Introduction
Bladder cancer (BC), the most frequently occurring
urogenital malignancy of the urinary tract worldwide, results in
substantial morbidity and mortality (1,2). In the
USA, bladder cancer is the fourth most common cancer, with 76,960
estimated new cases and 16,390 mortalities in 2016 (3). In addition to other carcinoma types,
such as cervical (4), prostate
(5) and ovarian cancer (6), the accumulation of inherited and somatic
mutations in oncogenes and tumor suppressor genes are believed to
be the reason for the occurrence, progression and metastasis of BC
(7–9).
Although a number of cancer-associated genes and cellular pathways
have been proven to be associated with the initiation and
development of BC (10,11), the accuracy of early diagnosis,
therapeutic and prognostic evaluation for BC remains low.
Consequently, investigating the molecular mechanisms, including the
proliferation, apoptosis and invasion of BC is crucial for the
progress of diagnostic and treatment strategies. With the
development of gene microarray technology, various advanced
techniques for assessing gene expression have been widely applied
in assessing tumor development and progression with lower expenses,
compared with past decades (12,13). In
previous years, numerous gene expression profiling studies on BC
have revealed hundreds of differentially expressed genes (DEGs) and
provided substantial functional information on gene regulatory
network analysis (14–16). However, comparative analysis of DEGs
reported by independent research appears to rarely display
substantial overlap (17), and no
reliable biomarker profile discriminating cancerous from
non-cancerous samples has been identified. At present, due to
bioinformatics methods, the data generated by microarray technology
have been analyzed in order to identify mRNA expression changes in
collected urothelial cells and to examine DEGs in BC and
non-cancerous urothelial cells (18).
However, the interactions among DEGs, particularly pathways in the
interaction network, remain to be elucidated.
In the present study, original data (data set
GSE31189) were downloaded from the Gene Expression Omnibus (GEO)
(19), in order to identify DEGs
between BC and non-cancerous urothelial cells. Then, functional
annotation and network analyses were performed to identify DEGs. By
analyzing the biological functions and networks of BC, the present
study may be useful to gain a better understanding of BC
development at a molecular level and explore the potential
candidate biomarkers for diagnosis, prognosis and treatment.
Materials and methods
Data source
The GSE31189 gene expression profile and its
corresponding platform annotation files were downloaded from the
GEO database. This data set was submitted by Professor Virginia
Urquidi on 3rd August 2011, last updated on 21st April 2017 and
stockpiled on the GPL570 platform (HG-U133_Plus_2) Affymetrix Human
Genome U133 Plus 2.0 Array (Affymetrix; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). This gene expression data consisted of the
data of 92 samples, including 52 BC and 40 non-cancerous urothelial
cells.
Data preprocessing and screening of
DEGs
The probes without annotation of the gene expression
profiles were filtered using the Affy package (20), which is based on the Bioconductor
principles of reproducibility, transparency and efficiency of
development (21). The probe ID for
each gene was then transformed into a gene symbol using the
Affymetrix Human Genome U133 Plus 2.0 Array annotation data
(hgu133plus2.db) and Genome wide annotation for Human, version: 3.7
(org. Hs.eg.db) packages from Bioconductor (http://www.bioconductor.org/). For a gene symbol
corresponding to multiple probe IDs, the average value of these
probes was calculated as the representative expression level of
this gene. The DEGs were screened by using the Linear Models for
Microarray Analysis package in R software (R ×64 3.3.3) (22) with cut-off criteria of P<0.05 and
|log2 fold-change (FC)|>0.5.
Functional annotation and pathway
enrichment
Gene Ontology analysis (GO) is a common useful
approach for annotating genes and gene products and for identifying
characteristic biological phenomena for high-throughput genome or
transcriptome data (23,24). To describe gene product attributes, GO
provides three categories of defined terms, including biological
process (BP), cellular component (CC) and molecular function (MF)
categories (25). Kyoto Encyclopedia
of Genes and Genomes (KEGG) is an integrated database resource for
the systematic analysis of gene functions, linking genomic
information with higher-order functional information (26). The two analyses were available in the
Database for Annotation, Visualization and Integrated Discovery
(DAVID; https://david.ncifcrf.gov/; date of
access, 8/02/2018), which is a bioinformatics data resource
composed of an integrated biology knowledge base and analysis tools
to extract biological meanings from large quantities of genes and
protein collections through a novel agglomeration algorithm
(27). In the present study, GO term
analysis and KEGG pathway analysis were performed using the DAVID
online tool. P<0.05 was set as the cut-off criterion.
