Introduction
Bladder cancer is reported to be one of the four
most commonly occurring genitourinary tumors in China (1), and is one of the most expensive types of
cancer to treat due to the essential life-long surveillance
involving upper tract imaging, urinary cytology and cystoscopy
(2). The disease usually presents in
two distinct forms: Non-muscle-invasive tumors in clinical stages
Ta and T1 and muscle-invasive cancer occurring in clinical stages
T2-4 (3). As bladder cancer may
result from numerous processes with accumulation of genetic and
epigenetic changes, high-throughput technologies have been used to
generate numerous genetic and genomic datasets to uncover disease
causal genes and their actions involved in the initiation and
development of bladder cancer (4,5). A number
of studies have documented a strong link between microRNA
(miRNA/miR) function and cancer pathogenesis, including bladder
cancer (6–8).
miRNAs represent a class of naturally non-coding
RNAs (21–23 nucleotides in length), which act as
post-transcriptional silencing modulators of target genes either by
negatively regulating specific target mRNAs or by inhibiting target
protein synthesis (9,10). A previous study reported a potential
diagnostic role of miRNAs, as the miRNAs-profiling revealed a more
accurate diagnostic effect compared with the mRNAs-classifiers
(7). miRNAs have received increasing
attention in cancer genomic research and the novel oncogenic or
tumor suppression functions of miRNAs have been revealed in a
clinical study (6). Certain studies
have provided a detailed insight into regulatory networks involving
bladder cancer by integrated analysis of miRNA and mRNA data
(5,7).
Altered miRNA expression levels have been reported in bladder
cancer (3,11); however, the target genes of these
miRNAs have not yet been fully elucidated, particularly in bladder
cancer, and further analysis is required to elucidate the
subsequent processes.
Biological interpretation of large gene lists
derived from microarray analysis may require researchers to select
the most useful genes for further investigation. The Database for
Annotation, Visualization and Integration Discovery (DAVID) has
emerged as a publicly available high-throughput functional
annotation tool (12). Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis (13) and Gene Ontology (GO)
term analysis (14) are able to
identify the main functional and metabolic signaling pathways of
differentially expressed genes (DEGs). The present study used KEGG
pathways (http://www.genome.jp/kegg/pathway.html) and GO term
(http://geneontology.org/) analysis for enrichment
analysis.
In addition, genes with similar functions have been
reported to interact with each other closely, as presented in the
protein-protein interaction (PPI) network, which provides a global
picture used to understand molecular mechanisms and biological
processes of diseases, in particular to analyze cancer (15,16). The
PPI network has been used to systematically analyze and compare the
disease genes that would otherwise not be identified by single gene
analysis (17), in order to gain
insights into distinct topological features of cancer genes using
cancer-associated sub-networks. Hub nodes are known to be the most
important nodes in the PPI network, as the corresponding proteins
are important proteins in metabolic networks. In the present study,
disease dysregulated genes were mapped into the PPI network, where
proteins are represented as nodes and interactions as edges, in
order to understand key biological processes involved in bladder
cancer in a global sense.
miRNAs exert their function through translational
repression or degradation of mRNA targets, and of particular note
is the fact that no study has yet systematically analyzed the
significant mechanisms underlying miRNA targets contributing to
bladder cancer incidence. In the present study, numerous
computational methods were used to analyze microarray data of
miRNAs and mRNAs between bladder cancer and normal samples, in an
attempt to describe the variation of miRNA targets and the
potential mechanisms involved in the development of bladder
cancer.
Materials and methods
miRNA and mRNA expression profiling
data
Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), which is
established at the National Center for Biotechnology Information,
provides high-throughput gene expression data from the research
community worldwide. The microarray expression profiling data of
miRNAs and mRNAs in bladder urothelial carcinoma (‘case’) and
normal bladder tissue samples (‘normal’) were downloaded from the
GEO database (dataset GSE40355). Detailed messages were accessible
as follows: Microarray data of miRNAs included 8 normal
(GSM881439-GSM881446) and 8 case chips (GSM881455-GSM881462); and
microarray data of mRNAs included 8 normal (GSM991923-GSM991930)
and 8 case chips (GSM991939-GSM991946). The expression levels of
miRNAs and mRNAs were previously studied in these samples on two
platforms: GPL8227 Agilent-019118 Human miRNA Microarray 2.0 G4470B
(miRNA ID version) and GPL13497 Agilent-026652 Whole Human Genome
Microarray 4×44K v2 (Probe Name version) (both Agilent
Technologies, Inc., Santa Clara, CA, USA) (18).
