Introduction
Cervical cancer is the third most death-related
cancer in women worldwide and a consequence of persistent infection
with high-risk human papillomaviruses. Neoplastic progression to
cancer takes years or decades and develops from low-grade cervical
intraepithelial neoplasia (CIN1) through high-grade lesions, CIN2
and CIN3 (carcinoma in situ) (1). Cervical cancer treatment depends on FIGO
tumor stages and includes surgery, chemo-, radio- or
chemoradiotherapy. For more than 50 years, radiation therapy was
the standard treatment for patients with locally advanced cervical
carcinoma but patients with advanced stage >IIB disease were
cured only in 35–45% of cases with radiation therapy alone
(2–4).
According to the European clinical guidelines since 1999 locally
advanced cervical carcinomas (FIGO>IIB) are treated with
simultaneous cisplatin-based chemoradiotherapy (CCRT) (5). CCRT has become the standard treatment
for locally advanced cervical carcinoma in North America and Europe
(5) and several studies have
demonstrated a 40–60% reduction in the relative risk of recurrence
and a 30–50% reduction of the risk of death with CCRT (6–8).
Nevertheless, resistance to non-surgical therapies is still a major
challenge (9). For patients who do
not respond to standard therapies, new strategies are needed.
We recently showed that cervical cancer cells can be
sensitized for chemotherapeutic drug induced cell death. We found
that pre-treatment of cervical carcinoma cells with Oncostatin M
(OSM) resulted in enhanced responsiveness of the cells to
chemotherapeutic drugs (10). OSM is
a member of the IL6-type cytokine family (11) and binds to the OSM receptor-b which
then associates with the receptor chain gp130. The recruitment of
Janus kinases leads to subsequent signal transducer and activator
of transcription 3 (STAT3)-phosphorylation at tyrosine 705
(12). We clarified the molecular
mechanism responsible for cell death sensitization, which was
dependent on the STAT3/IRF1 signaling pathway. This was unexpected
because in cervical cancer patients in situ the STAT3
activation is weak or absent (10).
This is in contrast to other malignancies, where STAT3 is
constitutively active and is a considered anti-apoptotic factor
(13–15).
Because CCRT is more frequently applied than
neoadjuvant chemotherapy, we were interested in the impact of OSM
pre-treatment on the responsiveness of cervical cancer cell to both
irradiation and chemoradiotherapy in this study. We found varying
sensitivities or even resistance of different cervical cancer cells
toward irradiation alone. Notably, OSM pre-treatment sensitized all
tested cervical cancer cells, including the irradiation resistant
cells, for CCRT-induced cell death.
Materials and methods
Cells and cell culture
HPV16-positive CaSki [ATCC CRL-1550; (16)] or HPV18-positive cervical carcinoma
cell lines SW756 [ATCC CRL-10302; (17)] and HeLa [ATCC CCL-2; (18)] were obtained from M. von
Knebel-Doeberitz (Heidelberg, Germany) before 2000. Cells were
authenticated by qRT-PCR for HPV16 or HPV18 E6 and E7 expression.
Cells were cultured at a density of 1×106 in DMEM
supplemented with 10% heat-inactivated fetal calf serum (FCS), 100
U/ml penicillin, 0.1 mg/ml streptomycin, 1 mM sodium pyruvate and 2
mM L-alanyl-L-glutamin (all from PAA, Pasching, Austria). The more
recently generated cervical cancer cells 808 (HPV18-positive) and
879 (HPV16-positive) were obtained from P. L. Stern, cultured as
previously described (19) and last
tested by short tandem repeat profiling in 2014. All cells were
tested for mycoplasma infection once per month.
Plasmids and transfections
The vectors pCAGGS and pCAGGS-STAT3F were kindly
provided by Dr. K. Nakajima, Osaka City University, Japan and Dr.
M. Hibi, Center for Developmental Biology, Kobe, Japan (12). For stable transfections HeLa cells
were seeded into 10 cm culture dishes at a density of
8×105 cells/dish and transfected after 24 h with 300 ng
linearized (PvuI) pCAGGS or pCAGGS-STAT3F and FuGene 6 (Roche,
Mannheim, Germany) according to the manufacturer's guidelines.
Clones were selected with 100 µg/ml G418 and analyzed for
inhibition of STAT3 activation by western blot analysis.
Protein expression analysis by western
blot
HeLa cells stably expressing pCAGGS or pCAGGS-STAT3F
were seeded in 6 cm culture dishes at a density of
1.5×106 cells/dish. 24 h later they were incubated with
medium or 10 ng/ml OSM (PeproTech, Hamburg, Germany) for 15 min.
Stimulated cells were resuspended in sample buffer (62.5 mM
Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 100 mM DTT) and equal
amounts of protein were analyzed using Abs directed against
pTyr705-STAT3 (Cell Signaling Technology, Inc., Danvers, MA, USA),
STAT3 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) or β-actin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Secondary Abs
(Sigma-Aldrich; Merck KGaA) and ECL reagent (Roche) were used for
standardized detection with ChemiDoc XRS+ Molecular Imager.