PPI network construction and analysis
of modules
Search Tool for the Retrieval of Interacting Genes
(STRING) database (http://string-db.org/) is an online software designed
to assess protein-protein interaction (PPI) information, including
direct (physical) and indirect (functional) associations (28). In the present study, the DEGs were
mapped using STRING to evaluate the PPI information with a combined
score of >0.4 set as the cut-off criterion. Then, the PPI
network was visualized using Cytoscape 3.5.0 (29). To screen the hub genes, a node degree
of ≥10 was selected as the threshold. Furthermore, the Molecular
Complex Detection (MCODE) plug-in was used to screen modules of hub
genes from the PPI network with degree threshold=10, haircut on,
node score cut-off=0.2, k-core=2 and maximum depth=100 (30). The genes in the significant modules
were further mapped to GO terms and KEGG pathways for functional
analysis.
PrognoScan database analysis
The overall survival (OS) rate of mRNA expression
was assessed using an online database, PrognoScan (http://www.abren.net/PrognoScan/; keywords: NOD2,
S100A9, CXCL1, CXCR2; date of access, 8/02/2018), which is a
platform used for evaluating potential tumor markers and
therapeutic targets. To evaluate the OS rate of patients with
breast cancer, patient samples were divided into two groups by
median expression [high vs. low expression; Threshold: NOD (0.87),
S100A9 (0.77), CXCL1 (0.68), CXCR2 (0.55)] and analyzed using
PrognoScan, with a hazard ratio with 95% confidence intervals and
Cox's Proportional-Hazards Model.
Results
Data processing and DEG screening
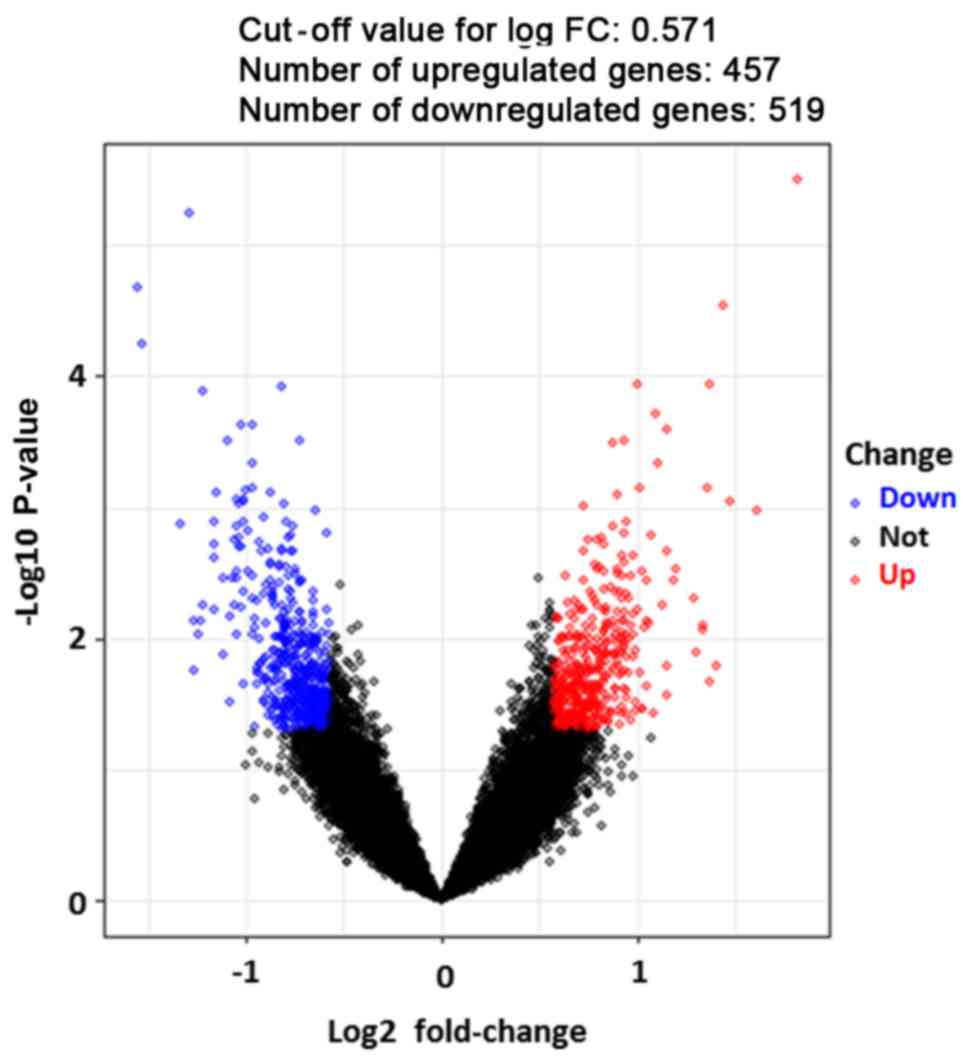
With a criteria of the false discovery ratio
<0.05 and |log2 FC|≥0.571, a total of 976 DEGs (when
compared with those of the noncancerous urothelial cells) were
identified in the BC samples, including 457 upregulated genes and
519 downregulated genes (Fig. 1).