Identification of differentially
expressed miRNAs and genes
The raw microarray expression data of miRNAs and
mRNAs downloaded as Series Matrix files from the GEO database were
mapped to the corresponding genes according to the SOFT formatted
family files from GEO database. For each sample, the expression
values of all probes for the same miRNA or mRNA were reduced to a
single value by calculating the average expression value. Bayesian
method was used for multiple comparisons under the control of false
discovery rate (FDR) (19). Limma
package in R software (version 3.24.15, https://www.bioconductor.org/packages/3.1/bioc/html/limma.html;
Bioconductor; Fred Hutchinson Cancer Reearch Center, Seattle, WA,
USA) (20), a linear regression
model, was used to identify differentially expressed miRNAs and
genes with the cut-off criteria of |log fold change (FC)|>1,
P<0.05 and FDR<0.01 (21).
Identification of disease dysregulated
genes
To identify the disease miRNAs associated with
bladder cancer, the selected differentially expressed miRNAs were
mapped to the online software, Human microRNA Disease Database
(HMDD; version 2.0; 2014.06.14 update; http://www.cuilab.cn/hmdd). As the functions of miRNAs
were dependent on its target genes (9), the target genes of disease miRNAs were
screened using target prediction programs. A single miRNA can
regulate numerous mRNAs and a single mRNA may be targeted by
different miRNAs. In order to ensure a high level of accuracy, five
target prediction programs, including miRanda http://www.microrna.org), MirTarget2 (http://mirdb.org/miRDB), PicTar (http://www.pictar.org/), PITA (http://genie.weizmann.ac.il/pubs/mir07/mir07_data.html)
and TargetScan (http://www.targetscan.org/) were used to screen
potential targets of miRNAs (22). To
decrease the number of false-positive results, putative target
genes predicted by at least 3 programs of the five were accepted as
the disease miRNAs targets.
Finally, to identify which differentially expressed
miRNAs targets were dysregulated in bladder cancer, the present
study selected the intersection of disease miRNA targets and DEGs
via the mRNAs chip data obtained above. The common genes were
considered as the disease dysregulated genes for further
analysis.
Enrichment analysis and
cancer-associated gene screening
To further understand gene functions and identifying
pathways closely associated with disease dysregulated genes, the
functional annotation of associated genes was performed using the
online software, DAVID. The present study used KEGG pathway and GO
term analysis for the following enrichment analysis of disease
dysregulated genes to identify the function and pathway alterations
involved in bladder cancer, with P<0.05 used as the cut-off
criterion. Furthermore, DAVID was used to perform OMIM_DISEASE
analysis, in order to distinguish cancer-associated genes from the
disease dysregulated genes.
PPI network and miRNA-gene regulatory
network
Highly complex cellular functions are the results of
tightly regulated interactions of groups of proteins encoded by
genes. Interactions between proteins possess a crucial role in
modern biology, thus a rapidly growing number of technologies have
been developed for the global charting of the PPI network. The
Search Tool for the Retrieval of Interacting Genes (STRING), an
online database resource (http://string-db.org/), provides uniquely
comprehensive information for assembling, evaluating and
disseminating PPI in a user-friendly manner (23).
In the present study, STRING was used to screen the
PPI of disease dysregulated genes, which was then visualized using
Cytoscape software (http://cytoscape.org/; version 3.2.1; National
Institute of General Medical Sciences, Seattle, WA, USA). The total
connectivity degree of each node in the network was calculated
using Cytoscape software and only the nodes with ≥30 degrees were
selected as the important nodes (Hubs) of PPI. Additionally, the
integrated miRNA-disease dysregulated genes regulatory network was
also constructed using Cytoscape software, followed by network
module screening using the threshold of P<0.05 by employing the
clusterONE plugins in Cytoscape (24). Subsequently, protein domain enrichment
analysis was performed on the screened modules using the InterPro
database (25).
Results
Differentially expressed clustered
miRNAs and genes
Based on the Limma package in R language, under
cut-off criteria of |logFC|>1, P<0.05 and FDR<0.01, 112
miRNAs and 2,290 genes were identified to be differentially
expressed in bladder cancer samples compared with the control
groups. There were 56 upregulated and 56 downregulated miRNAs; as
well as 1,444 upregulated and 846 downregulated DEGs.
Target screening of the differentially
expressed miRNAs
Firstly, differentially expressed miRNAs associated
with bladder cancer were screened from the 112 miRNAs using the
HMDD database and 71 bladder cancer-associated miRNAs were obtained
from this procedure, including hsa-miR-143, hsa-miR-205,
hsa-miR-21, hsa-miR-93, hsa-miR-145 and hsa-miR-143. Subsequently,
targets of the 71 disease miRNAs were identified using five target
prediction programs, including miRanda, MirTarget2, PicTar, PITA
and Target. For each target selection, the present study selected
the target genes of miRNAs coexisting in at least three of the
databases.