Quantification was done with the Quantity One analysis software
(both Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Irradiation
Cervical carcinoma cells were seeded in flat-bottom
microtiter plates at a density of 1×104 cells/well. 24 h
later cells received single-dose of irradiation (2, 4, 6, 8 Gy)
using a linear accelerator (Oncor™; Siemens AG, Munich, Germany) as
indicated. Separate plates were used for each irradiation dose. The
plates were covered by 2 cm thick plexiglass leaf to improve photon
dose homogeneity. The radiation characteristics were as follows:
Size of the radiation field 30×30 cm; collimator angle 0°; gantry
angle 0°; source surface distance 208 cm; beam energy 6 MV photons;
dose-rate 2 Gy/min. Computed-tomography-based three-dimensional
dose calculations were made with the Pinnacle™ planning system
(Philips Radiation Oncology Systems; Philips Medical Systems,
Fitchburg, WI, USA) previously.
Stimulation experiments and
cytotoxicity assays
Cervical carcinoma cells were seeded in a
flat-bottom microtiter plates at a density of 1×104
cells/well and incubated for 24 h. Cervical carcinoma cells were
stimulated with 10 ng/ml OSM (PeproTech, Hamburg, Germany) for 2 h
or medium as a control. For irradiation experiments cells were
subsequently irradiated with increasing irradiation doses (0–8 Gy)
as described above. For chemotherapy experiments cells were
stimulated with medium or OSM and challenged with a cisplatin
concentration of 0.975 µg/ml (Hexal, Holzkirchen, Germany) for 2 h.
In chemoradiotherapy experiments cells were stimulated with medium
or OSM, challenged with a cisplatin concentration of 0.975 µg/ml
(Hexal) for 2 h and subsequent irradiated with a dose of 6 Gy. In
all experiments cell viability was assessed 48 h later by the
neutral red uptake method as described previously (10).
Statistical analysis
All statistical analyses were performed using the
GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA)
program. To evaluate the statistical differences between multiple
groups, one-way analysis of variance with Bonferroni post hoc test
was applied. P<0.05 was considered to indicate a statistically
significant difference.
Results
Heterogeneous radiosensitivity of
cervical carcinoma cells
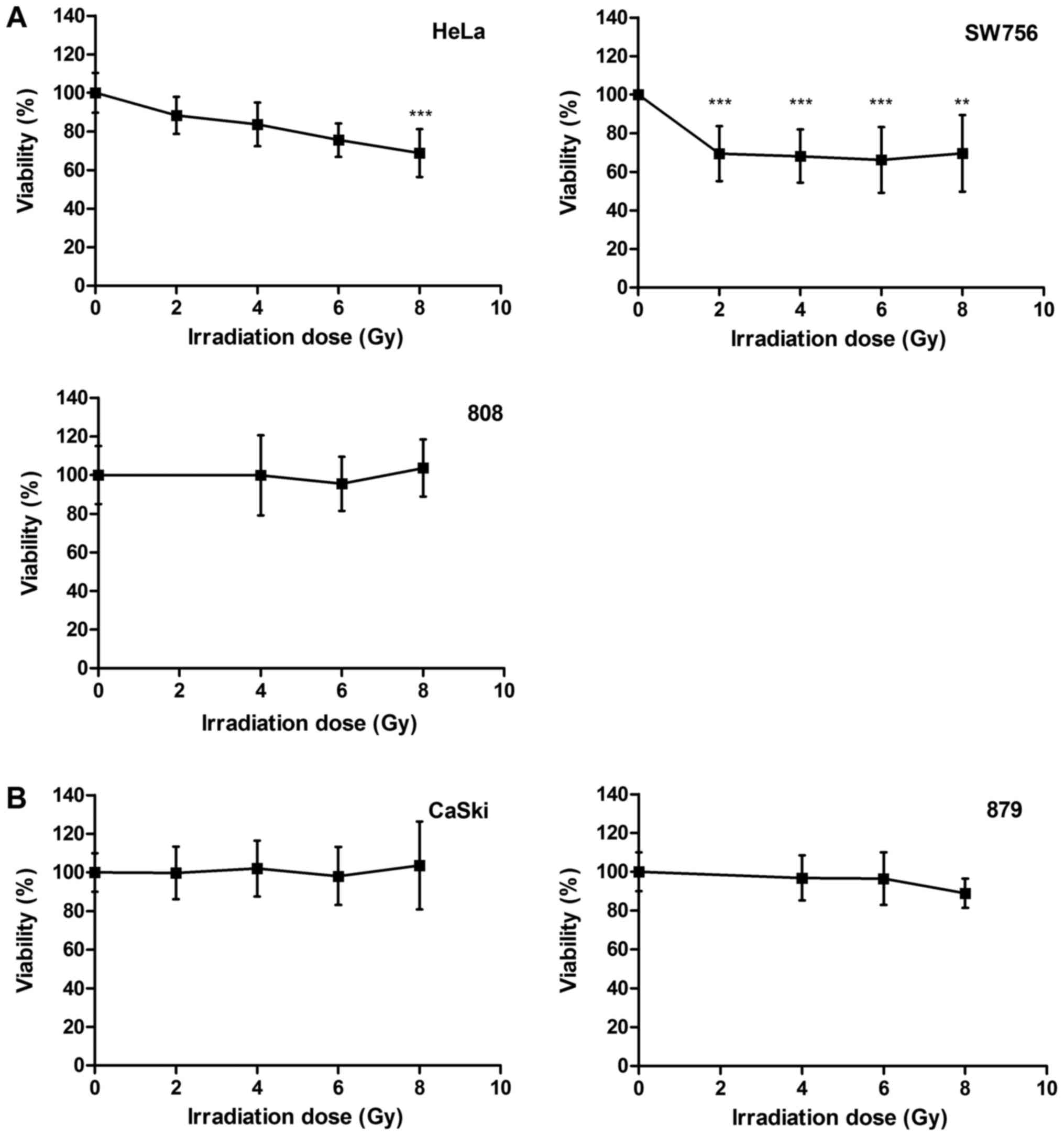
We compared the radiosensitivity of different
cervical carcinoma cells. HPV18-positive cell lines HeLa and SW756,
HPV16-positive cell line CaSki and the more recently generated
cervical cancer cells 808 (HPV18-positive) and 879 (HPV16-positive)
were treated with increasing doses of irradiation (0–8 Gy). After
irradiation HeLa cells died in a dose dependent manner up to 24.5%
at a dose of 6 Gy (P<0.001) and up to 32% at a dose of 8 Gy
(P<0.001; Fig. 1A, left panel). In
SW756 cells cell death was observed in 31% for 2 Gy (P<0.001).
