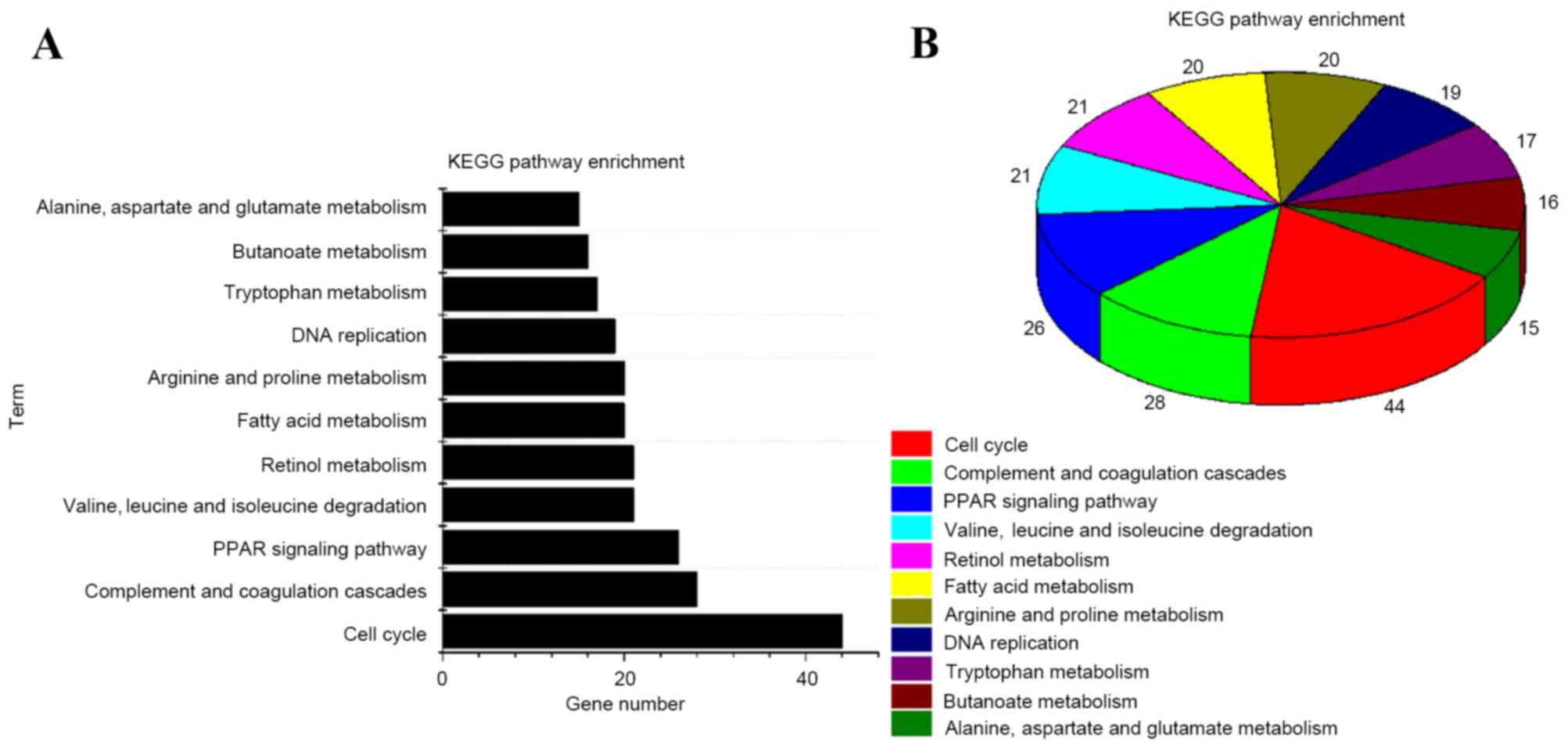
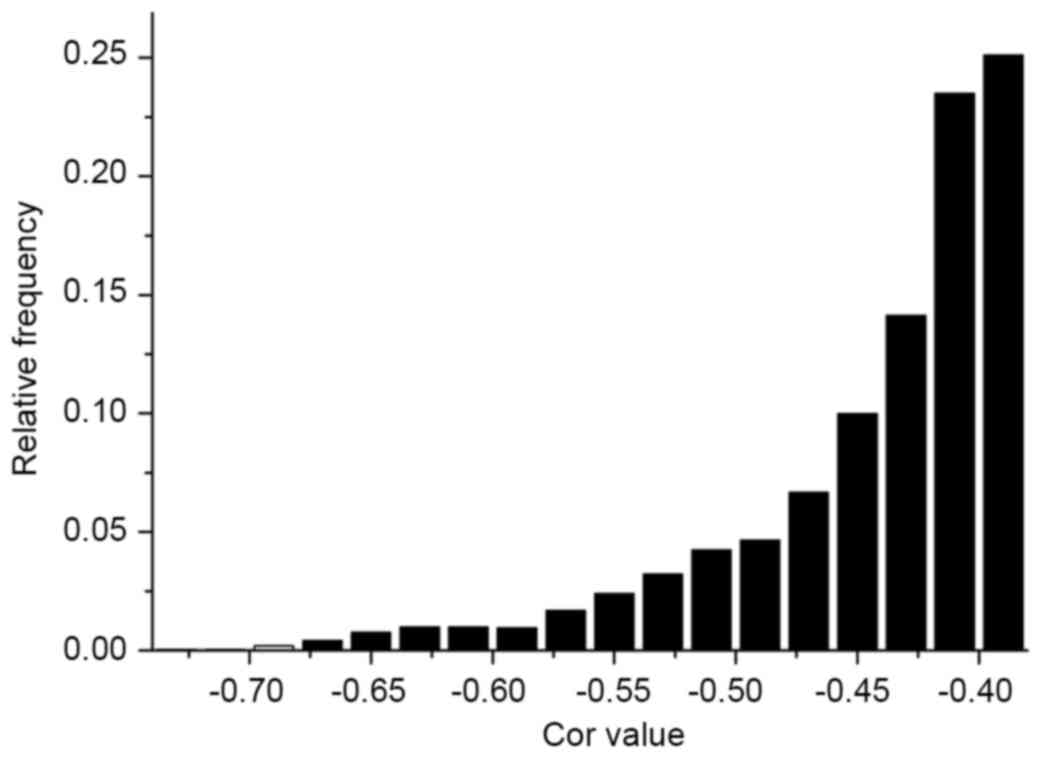
|
1
|
Lim L, Balakrishnan A, Huskey N, Jones KD,
Jodari M, Ng R, Song G, Riordan J, Anderton B, Cheung ST, et al:
MicroRNA-494 within an oncogenic microRNA megacluster regulates
G1/S transition in liver tumorigenesis through suppression of
mutated in colorectal cancer. Hepatology. 59:202–215. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li H, An J, Wu M, Zheng Q, Gui X, Li T, Pu
H and Lu D: LncRNA HOTAIR promotes human liver cancer stem cell
malignant growth through downregulation of SETD2. Oncotarget.
6:27847–27864. 2014.
|
|
3
|
Huang JL, Zheng L, Hu YW and Wang Q:
Characteristics of long non-coding RNA and its relation to
hepatocellular carcinoma. Carcinogenesis. 35:507–514. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu W and Kan X: Neural network cascade
optimizes microRNA biomarker selection for nasopharyngeal cancer
prognosis. PLoS One. 9:e1105372014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kota J, Chivukula RR, O'Donnell KA,
Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P,
Torbenson M, Clark KR, et al: Therapeutic microRNA delivery
suppresses tumorigenesis in a murine liver cancer model. Cell.
137:1005–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gutschner T and Diederichs S: The
hallmarks of cancer: A long non-coding RNA point of view. RNA Biol.
9:703–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen
X, Zhang Q, Yan G and Cui Q: LncRNA disease: A database for
long-non-coding RNA-associated diseases. Nucleic Acids Res.
41:D983–D986. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Geisler S and Coller J: RNA in unexpected
places: Long non-coding RNA functions in diverse cellular contexts.