Identification of disease dysregulated
genes
A total of 573 disease dysregulated genes associated
with bladder cancer were identified by taking the intersection of
disease miRNA targets and 2,290 differentially expressed genes
corresponding to mRNA datasets.
Enrichment analysis of disease
dysregulated genes
To analyze the biological functions of disease
dysregulated genes in bladder cancer, GO and KEGG pathway
enrichment analyses were performed and the results are presented in
Tables I and II, respectively. Following combination of
the results from the GO and KEGG pathway analyses, it was revealed
that the most marked function and pathway involved in bladder
cancer were muscle organ development and vascular smooth muscle
contraction, respectively. Furthermore, DAVID was used to perform
OMIM_DISEASE analysis, and a set of six target genes [frizzled
class receptor 8 (FZD8), sacsin molecular chaperone (SACS), calcium
voltage-gated channel auxiliary subunit β2 (CACNB2), peptidase
inhibitor 15 (PI15) and catenin α2 (CTNNA2)] were identified to
closely associate with the functional term of disease of cancer,
suggesting a potential association with bladder cancer.
 | Table I.Top five enriched GO terms. |
Table I.
Top five enriched GO terms.
| Category | Term | Count | P-value |
|---|
| GOTERM_BP_FAT | GO:0007517~muscle
organ development | 34 |
3.83×10−14 |
| GOTERM_BP_FAT | GO:0042692~muscle
cell differentiation | 22 |
2.54×10−10 |
| GOTERM_BP_FAT | GO:0014706~striated
muscle tissue development | 21 |
1.23×10−9 |
| GOTERM_BP_FAT | GO:0060537~muscle
tissue development | 21 |
3.00×10−9 |
| GOTERM_BP_FAT |
GO:0007167~enzyme-linked receptor protein
signaling pathway | 35 |
5.26×10−9 |
| Table II.Top five enriched KEGG pathways. |
Table II.
Top five enriched KEGG pathways.
| Category | Term | Count | P-value |
|---|
| KEGG_PATHWAY | hsa04270: Vascular
smooth muscle contraction | 15 |
2.45×10−5 |
| KEGG_PATHWAY | hsa04360: Axon
guidance | 15 |
1.19×10−4 |
| KEGG_PATHWAY | hsa04510: Focal
adhesion | 19 |
1.61×10−4 |
| KEGG_PATHWAY | hsa04310: Wnt
signaling pathway | 15 |
6.22×10−4 |
| KEGG_PATHWAY | hsa04020: Calcium