Again, higher irradiation doses (4–8 Gy) did not enhance
radiosensitivity of these cells (Fig.
1A, right panel). 808 cells (Fig.
1A, lower panel), CaSki cells and 879 cells (Fig.1B) were almost completely resistant to
irradiation in our experiments. Thus, the cervical carcinoma cells
used in this study showed a heterogeneous responsiveness to
irradiation.
OSM signaling sensitizes cervical
carcinoma cells for CCRT-induced cell death
We previously described that OSM signaling
sensitized cervical carcinoma cells to chemotherapeutic
drug-induced cell death (10). Here
we investigated the impact of OSM pre-treatment in cervical
carcinoma cells on radio- or concurrent chemoradiotherapy. Cervical
carcinoma cells were pre-treated with 10 ng/ml OSM or medium for 2
h. To mimic radio-, chemo- or chemoradiotherapy in vitro,
cells were challenged with medium or 0.975 µg/ml cisplatin for 2 h
and irradiated with a dose of 6 Gy or left untreated. The low
cisplatin concentration of 0.975 µg/ml was selected to minimize its
own effects on cancer cell viability but preserving its role as a
radio sensitizer in chemoradiotherapy experiments. Cell viability
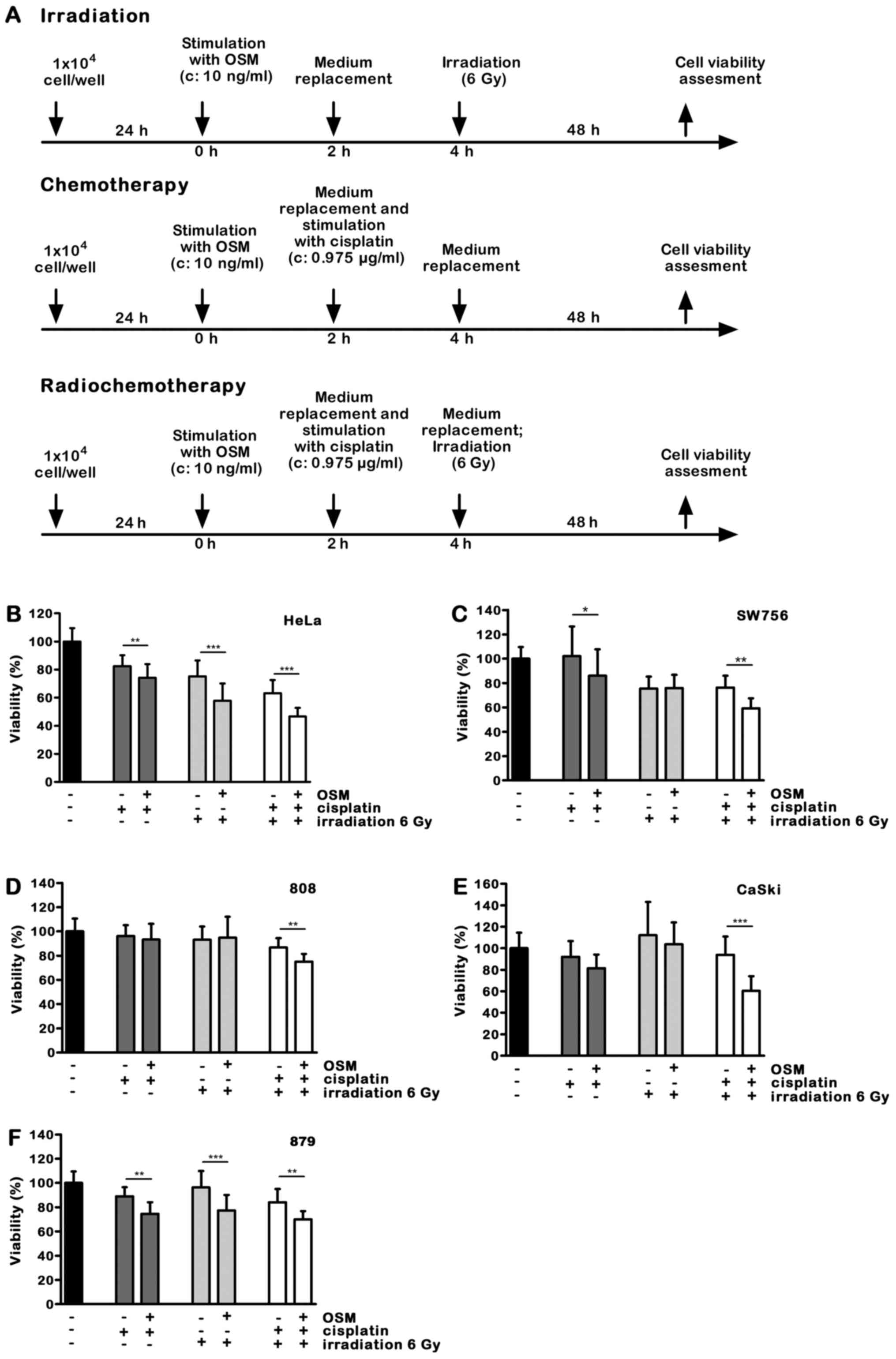
was assessed after 48 h. A time schedule of the experimental
procedure is shown in Fig. 2A.