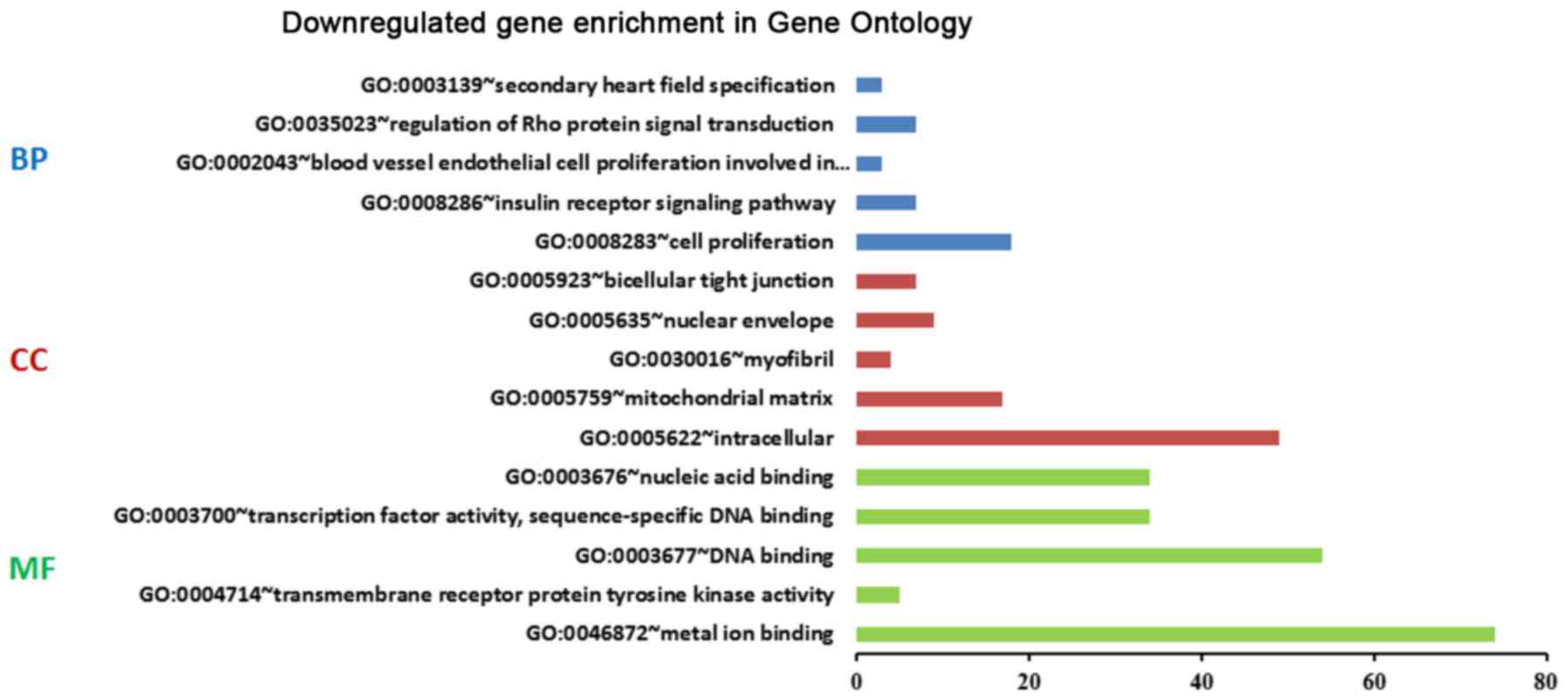
Functional enrichment of DEGs
DAVID analysis was performed to predict the
potential functions and mechanisms of BC by mapping the upregulated
and downregulated genes using GO terms and the KEGG pathways. The
top five significant GO terms of the BP, CC and MF categories
enriched by the up- and downregulated DEGs were identified
(Figs. 2 and 3). The results demonstrated that the
upregulated genes were mainly involved in the negative regulation
of the apoptotic process in the BP category, constituted integral
components of the plasma membrane in the CC category, and were
mainly involved in protein binding in the MF category (Fig. 2), whereas the downregulated genes were
mainly associated with cell proliferation in the BP category, were
mainly intracellular in the CC category, and were mainly involved