signaling pathway | 15 |
2.74×10−3 |
PPI network and integrated
miRNA-target network construction
In order to gain further insights into the changes
of biological pathways in bladder cancer, the present study used
Cytoscape to identify the PPI of disease dysregulated genes
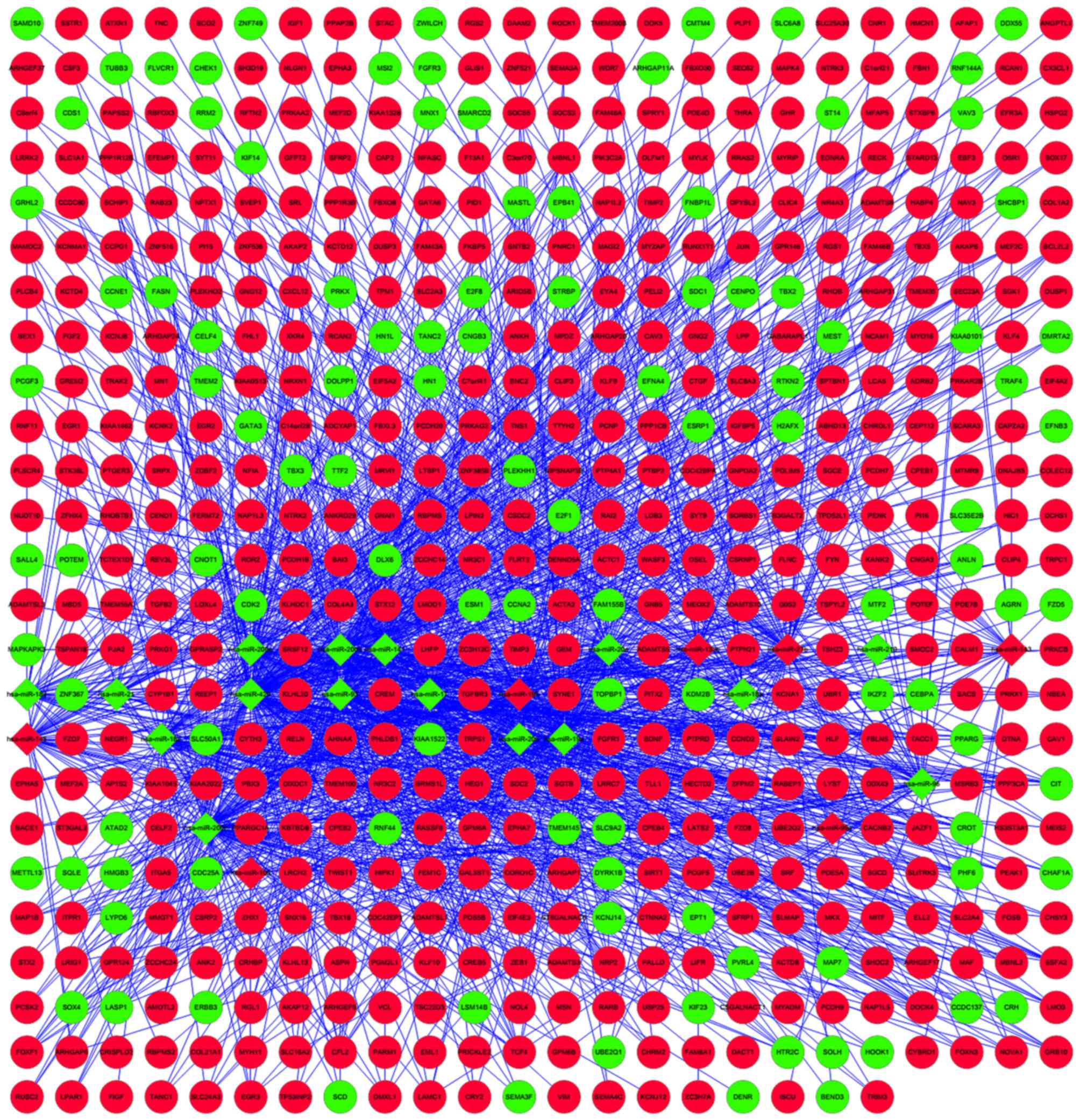
(Fig. 1). The upregulated calmodulin
1 (CALM1), jun proto-oncogene (JUN) and insulin-like factor 1
(IGF1) were key hub nodes in the PPI network. Cytoscape was also
used to perform regulatory network construction of disease
dysregulated genes and the corresponding miRNAs, as shown in
Fig. 2.
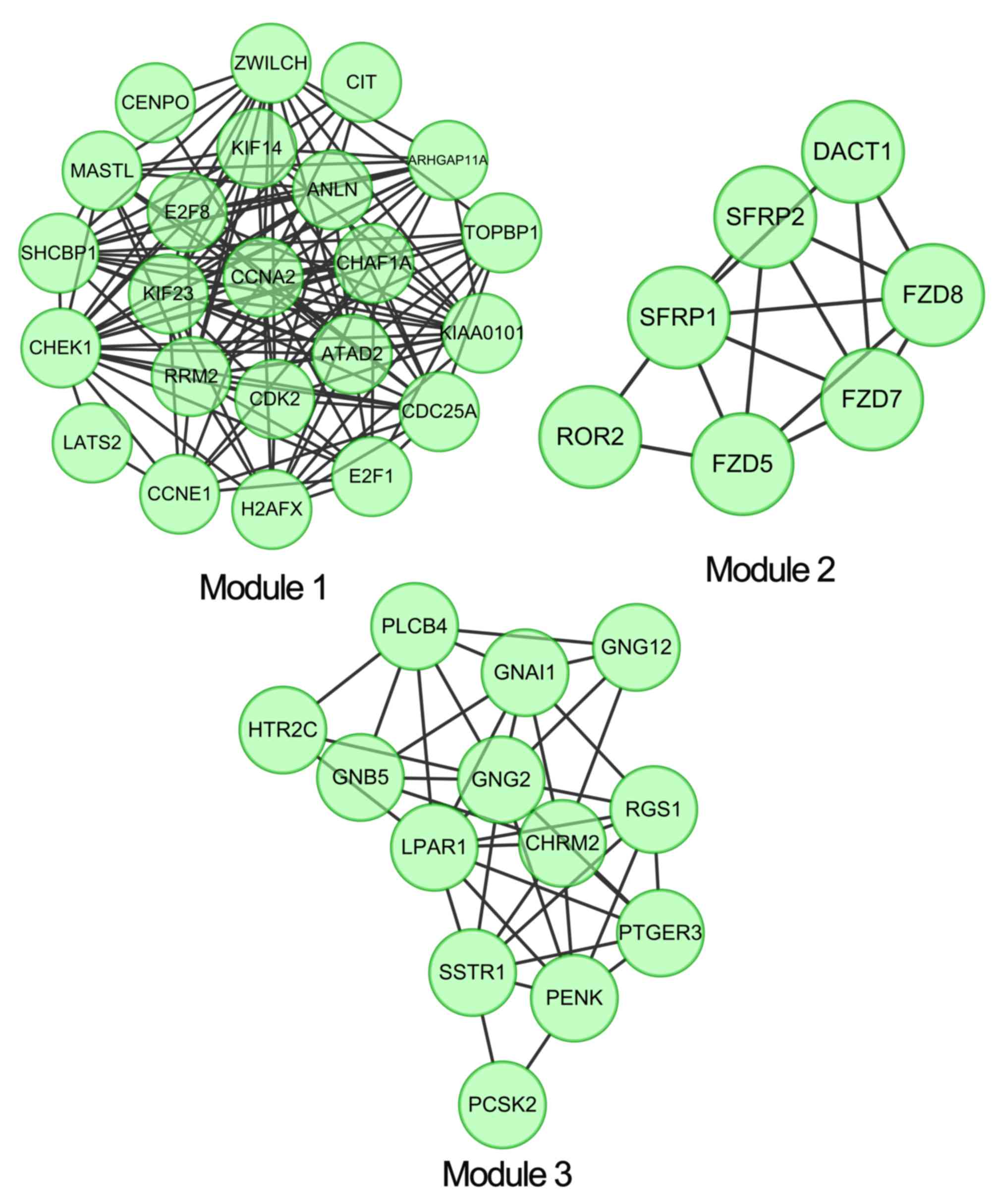
Module screening and subsequent
protein domain enrichment analysis
ClusterONE plugins were used to perform module
clustering analysis in the PPI network, with a criterion of
P<0.05. A total of three clustering module were identified:
Module 1 (density, 0.561; quality, 0.732; P=1.582×10−7),
module 2 (density, 0.714; quality, 0.625; P=0.002) and module 3
(density, 0.526; quality, 0.512; P=0.004) (Fig. 3). The protein domain enrichment
analysis revealed that there were no significant enrichment results
in module 2 and module 3, whereas in module 1 the contained disease
dysregulated genes, including SHC SH2-domain binding protein 1
(SHCBP1), were mainly enriched at IPR008271: Serine/threonine
protein kinase active site (Table
III).
| Table III.Protein Domain enrichment analysis on
module 1 using the InterPro database. |
Table III.
Protein Domain enrichment analysis on
module 1 using the InterPro database.
| Category | Term | Count | P-value |
|---|
| INTERPRO | IPR008271:
Serine/threonine protein kinase, active site | 5 |
7.42×10−4 |
| INTERPRO | IPR017442:
Serine/threonine protein kinase-related | 5 |
7.82×10−4 |
| INTERPRO | IPR017441: Protein
kinase, ATP binding site | 5 |
1.88×10−3 |
| INTERPRO | IPR000961:
AGC-kinase, C-terminal | 3 |
1.96×10−3 |
| INTERPRO | IPR000719: Protein
kinase, core | 5 |
2.22×10−3 |
| INTERPRO | IPR002290:
Serine/threonine protein kinase | 4 |
3.48×10−3 |
| INTERPRO | IPR015633: E2F
Family | 2 |
9.57×10−3 |
| INTERPRO | IPR003316:
Transcription factor E2F/dimerization partner | 2 |
1.31×10−2 |
| INTERPRO | IPR014400: Cyclin,
A/B/D/E | 2 |
1.43×10−2 |
| INTERPRO | IPR004367: Cyclin,
C-terminal | 2 |
1.67×10−2 |
| INTERPRO | IPR006671: Cyclin,
N-terminal | 2 |
3.89×10−2 |
| INTERPRO | IPR013763:
Cyclin-related | 2 |
4.35×10−2 |
| bINTERPRO | IPR006670:
Cyclin | 2 |
4.70×10−2 |
| INTERPRO | IPR017892: Protein
kinase, C-terminal | 2 |
4.70×10−2 |
| INTERPRO | IPR001752: Kinesin,
motor region | 2 |
4.93×10−2 |
| INTERPRO | IPR019821: Kinesin,
motor region, conserved site | 2 |
4.93×10−2 |
Discussion
Recently, emerging bioinformatic methods have
accelerated the progress made to identify the mechanisms involved
in bladder cancer development at the molecular level. miRNAs were
reported to regulate the activity of ~30% of all protein-coding
genes in the human genome (26),
whereas the deregulation of miRNAs and their targeted mRNAs may
serve an important function in the development of cancer. It was
previously revealed that aberrantly expressed miRNAs served
significant roles in the initiation, development and metastasis of
bladder cancer (3,27). Previous studies have identified the
large set of differentially expressed miRNAs between bladder cancer
and normal samples (11,27,28). The
present study further analyzed the expression profile data of
miRNAs and mRNAs, and identified a total of 573 differentially
expressed targets of the differentially expressed miRNAs in bladder
cancer. These genes provided a global perspective for understanding
the mechanism underlying bladder tumorigenesis. In addition, the
results of functional enrichment analysis indicated that the genes
were most significantly associated with the GO term of muscle organ
development and vascular smooth muscle contraction pathway. Genes,
including FZD8, EYA transcriptional coactivator and phosphatase 4
(EYA4), SACS, CACNB2, PI15 and CTNNA2 in the PPI network, were
demonstrated to be potentially associated with bladder cancer by
OMIM_DISEASE analysis. Furthermore, CALM1, JUN and IGF1 were
important hub nodes in the PPI network with high degrees of
connectivity.