Nat Rev Mol Cell Boil. 14:699–712. 2013. View Article : Google Scholar
|
|
9
|
Ellis BC, Graham LD and Molloy PL: CRNDE,
a long non-coding RNA responsive to insulin/IGF signaling,
regulates genes involved in central metabolism. Biochim Biophys
Acta. 1843:372–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Juan L, Wang G, Radovich M, Schneider BP,
Clare SE, Wang Y and Liu Y: Potential roles of microRNAs in
regulating long intergenic noncoding RNAs. BMC Med Genomics. 6
Suppl 1:S72013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grimson A, Farh KK, Johnston WK,
Garrett-Engele P, Lim LP and Bartel DP: MicroRNA targeting
specificity in mammals: Determinants beyond seed pairing. Mol Cell.
27:91–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The microRNA.org resource: Targets and expression.
Nucleic Acids Res. 36:D149–D153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X: miRDB: A microRNA target
prediction and functional annotation database with a wiki
interface. RNA. 14:1012–1017. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krek A, Grün D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky N: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Volders PJ, Helsens K, Wang X, Menten B,
Martens L, Gevaert K, Vandesompele J and Mestdagh P: LNCipedia: A
database for annotated human lncRNA transcript sequences and
structures. Nucleic Acids Res. 41:D246–D251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Volders PJ, Verheggen K, Menschaert G,
Vandepoele K, Martens L, Vandesompele J and Mestdagh P: An update
on LNCipedia: A database for annotated human lncRNA sequences.
Nucleic Acids Res. 43:4363–4364. 2014. View Article : Google Scholar
|
|
21
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T
and Yamanishi Y: KEGG for linking genomes to life and the
environment. Nucleic Acids Res. 36:D480–D484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Braun R, Finney R, Yan C, Chen QR, Hu Y,
Edmonson M, Meerzaman D and Buetow K: Discovery analysis of TCGA
data reveals association between germline genotype and survival in
ovarian cancer patients. PLoS One. 8:e550372013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim YK and Kim VN: Processing of intronic
microRNAs. EMBO J. 26:775–783. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ling H, Fabbri M and Calin GA: MicroRNAs
and other non-coding RNAs as targets for anticancer drug
development. Nat Rev Drug Discov. 12:847–865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang L, Wong CM, Ying Q, Fan DN, Huang S,
Ding J, Yao J, Yan M, Li J, Yao M, et al: MicroRNA-125b
suppressesed human liver cancer cell proliferation and metastasis
by directly targeting oncogene LIN28B2. Hepatology. 52:1731–1740.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu AM, Poon RT and Luk JM: MicroRNA-375
targets Hippo-signaling effector YAP in liver cancer and inhibits
tumor properties. Biochem Biophys Res Commun. 394:623–627. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He XX, Chang Y, Meng FY, Wang MY, Xie QH,
Tang F, Li PY, Song YH and Lin JS: MicroRNA-375 targets AEG-1 in
hepatocellular carcinoma and suppresses liver cancer cell growth in
vitro and in vivo. Oncogene. 31:3357–3369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He JH, Zhang JZ, Han ZP, Wang L, Lv YB and
Li YG: Reciprocal regulation of PCGEM1 and miR-145 promote
proliferation of LNCaP prostate cancer cells. J Exp Clin Cancer
Res. 33:722014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bayoumi AS, Sayed A, Broskova Z, Teoh JP,
Wilson J, Su H, Tang YL and Kim IM: Crosstalk between long
noncoding RNAs and MicroRNAs in health and disease. Int J Mol Sci.
17:3562016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He X, Tan X, Wang X, Jin H, Liu L, Ma L,
Yu H and Fan Z: C-Myc-activated long noncoding RNA CCAT1 promotes
colon cancer cell proliferation and invasion. Tumour Biol.
35:12181–12188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cesana M, Cacchiarelli D, Legnini I,
Santini T, Sthandier O, Chinappi M, Tramontano A and Bozzoni I: A
long noncoding RNA controls muscle differentiation by functioning
as a competing endogenous RNA. Cell. 147:358–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang K, Liu F, Zhou LY, Long B, Yuan SM,
Wang Y, Liu CY, Sun T, Zhang XJ and Li PF: The long noncoding RNA
CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res.
114:1377–1388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y,
Chen N, Sun F and Fan Q: CREB up-regulates long non-coding RNA,
HULC expression through interaction with microRNA-372 in liver
cancer. Nucleic Acids Res. 38:5366–5383. 2010. View Article : Google Scholar : PubMed/NCBI
|