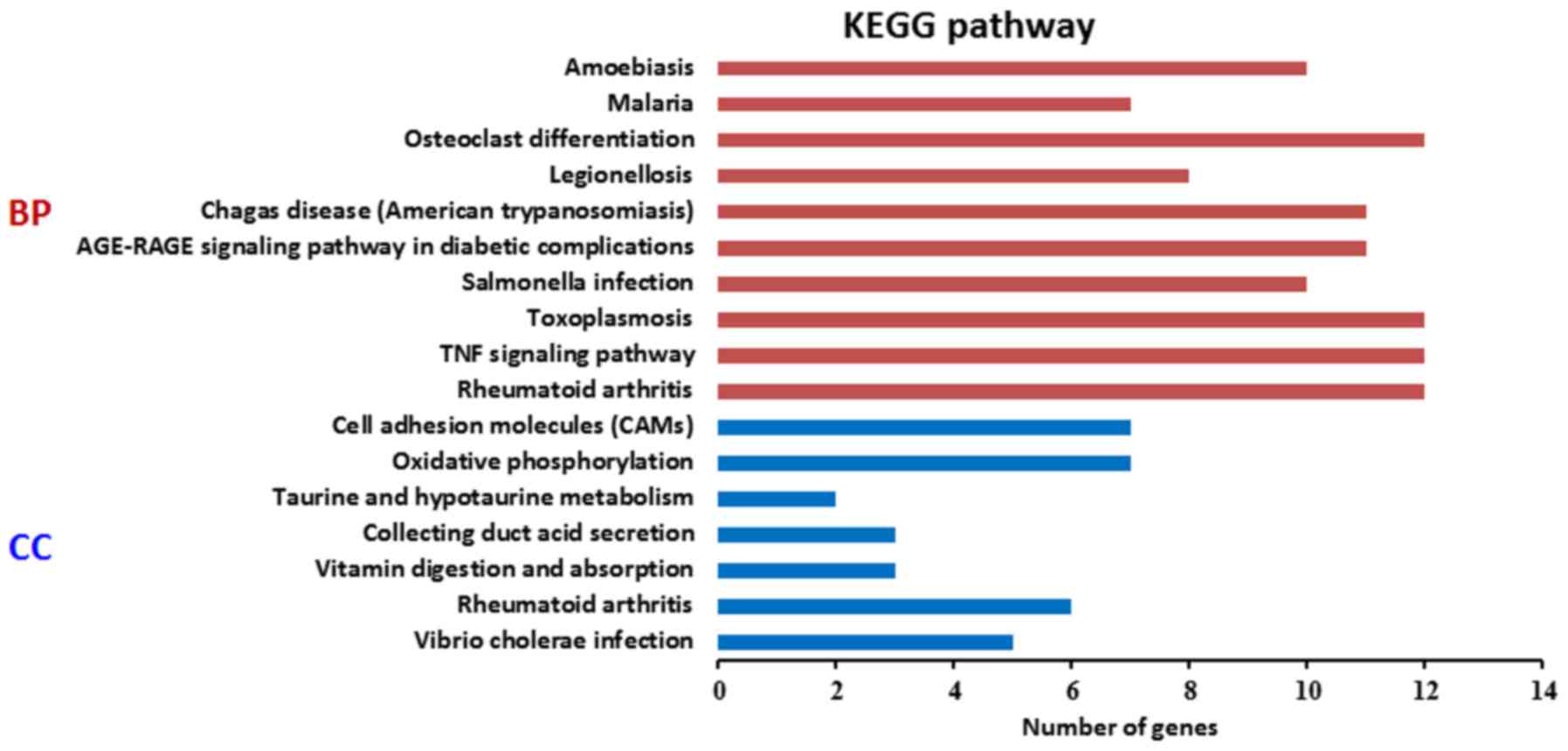
in metal ion binding in the MF category (Fig. 3). The results for the KEGG pathway
enrichment are shown in Fig. 4, which
indicated that the upregulated genes were significantly enriched in
amoebiasis, malaria, osteoclast differentiation, Legionellosis,
Chagas disease, advanced glycation end product-receptor for AGE
signaling pathway in diabetic complications, salmonella infection,
toxoplasmosis, tumor necrosis factor (TNF) signaling pathway and
rheumatoid arthritis pathways, while the downregulated genes were
mainly enriched in cell adhesion molecules, oxidative
phosphorylation, taurine and hypotaurine metabolism, collecting
duct acid secretion, vitamin digestion and absorption, rheumatoid
arthritis and vibrio cholerae infection pathways.