Previous evidence suggested that there were two
distinct and overlapping stages of bladder cancer:
Non-muscle-invasive and progressive muscle-invasive tumors
(3). GO term and KEGG pathway
enrichment analysis of disease dysregulated genes revealed that
muscle organ development and vascular smooth muscle contraction,
respectively, were involved in bladder cancer, which suggested a
vital role of normal muscle function in preventing bladder cancer
progression. On the molecular level, a previous study reported the
participation of tumor-initiating cells, i.e., cancer stem cells,
in urothelial cancer (29). FZD8,
EYA4, SACS, CACNB2, PI15 and CTNNA2 were demonstrated to be the
cancer-associated target genes of differential miRNAs. Tumor
suppression miRNAs targeting FZDs were reported to participate in
the suppression of cell growth and metastasis via the Wnt signaling
pathway, which served important roles in tumorigenesis (30). EYA4, PI15 and CTNNA2 functioned as
tumor suppressors in the tumorigenesis of Barrett's esophageal
cancer (31), glioblastoma (32) and laryngeal carcinomas (33), respectively. CACNB2 was downregulated
in breast cancer cells, thereby causing poor signaling by
negatively affecting the calcium metabolism (34). SACS targeted by miR-223 was reported
to be upregulated in urothelial carcinoma in comparison to that in
the normal mucosa (11). Notably, in
the present study, the expression levels of 6 miRNA targets were
revealed to be markedly increased in bladder cancer compared with
those in the normal samples, which implied the potential
involvement in bladder cancer development.
In order to further understand the oncogenic
mechanisms underlying the screened miRNA targets involved in
bladder cancer, the present study performed PPI analysis and
identified three hub nodes: CALM1, JUN and IGF1. PPI networks are
typically scale-free, in which hubs are ‘highly connected’ and tend
to correspond to essential biological genes (35). IGF signaling, possessing an important
effect on cell proliferation and differentiation, was implicated in
numerous types of cancer and was thus considered to be a
therapeutic target in cancer treatment (36). Dysregulated miRNAs targets were
demonstrated to be involved in IGF1R signaling, as well as in its
downstream antipoptotic signaling pathways, including
phophoinisitide-3 kinase (PI3K)/protein kinase B (Akt)/mechanistic
target of rapamycin (mTOR), in bladder urothelial carcinoma
(37). Notably, the activation of the
PI3K/Akt/mTOR signaling pathway was reported to be a candidate
driver of the muscle-invasive phenotype of bladder cancer (38). In the present study, the upregulation
of IGF1 suggested possible active IGF1R signaling and subsequent
cascade dysregulation in bladder cancer. CALM, a ubiquitous
Ca2+ receptor protein interacting with hundreds of
different target proteins, revealed upregulated expression levels
in numerous tumor cells in comparison with that in cells from
normal tissues. CALM antagonists may inhibit tumor cells growth of
different origins by affecting the CALM-dependent mechanisms
(39). JUN, which is important for
cell proliferation, survival and apoptosis, was reported to be a
crucial contributing factor for tumorigenesis due to its
overexpression in numerous types of human cancer (40). The results of the present study
revealed that the increased expression levels of IGF1, CALM1 and
JUN in bladder cancer may be potentially effective therapeutic
targets for bladder cancer treatment.
In addition, the serine/threonine protein kinase
active site was revealed to be enriched in genes contained in
module 1, which was extracted from the PPI network. The human
protein kinase family, consisting of 518 genes, was classified as
protein-serine/threonine kinases, protein-tyrosine kinases and
tyrosine-kinase-like proteins based on the nature of the
phosphorylated-hydroxy group (41,42). The
protein-serine/threonine kinases that participated in the
RAS/RAF/MEK/extracellular signal-regulated kinase (ERK) signaling
pathway were reported to be associated with retroviral oncogenes
and were thereby attractive cancer drug targets (43–45).