While the low cisplatin dose of 0.975 µg/ml alone
had only a minor effect on cell viability, OSM pretreatment for 2 h
further increased cell death induction in 4 out of 5 cell lines. In
808 cells the selected combination of OSM and cisplatin had no
effect on cell viability (Fig. 2B-F;
dark grey bars).
We then studied the impact of OSM stimulation on
irradiation induced cell death. Cell viability of HeLa cells
irradiated with a dose of 6 Gy was 75.6%. OSM sensitized the HPV18
positive HeLa cells for irradiation-induced cell death (Fig. 2B, 18%, light grey bars; P<0.001)),
whereas in SW756 and 808 cells OSM pretreatment did not affect cell
viability in combination with irradiation (Fig. 2C and D). OSM pretreatment sensitized
the HPV16 positive cell line CaSki only slightly (7%; Fig. 2E), whereas it significantly sensitized
the 879 cells for irradiation-induced cell death (Fig. 2F; 19.1%, light grey bars; P<0.001).
Thus, pre-treatment with OSM significantly sensitized two of five
tested cervical cancer cells for irradiation-induced cell death.
Furthermore, in the completely radioresistant 879 cells OSM
pretreatment was sufficient to sensitize for irradiation.
Concurrent chemoradiotherapy is the standard
treatment for advanced cervical cancers with FIGO>IIB. For this
reason we analyzed the impact of OSM pretreatment on
chemoradiotherapy-induced cell death. The HPV18 positive cell lines
HeLa and SW756 as well as the more recently generated cells 808
were killed significantly more by OSM stimulation (Fig. 2B-D; 12–17%, white bars; P<0.01).
OSM significantly sensitized the HPV16-positive CaSki cells for
chemoradiotherapy-induced cell death up to 33% (Fig. 2E; white bars; P<0.001) and the more
recently generated 879 cells, that were radio-resistant in our
experiments, for enhanced cell death after combined
chemoradiotherapy treatment (Fig. 2F;
white bars; P<0.01).
Taken together, OSM stimulation of all five tested
cervical carcinoma cells sensitized these cells for
chemoradiotherapy-induced cell death. Notably, OSM treatment
induced responsiveness to chemoradiotherapy-induced cell death in
the irradiation-resistant cells 808, CaSki and 879. Table I summarizes our findings.
 | Table I.OSM-induced cell death sensitization
in different HPV-18- and HPV16-positive cervical cancer cells
toward irradiation, chemo- and chemoradiotherapy. |
Table I.
OSM-induced cell death sensitization
in different HPV-18- and HPV16-positive cervical cancer cells
toward irradiation, chemo- and chemoradiotherapy.