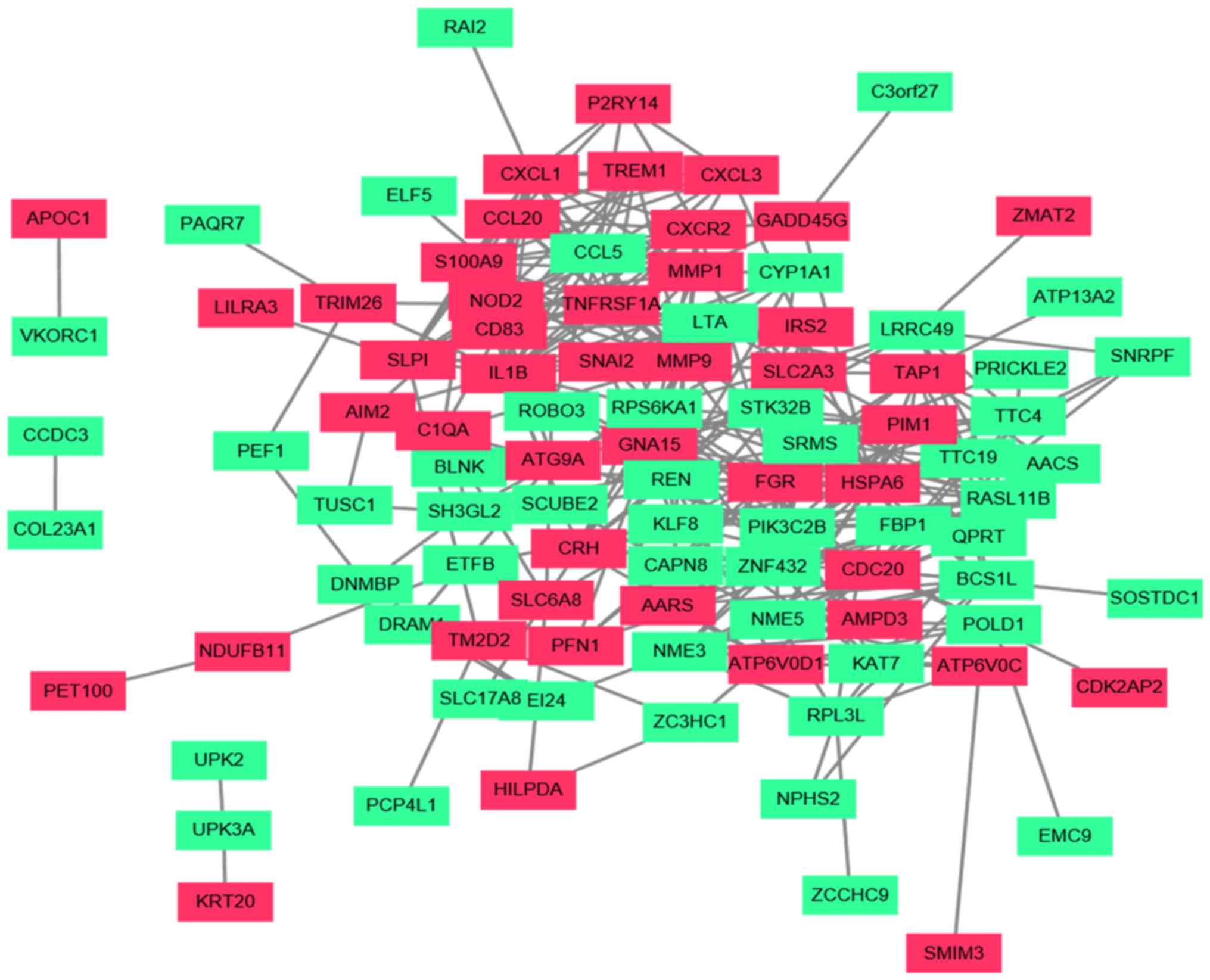
Construction of a PPI network and
functional analysis for key genes
The Cytoscape tool visually constructed the PPI
network with 98 nodes and 321 edges, which were predicted using
STRING with a PPI score of >0.4 (Fig.
5). In the PPI network, 28 nodes with a degree of ≥10 were
regarded as key genes (Table I),
including interleukin (IL)-1B, matrix metalloproteinase (MMP)9,
heat shock protein family A (Hsp70) member 6, FGR proto-oncogene,
Src family tyrosine kinase, ribosomal protein S6 kinase A1, TNF
receptor superfamily member 1A, RAS-like family 11 member B, MMP1,
chemokine ligand (CCL)5, C-X-C motif chemokine receptor 2 (CXCR2),
C-X-C motif chemokine ligand 1 (CXCL1), Pim-1 proto-oncogene,
serine/threonine kinase, nucleotide binding oligomerization domain