SHCBP1, a dysregulated gene in module 1, was previously
demonstrated to regulate the expression of activated ERK1/2 and
thus be necessary for the RAS/MEK/ERK signaling pathway (46). In the present study, the dysregulation
of the serine/threonine protein kinase presented in module 1 may
suggest abnormal RAS/MEK/ERK transduction signaling involved in
bladder cancer progression.
In summary, the findings of the present study
suggested the potential therapeutic ability of the IGF signaling
pathway and RAS/MEK/ERK transduction signaling in bladder cancer,
as well as bladder cancer-associated oncogenes, including FZD8,
EYA4, SACS, CACNB2, PI15 and CTNNA2. Notably, CALM1, JUN and IGF1,
which were identified as hubs in the PPI network, may be important
therapeutic targets for bladder cancer intervention. However,
further research, both epidemiological and mechanistic, is required
to further validate the results and clarify the underlying
molecular mechanisms.
Acknowledgements
The present study was supported by the Special Fund
for Medical Service of Jilin Finance Department (grant no.
SCZSY201507).
Glossary
Abbreviations
Abbreviations:
|
HMDD
|
Human microRNA Disease Database
|
|
DAVID
|
Database for Annotation, Visualization
and Integration Discovery
|
|
PPI
|
protein-protein interaction
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GO
|
Gene Ontology
|
|
GEO
|
Gene Expression Omnibus
|
|
FDR
|
false discovery rate
|
|
FC
|
fold-change
|
|
DEGs
|
differentially expressed genes
|
|
STRING
|
Search Tool for the Retrieval of
Interacting Genes
|
|
CALM
|
calmodulin
|
References
|
1
|
Fangliu G and Yuli L: Changing status of
genitourinary cancer in recent 50 years. Chin J Urol. 2:88–90.
2002.
|
|
2
|
Gaston KE and Grossman HB: Proteomic
assays for the detection of urothelial cancer. Methods Mol Biol.
641:303–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dyrskjøt L, Ostenfeld MS, Bramsen JB,
Silahtaroglu AN, Lamy P, Ramanathan R, Fristrup N, Jensen JL,
Andersen CL, Zieger K, et al: Genomic profiling of microRNAs in
bladder cancer: miR-129 is associated with poor outcome and
promotes cell death in vitro. Cancer Res. 69:4851–4860. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dyrskjot L, Zieger K and Orntoft TF:
Recent advances in high-throughput molecular marker identification
for superficial and invasive bladder cancers. Front Biosci.
12:2063–2073. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fendler A, Stephan C, Yousef GM and Jung
K: MicroRNAs as regulators of signal transduction in urological
tumors. Clin Chem. 57:954–968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu W, Sun M, Zou GM and Chen J: MicroRNA
and cancer: Current status and prospective. Int J Cancer.
120:953–960. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X, Chen J, Hu X, Huang Y, Li Z, Zhou L,
Tian Z, Ma H, Wu Z, Chen M, et al: Comparative mRNA and microRNA
expression profiling of three genitourinary cancers reveals common
hallmarks and cancer-specific molecular events. PLoS One.
6:e225702011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo AY, Sun J, Jia P and Zhao Z: A novel
microRNA and transcription factor mediated regulatory network in
schizophrenia. BMC Syst Biol. 4:102010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gottardo F, Liu CG, Ferracin M, Calin GA,
Fassan M, Bassi P, Sevignani C, Byrne D, Negrini M, Pagano F, et
al: Micro-RNA profiling in kidney and bladder cancers. Urol Oncol.
25:387–392. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al: The gene ontology (GO) database and informatics resource.
Nucleic Acids Res. 32:(Database Issue). D258–D261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kar G, Gursoy A and Keskin O: Human cancer
protein-protein interaction network: A structural perspective. PLoS
Comput Biol. 5:e10006012009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song J and Singh M: How and when should
interactome-derived clusters be used to predict functional modules
and protein function? Bioinformatics. 25:3143–3150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia P, Kao CF, Kuo PH and Zhao Z: A
comprehensive network and pathway analysis of candidate genes in
major depressive disorder. BMC Syst Biol. 5 Suppl 3:S122011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hecker N, Stephan C, Mollenkopf HJ, Jung
K, Preissner R and Meyer HA: A new algorithm for integrated
analysis of miRNA-mRNA interactions based on individual
classification reveals insights into bladder cancer. PLoS One.
8:e645432013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Efron B and Tibshirani R: Empirical bayes
methods and false discovery rates for microarrays. Genet Epidemiol.
23:70–86. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Polpitiya AD, Qian WJ, Jaitly N, Petyuk
VA, Adkins JN, Camp DG II, Anderson GA and Smith RD: DAnTE: A
statistical tool for quantitative analysis of -omics data.