|
|
| Response toward
combined OSM pretreatment and |
|---|
|
|
|
|
|---|
| HPV status | Cellsa | Irradiation | Chemotherapy |
Chemoradiotherapy |
|---|
| HPV18-positive | HeLa | ++ | + | ++ |
|
| SW756 | − | + | ++ |
|
| 808 | − | − | ++ |
| HPV16-positive | CaSki | + | + | +++ |
|
| 879 | ++ | ++ | ++ |
STAT3 mediates sensitization for
CCRT-induced cell death by OSM signaling in cervical cancer
cells
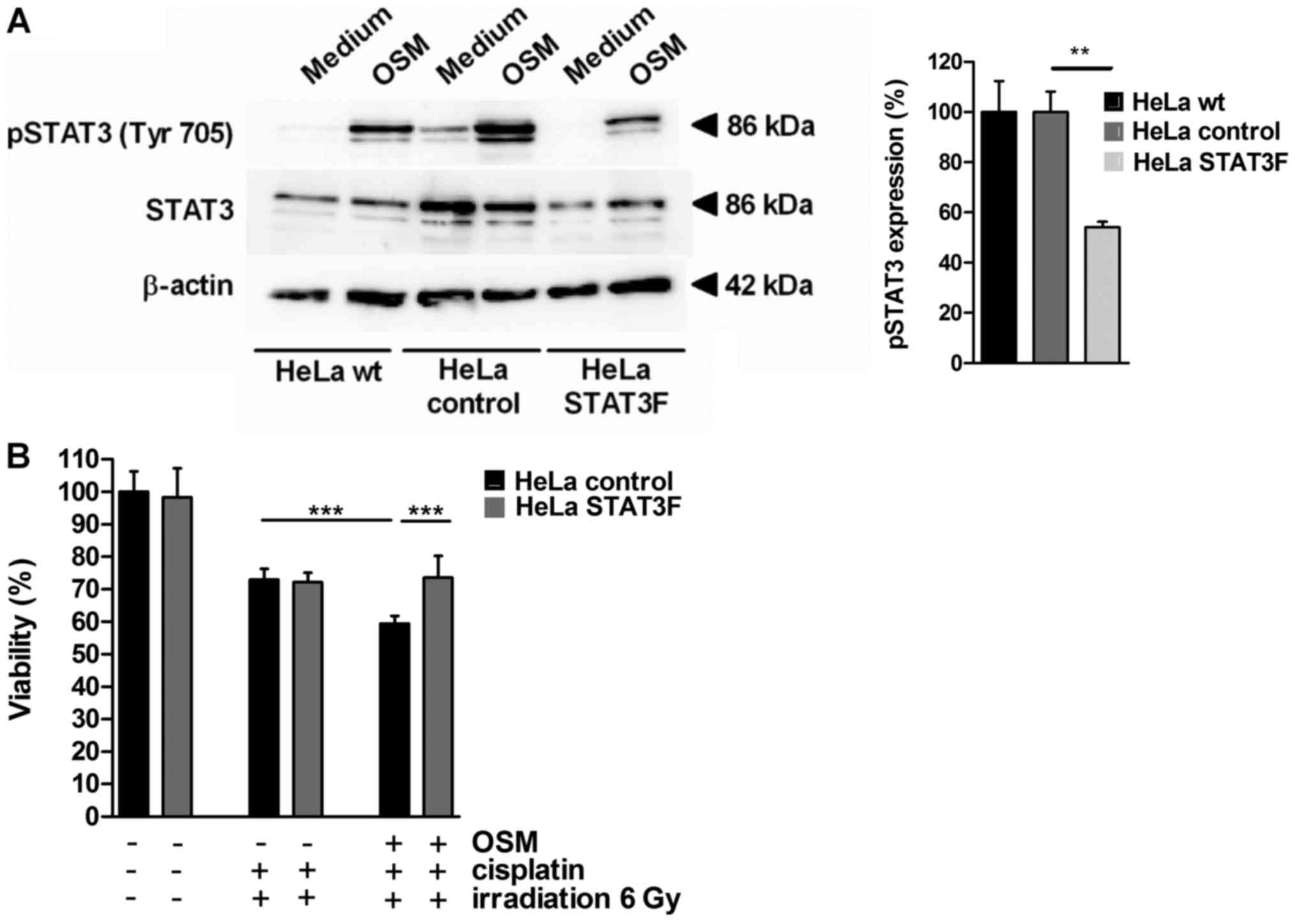
To investigate the molecular mechanism for
CCRT-induced cell death sensitization by OSM in cervical cancer
cells, HeLa cells were stably transfected with a dominant-negative
version of STAT3 interfering with phosphorylation at Tyr705
(dnSTAT3-Y705F, HeLa STAT3F) or the empty vector as a control (HeLa
control). OSM stimulation led to STAT3 phosphorylation at
Tyrosin705 in HeLa wt and HeLa control cells (Fig. 3A) while in HeLa cells stably
expressing STAT3F the pSTAT3 (Tyr705) phosphorylation was
significantly decreased (45% reduction). In cell viability assays
OSM pretreatment sensitized HeLa control cells for CCRT-induced
cell death (18.7%, black bars, Fig.
3B). STAT3F overexpression completely abolished OSM-mediated
sensitization (grey bars; P<0.001). Thus, our results provide
evidence that chemoradiosensitisation by OSM depends on the pSTAT3
(Tyr705) signaling pathway in cervical cancer cells.
Discussion
Resistance of cervical cancer patients toward
platinum-based radio/chemotherapy is a major clinical problem
(9). For patients who do not respond
to standard therapies new therapeutic strategies are needed. In our
study we analyzed the impact of OSM pretreatment on the response of
cervical cancer cells to radio/chemotherapy. Cervical cancer cells
responded heterogeneously toward irradiation alone, three of the
tested cells were resistant in our experiments. However, OSM
pretreatment improved chemosensitivity for irradiation in all
cervical cancer cells and even rendered two cell lines susceptible
for irradiation that were otherwise completely resistant.
Over the past years, improved understanding in
cancer pathogenesis gave rise to new treatment options to support
standard cancer therapies based on surgery or radio/chemotherapy.