containing 2 (NOD2), CCL20, src-related kinase lacking C-terminal
regulatory tyrosine and N-terminal myristylation sites, serine
threonine kinase 21B, S100 calcium binding protein A9 (S100A9),
Kruppel-like factor 8, phosphatidylinositol-4-phosphate 3-kinase
catalytic subunit type 2 β, cluster of differentiation 83, renin, G
protein subunit α 15, transporter 1, ATP binding cassette subfamily
B member, secretory leukocyte peptidase inhibitor, NME/NM23 family
member 5, BCS homolog, ubiquinol-cytochome c reductase complex
chaperone, cell division cycle 20 and NME/NM23 nucleoside
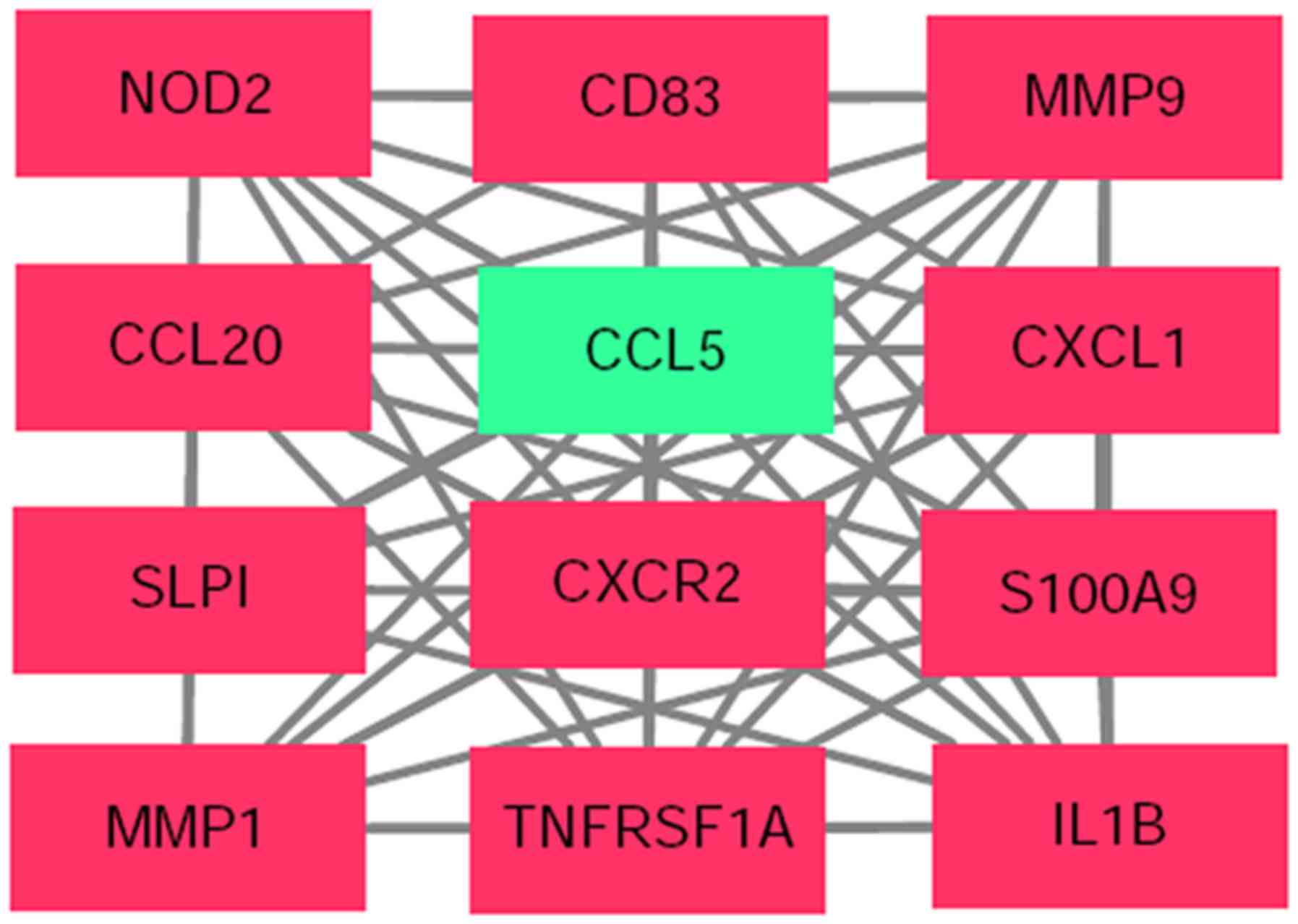
diphosphate kinase 3. One module including 12 nodes and 62 edges
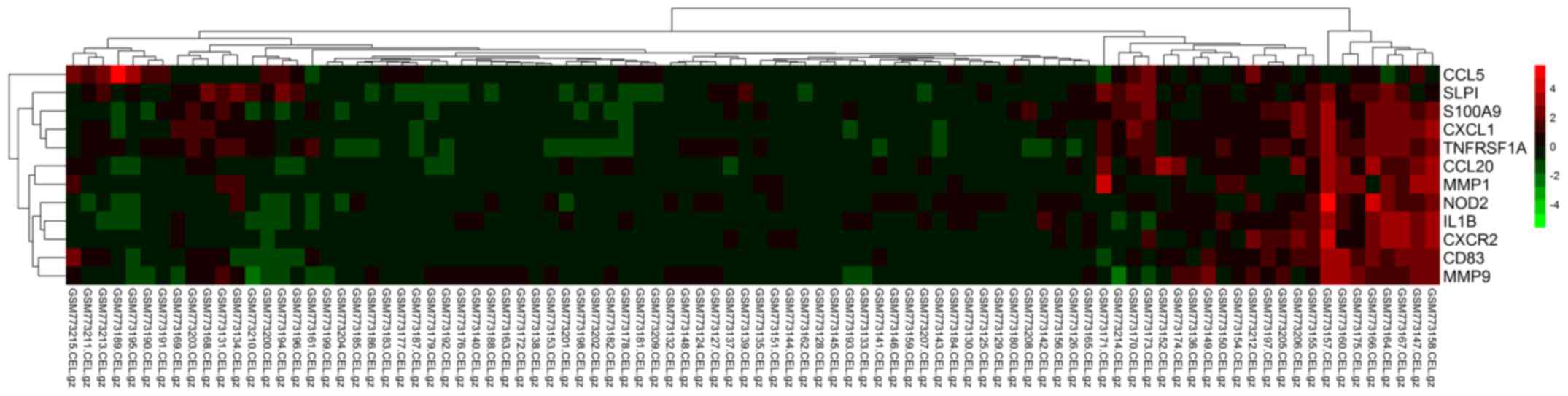
was obtained using MCODE (Fig. 6).