Bioinformatics. 24:1556–1558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saito R, Smoot ME, Ono K, Ruscheinski J,
Wang PL, Lotia S, Pico AR, Bader GD and Ideker T: A travel guide to
Cytoscape plugins. Nat Methods. 9:1069–1076. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:(Database Issue). D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9. 1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
4137:(Database Issue). D808–D815. 2013.
|
|
24
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Apweiler R, Attwood TK, Bairoch A, Bateman
A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD,
et al: The InterPro database, an integrated documentation resource
for protein families, domains and functional sites. Nucleic Acids
Res. 29:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Itesako T, Seki N, Yoshino H, Chiyomaru T,
Yamasaki T, Hidaka H, Yonezawa T, Nohata N, Kinoshita T, Nakagawa M
and Enokida H: The MicroRNA expression signature of bladder cancer
by deep sequencing: The functional significance of the miR-195/497
cluster. PLoS One. 9:e843112014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han Y, Chen J, Zhao X, Liang C, Wang Y,
Sun L, Jiang Z, Zhang Z, Yang R, Chen J, et al: MicroRNA expression
signatures of bladder cancer revealed by deep sequencing. PLoS One.
6:e182862011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McConkey DJ, Lee S, Choi W, Tran M,
Majewski T, Lee S, Siefker-Radtke A, Dinney C and Czerniak B:
Molecular genetics of bladder cancer: Emerging mechanisms of tumor
initiation and progression. Urol Oncol. 28:429–440. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ueno K, Hirata H, Hinoda Y and Dahiya R:
Frizzled homolog proteins, microRNAs and Wnt signaling in cancer.
Int J Cancer. 132:1731–1740. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zou H, Osborn NK, Harrington JJ, Klatt KK,
Molina JR, Burgart LJ and Ahlquist DA: Frequent methylation of eyes
absent 4 gene in Barrett's esophagus and esophageal adenocarcinoma.
Cancer Epidemiol Biomarkers Prev. 14:830–834. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gibbs GM, Roelants K and O'Bryan MK: The
CAP superfamily: Cysteine-rich secretory proteins, antigen 5, and
pathogenesis-related 1 proteins-roles in reproduction, cancer, and
immune defense. Endocr Rev. 29:865–897. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fanjul-Fernández M, Quesada V, Cabanillas
R, Cadiñanos J, Fontanil T, Obaya A, Ramsay AJ, Llorente JL,
Astudillo A, Cal S and López-Otín C: Cell-cell adhesion genes
CTNNA2 and CTNNA3 are tumour suppressors frequently mutated in
laryngeal carcinomas. Nat Commun. 4:25312013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rai A, Menon AV and Jalan S: Randomness
and preserved patterns in cancer network. Sci Rep. 4:63682014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
He X and Zhang J: Why do hubs tend to be
essential in protein networks? PLoS Genet. 2:e882006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tognon CE and Sorensen PH: Targeting the
insulin-like growth factor 1 receptor (IGF1R) signaling pathway for
cancer therapy. Expert Opin Ther Targets. 16:33–48. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tsai TF, Lin YC, Chen HE, Chou KY, Lin JF
and Hwang TI: Involvement of the insulin-like growth factor I
receptor and its downstream antiapoptotic signaling pathway is
revealed by dysregulated microRNAs in bladder carcinoma. Urological
Sci. 25:58–54. 2014. View Article : Google Scholar
|
|
38
|
Puzio-Kuter AM, Castillo-Martin M, Kinkade
CW, Wang X, Shen TH, Matos T, Shen MM, Cordon-Cardo C and
Abate-Shen C: Inactivation of p53 and Pten promotes invasive
bladder cancer. Genes Dev. 23:675–680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Berchtold MW and Villalobo A: The many
faces of calmodulin in cell proliferation, programmed cell death,
autophagy, and cancer. Biochim Biophys Acta. 1843:398–435. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lopez-Bergami P, Lau E and Ronai Z:
Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat
Rev Cancer. 10:65–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roskoski R Jr: RAF
protein-serine/threonine kinases: Structure and regulation. Biochem
Biophys Res Commun. 399:313–317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zebisch A and Troppmair J: Back to the
roots: The remarkable RAF oncogene story. Cell Mol Life Sci.
63:1314–1330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brault L, Gasser C, Bracher F, Huber K,
Knapp S and Schwaller J: PIM serine/threonine kinases in the
pathogenesis and therapy of hematologic malignancies and solid
cancers. Haematologica. 95:1004–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tao HC, Wang HX, Dai M, Gu CY, Wang Q, Han
ZG and Cai B: Targeting SHCBP1 inhibits cell proliferation in human
hepatocellular carcinoma cells. Asian Pac J Cancer Prev.
14:5645–5650. 2013. View Article : Google Scholar : PubMed/NCBI
|