This includes therapies that target tumor angiogenesis and cancer
growth, as well as cancer immunotherapies that activate the patient
immune system to support antitumor immunity (20,21).
Targeted therapy strategies include several antibodies or
inhibitors to block essential biochemical pathways required for
tumor growth and survival, like EGFR, VEGF, BRAF or HER2 (21). Blockage of the inhibitory proteins
CTLA-4, PD-1 or the ligand PD1-L with specific antibodies,
checkpoint inhibitors (22), resulted
in clinical benefit in several tumor types (23–25).
In cervical cancer the only approved targeted
therapy so far is bevacizumab, an anti-VEGF antibody to inhibit
angiogenesis, in combination with a cisplatin-based chemotherapy in
patients with advanced, metastatic or recurrent cervical cancer
(26,27). There is a strong need for new
therapeutic strategies in cervical cancer patients because HPV
interferes with local immunity suppressing the expression of
inflammatory cytokines and chemokines in infected cells (28). Immunostimulatory cytokines like CCL2
and CCL20 are induced in the stromal compartment of invasive
cervical carcinoma but they are involved in the generation of a
pro-tumorigenic microenvironment (29–31). In
contrast, the regulator of the adaptive immunity interleukin-12 is
down-regulated in the cervical cancer microenvironment [own
unpublished data and (32,33)]. One immunotherapeutic strategy might
be usage of the synthetic viral dsRNA homolog
polyinosinic:polycytidylic acid (PolyIC) that can stimulate
necroptosis in cervical cancer cells expressing the kinase RIPK3
(34,35). Notably, this leads to enhanced
interleukin-12 production of dendritic cells (34).
Another strategy might be the application of cell
death sensitizers that employ the STAT3/IRF1 signaling pathway. We
have recently shown that stimulation of cervical cancer cells with
IL-6 in combination with the soluble IL-6R or OSM can potently
activate STAT3 which leads to IRF1 up-regulation (10). Patients with high expression of the
STAT3-regulated pro-apoptotic IRF1 in pretreatment cervical cancer
biopsy cells showed in fact significantly higher responses to
neoadjuvant chemo- and chemoradiotherapy (10). In line with this, the in vitro
results from this study confirmed that cell death sensitization
toward irradiation or chemoradiotherapy is induced by OSM
pre-treatment of cervical carcinoma cells. As the underlying
mechanism we identified the pSTAT3 (Tyr705) signaling pathway that
sensitized cervical cancer cells for CCRT-induced cell death as
shown via stable transfection of dominant-negative STAT3F (12). In this study we showed that OSM
pre-treatment improved chemosensitivity for irradiation in all
cervical cancer cells particularly in the initially radio-resistant
cells 808, CaSki and 879 with up to 33% cell death enhancement.
Sensitization by OSM stimulation for CCRT-induced cell death
occurred in all tested cervical cancer cells irrespectively whether
they were positive for HPV16 or HPV18. However, it appeared that
HPV16 positive cervical cancer cells showed a slightly higher
responsiveness towards OSM-mediated sensitization. This was
particularly the case for CaSki cells. It can be speculated that
their stronger response to OSM might be due to differences in the
interaction between HPV16 and the OSM/STAT3 signaling pathway.
Alternatively, the genetic or epigenetic alterations in these cells
might affect their OSM-responsiveness. This will be subject of
future studies.
OSM binds to the OSM receptor-β (OSM-R) which then
associates with the receptor chain gp130 to activate the STAT3
signaling pathway (12,36). Recent studies indicate that OSM-R is
overexpressed in advanced cervical squamous cell carcinomas making
the cells susceptible for OSM signals. However, high expression of
OSM-R in cervical cancers is associated with worse clinical outcome
and OSM signals were described to initiate several pro-tumorigenic
effects (37,38). For this reason, OSM-R is recommended
as a candidate for antibody-mediated inhibition to block
pro-malignant effects (38,39).
However, based on our findings [(10) and this study] blockage of OSM-R should
be employed with caution. Indeed, inhibition of the OSM-R might
block OSM-initiated pro-malignant effects but it would concurrently
prevent sensitization of cervical cancer cells to chemo- or
chemoradiotherapy. Thus OSM-R might have a dual role in cervical
cancers and this may have major implications for personalization of
cervical cancer therapy. In conclusion, based on our findings OSM
pre-treatment might be an interesting option to improve the
responsiveness of cervical cancer cells toward irradiation or
chemoradiotherapy particularly in radioresistant cells. OSM-R
blockage should therefore not be applied prior to irradiation or
chemoradiotherapy.
Acknowledgements
The authors would like to thank Mr. Georg Blaß
(Department of Radiotherapy and Radiation Oncology, Saarland
University, Homburg, Germany) for their assistance in the
irradiation experiments and Dr G.S. Stein (University of
Massachusetts Medical School, Worcester, MA, USA), Dr T. Hirano, Dr
M. Hibi (both Center for Developmental Biology, Kobe, Japan) and Dr
K. Nakajima (Osaka City University, Osaka, Japan) for providing the
cDNA constructs.