The heat map indicated that the 12 genes were able to distinguish
the two groups of cell samples, in that their expression was
distinctly different between the two groups (Fig. 7). GO term enrichment analysis
demonstrated that in the BP category, the genes in this module were
significantly involved in the chemokine-mediated signaling pathway,
fever generation, inflammatory responses, immune responses and the
positive regulation of cell division (Table II). The genes were significantly
enriched in the CC category were significantly part of the
extracellular space, autophagosome, cytoplasmic vesicle,
extracellular matrix and lysosome (Table
II). Finally, in the MF category, analysis revealed that the
genes were mainly associated with chemokine activity, IL-1 receptor
binding, C-C motif receptor chemokine receptor, cytokine activity
and metalloendopeptidase activity (Table
II). KEGG analysis revealed that the genes were mainly enriched
in the TNF signaling pathway, rheumatoid arthritis, prion diseases,
cytokine-cytokine receptor interaction and NOD-like receptor
signaling pathway (Table II).
 | Table I.Key nodes in the protein-protein
interaction network with a degree ≥10. |
Table I.
Key nodes in the protein-protein
interaction network with a degree ≥10.
| Gene | Degree of
change | Log2
fold-change |
|---|
| IL-1B | 23 | 1.211413 |
| MMP9 | 22 | 0.762970 |
| HSPA6 | 20 | 1.198230 |
| FGR | 18 | 0.674802 |
| RPS6KA1 | 18 | −0.692775 |
| TNFRSF1A | 17 | 0.601014 |
| RASL11B | 16 | −0.600296 |
| MMP1 | 15 | 0.743885 |
| CCL5 | 15 | −0.698948 |
| CXCR2 | 15 | 0.988178 |
| CXCL1 | 15 | 1.088843 |
| PIM1 | 14 | 0.686538 |
| NOD2 | 14 | 0.633431 |
| CCL20 | 14 | 0.677125 |
| SRMS | 13 | −0.619940 |
| STK21B | 13 | −0.598366 |
| S100A9 | 13 | 1.202516 |
| KLF8 | 13 | −0.585851 |
| PIK3c2B | 12 | −0.604130 |
| CD83 | 12 | 0.801585 |
| REN | 12 | −0.737372 |
| GNA15 | 12 | 0.647069 |
| TAP1 | 11 | 1.004478 |
| SLPI | 11 | 1.064963 |
| NME5 | 11 | −0.905805 |