Funding
This work was supported by a grant from the Deutsche
Krebshilfe (grant no. 109752) and the Saarland Staatskanzlei to SS
(grant no. WT/2-LFFP 14/15).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
Conception and design: BWR, SS; Development of
methodology: RS, BWR, JF, CR; Acquisition of data: RS, BWR; IJB,
EFS; Analysis and interpretation of data: RS, BWR, SS; Writing,
review, and/or revision of the manuscript: RS, BWR, CR, IJB, EFS,
SS; Final approval of the version to be published: RS, BWR, JF,
IJB, CR, EFS, SS; Administrative, technical, or material support:
IJB, EFS, CR, SS; Study supervision: SS.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hausen Zur H: Papillomaviruses in the
causation of human cancers-a brief historical account. Virology.
384:260–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barillot I, Horiot JC, Pigneux J, Schraub
S, Pourquier H, Daly N, Bolla M and Rozan R: Carcinoma of the
intact uterine cervix treated with radiotherapy alone: A French
cooperative study: Update and multivariate analysis of prognostics
factors. Int J Radiat Oncol Biol Phys. 38:969–978. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Logsdon MD and Eifel PJ: Figo IIIB
squamous cell carcinoma of the cervix: An analysis of prognostic
factors emphasizing the balance between external beam and
intracavitary radiation therapy. Int J Radiat Oncol Biol Phys.
43:763–775. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perez CA, Camel HM, Kuske RR, Kao MS,
Galakatos A, Hederman MA and Powers WE: Radiation therapy alone in
the treatment of carcinoma of the uterine cervix: A 20-year
experience. Gynecol Oncol. 23:127–140. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Monk BJ, Tewari KS and Koh WJ:
Multimodality therapy for locally advanced cervical carcinoma:
State of the art and future directions. J Clin Oncol. 25:2952–2965.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morris M, Eifel PJ, Lu J, Grigsby PW,
Levenback C, Stevens RE, Rotman M, Gershenson DM and Mutch DG:
Pelvic radiation with concurrent chemotherapy compared with pelvic
and para-aortic radiation for high-risk cervical cancer. N Engl J
Med. 340:1137–1143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keys HM, Bundy BN, Stehman FB, Muderspach
LI, Chafe WE, Suggs CL III, Walker JL and Gersell D: Cisplatin,
radiation, and adjuvant hysterectomy compared with radiation and
adjuvant hysterectomy for bulky stage IB cervical carcinoma. N Engl
J Med. 340:1154–1161. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rose PG, Bundy BN, Watkins EB, Thigpen JT,
Deppe G, Maiman MA, Clarke-Pearson DL and Insalaco S: Concurrent
cisplatin-based radiotherapy and chemotherapy for locally advanced
cervical cancer. N Engl J Med. 340:1144–1153. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peltenburg LT: Radiosensitivity of tumor
cells. Oncogenes and apoptosis. Q J Nucl Med. 44:355–364.
2000.PubMed/NCBI
|
|
10
|
Walch-Ruckheim B, Pahne-Zeppenfeld J,
Fischbach J, Wickenhauser C, Horn LC, Tharun L, Büttner R, Mallmann
P, Stern P, Kim YJ, et al: STAT3/IRF1 Pathway activation sensitizes
cervical cancer cells to chemotherapeutic drugs. Cancer Res.
76:3872–3883. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taniguchi K and Karin M: IL-6 and related
cytokines as the critical lynchpins between inflammation and
cancer. Semin Immunol. 26:54–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakajima K, Yamanaka Y, Nakae K, Kojima H,
Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M and Hirano T: A
central role for Stat3 in IL-6-induced regulation of growth and
differentiation in M1 leukemia cells. Embo J. 15:3651–3658.
1996.PubMed/NCBI
|
|
13
|
Wei LH, Kuo ML, Chen CA, Chou CH, Cheng
WF, Chang MC, Su JL and Hsieh CY: The anti-apoptotic role of
interleukin-6 in human cervical cancer is mediated by up-regulation
of Mcl-1 through a PI 3-K/Akt pathway. Oncogene. 20:5799–5809.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jee SH, Chiu HC, Tsai TF, Tsai WL, Liao
YH, Chu CY and Kuo ML: The phosphotidyl inositol 3-kinase/Akt
signal pathway is involved in interleukin-6-mediated Mcl-1
upregulation and anti-apoptosis activity in basal cell carcinoma
cells. J Invest Dermatol. 119:1121–1127. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen CL, Hsieh FC, Lieblein JC, Brown J,
Chan C, Wallace JA, Cheng G, Hall BM and Lin J: Stat3 activation in
human endometrial and cervical cancers. Br J Cancer. 96:591–599.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pattillo RA, Hussa RO, Story MT, Ruckert
AC, Shalaby MR and Mattingly RF: Tumor antigen and human chorionic
gonadotropin in CaSki cells: A new epidermoid cervical cancer cell
line. Science. 196:1456–1458. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Freedman RS, Bowen JM, Leibovitz A, Pathak
S, Siciliano MJ, Gallager HS and Giovanella BC: Characterization of
a cell line (SW756) derived from a human squamous carcinoma of the
uterine cervix. In vitro. 18:719–726. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jones HW Jr, McKusick VA, Harper PS and
Wuu KD: George Otto Gey. (1899–1970). The HeLa cell and a
reappraisal of its origin. Obstet Gynecol. 38:945–949.
1971.PubMed/NCBI
|
|
19
|
Brady CS, Bartholomew JS, Burt DJ,
Duggan-Keen MF, Glenville S, Telford N, Little AM, Davidson JA,
Jimenez P, Ruiz-Cabello F, et al: Multiple mechanisms underlie HLA
dysregulation in cervical cancer. Tissue Antigens. 55:401–411.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vanneman M and Dranoff G: Combining
immunotherapy and targeted therapies in cancer treatment. Nat Rev
Cancer. 12:237–251. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hughes PE, Caenepeel S and Wu LC: Targeted
therapy and checkpoint immunotherapy combinations for the treatment
of cancer. Trends Immunol. 37:462–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Melero I, Berman DM, Aznar MA, Korman AJ,
Gracia Pérez JL and Haanen J: Evolving synergistic combinations of
targeted immunotherapies to combat cancer. Nat Rev Cancer.
15:457–472. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharma P and Allison JP: The future of
immune checkpoint therapy. Science. 348:56–61. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sharma P and Allison JP: Immune checkpoint
targeting in cancer therapy: Toward combination strategies with
curative potential. Cell. 161:205–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Topalian SL, Drake CG and Pardoll DM:
Immune checkpoint blockade: A common denominator approach to cancer
therapy. Cancer Cell. 27:450–461. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tewari KS, Sill MW, Long HJ III, Penson
RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao
MM, et al: Improved survival with bevacizumab in advanced cervical
cancer. N Engl J Med. 370:734–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsuda N, Watari H and Ushijima K:
Chemotherapy and molecular targeting therapy for recurrent cervical
cancer. Chin J Cancer Res. 28:241–253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karim R, Meyers C, Backendorf C, Ludigs K,
Offringa R, van Ommen GJ, Melief CJ, van der Burg SH and Boer JM:
Human papillomavirus deregulates the response of a cellular network
comprising of chemotactic and proinflammatory genes. PLoS One.
6:e178482011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schroer N, Pahne J, Walch B, Wickenhauser
C and Smola S: Molecular pathobiology of human cervical high-grade
lesions: Paracrine STAT3 activation in tumor-instructed myeloid
cells drives local MMP-9 expression. Cancer Res. 71:87–97. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pahne-Zeppenfeld J, Schröer N,
Walch-Rückheim B, Oldak M, Gorter A, Hegde S and Smola S: Cervical
cancer cell-derived interleukin-6 impairs CCR7-dependent migration
of MMP-9 expressing dendritic cells. Int J Cancer. 134:2061–2073.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Walch-Rückheim B, Mavrova R, Henning M,
Vicinus B, Kim YJ, Bohle RM, Juhasz-Böss I, Solomayer EF and Smola
S: Stromal fibroblasts induce CCL20 through IL6/C/EBPbeta to
support the recruitment of Th17 cells during cervical cancer
progression. Cancer Res. 75:5248–5259. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heusinkveld M, de Vos van Steenwijk PJ,
Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, van Hall T and
van der Burg SH: M2 macrophages induced by prostaglandin E2 and
IL-6 from cervical carcinoma are switched to activated M1
macrophages by CD4+ Th1 cells. J Immunol. 187:1157–1165. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zijlmans HJ, Punt S, Fleuren GJ, Trimbos
JB, Kenter GG and Gorter A: Role of IL-12p40 in cervical carcinoma.
Br J Cancer. 107:1956–1962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmidt SV, Seibert S, Walch-Rückheim B,
Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, Solomayer EF, Kim YJ,
Bohle RM and Smola S: RIPK3 expression in cervical cancer cells is
required for PolyIC-induced necroptosis, IL-1α release and
efficient paracrine dendritic cell activation. Oncotarget.
6:8635–8647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smola S: RIPK3-a predictive marker for
personalized immunotherapy? Oncoimmunology. 5:e10756952015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tanaka M and Miyajima A: Oncostatin M, a
multifunctional cytokine. Rev Physiol Biochem Pharmacol. 149:39–52.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Winder DM, Chattopadhyay A, Muralidhar B,
Bauer J, English WR, Zhang X, Karagavriilidou K, Roberts I, Pett
MR, Murphy G and Coleman N: Overexpression of the oncostatin M
receptor in cervical squamous cell carcinoma cells is associated
with a pro-angiogenic phenotype and increased cell motility and
invasiveness. J Pathol. 225:448–462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Caffarel MM and Coleman N: Oncostatin M
receptor is a novel therapeutic target in cervical squamous cell
carcinoma. J Pathol. 232:386–390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kucia-Tran JA, Tulkki V, Smith S, Scarpini
CG, Hughes K, Araujo AM, Yan KY, Botthof J, Pérez-Gómez E,
Quintanilla M, et al: Overexpression of the oncostatin-M receptor
in cervical squamous cell carcinoma is associated with
epithelial-mesenchymal transition and poor overall survival. Br J
Cancer. 115:212–222. 2016. View Article : Google Scholar : PubMed/NCBI
|