| BCS1L | 10 | −0.621588 |
| CDC20 | 10 | 0.646275 |
| NME3 | 10 | −0.709424 |
| Table II.Functional and pathway enrichment
analysis of the genes in the module. |
Table II.
Functional and pathway enrichment
analysis of the genes in the module.
| Category | Term | Count | % | P-value |
|---|
| GO BP |
GO:0070098-chemokine-mediated signaling
pathway | 3 | 40.4 |
1.09×10−04 |
| GO_BP | GO:0001660-fever
generation | 2 | 27.0 |
2.00×10−03 |
| GO_BP |
GO:0006954-inflammatory response | 3 | 40.4 |
3.14×10−03 |
| GO_BP | GO:0006955-immune
response | 3 | 40.4 |
3.21×10−03 |
| GO_BP | GO:0051781-positive
regulation of cell division | 2 | 27.0 |
5.48×10−03 |
| GO_CC |
GO:0005615-extracellular space | 5 | 47.4 |
1.86×10−04 |
| GO_CC |
GO:0005776-autophagosome | 2 | 27.0 |
1.67×10−02 |
| GO_CC |
GO:0031410-cytoplasmic vesicle | 2 | 27.0 |
3.19×10−02 |
| GO_CC |
GO:0031012-extracellular matrix | 2 | 27.0 |
3.66×10−02 |
| GO_CC |
GO:0005764-lysosome | 2 | 27.0 |
4.73×10−02 |
| GO_MF |
GO:0008009-chemokine activity | 3 | 40.4 |
1.16×10−04 |
| GO_MF |
GO:0005149-interleukin-1 receptor
binding | 2 | 27.0 |
4.78×10−03 |
| GO_MF | GO:0048020-C-C
motif receptor chemokine receptor binding | 2 | 27.0 |
1.01×10−02 |
| GO_MF | GO:0005125-cytokine
activity | 2 | 27.0 |
4.28×10−02 |
| GO_MF |
GO:0004222-metalloendopeptidase
activity | 2 | 27.0 |
4.95×10−02 |
| KEGG_PATHWAY | ecb04668: Tumor
necrosis factor signaling pathway | 6 | 80.9 |
4.39×10−09 |
| KEGG_PATHWAY | ecb05323:
Rheumatoid arthritis | 5 | 67.4 |
3.04×10−07 |
| KEGG_PATHWAY | ecb05020: Prion
diseases | 3 | 40.4 |
3.42×10−04 |
| KEGG_PATHWAY | ecb04060:
Cytokine-cytokine receptor interaction | 4 | 53.9 |
5.48×10−04 |
| KEGG_PATHWAY | ecb04621:
Nucleotide oligomerization domain-like receptor signaling
pathway | 3 | 40.4 |
8.04×10−04 |
Association between 12 key genes and
prognosis in patients with BC
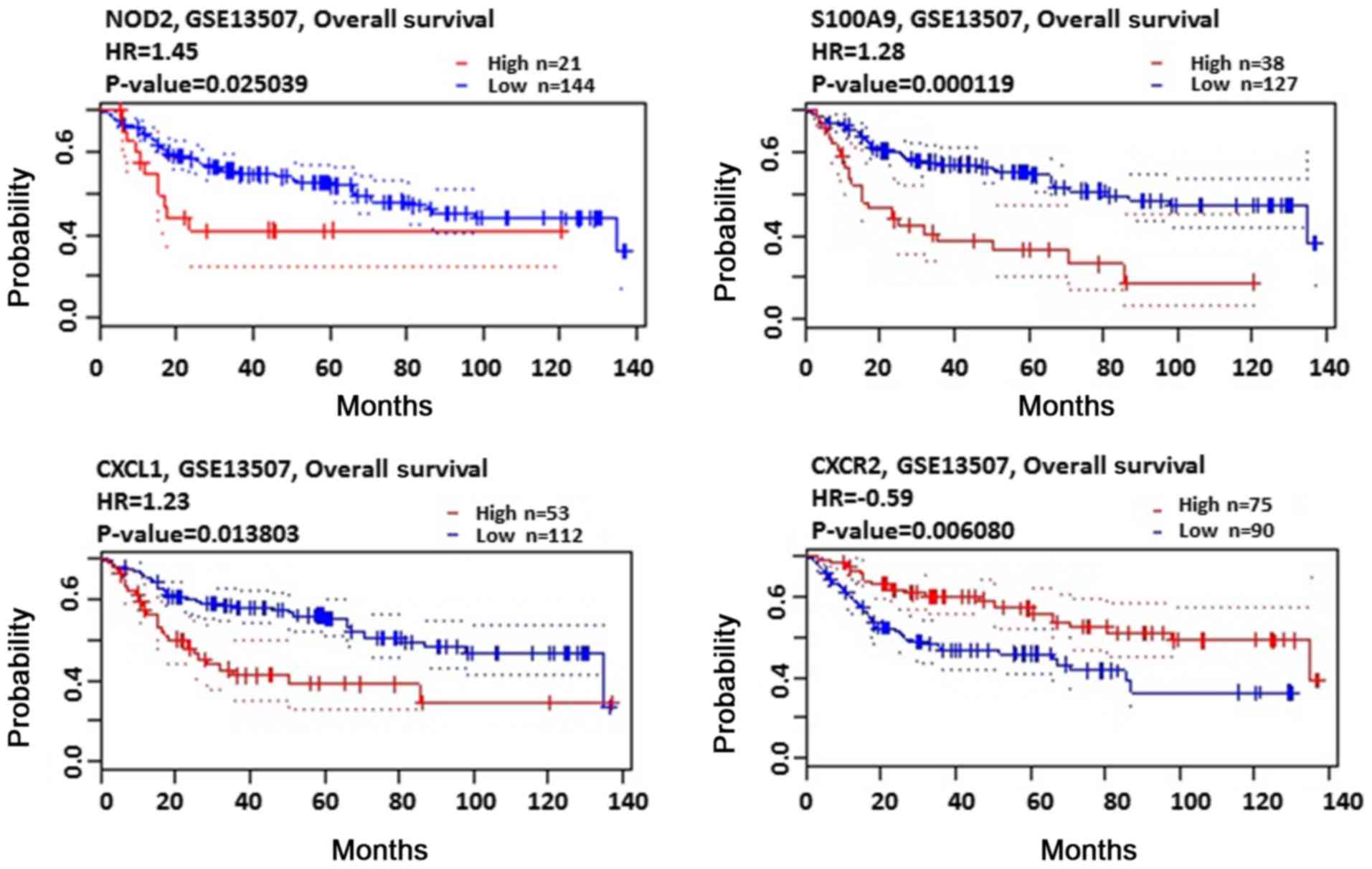
The PrognoScan database was used to perform the
association analysis of mRNA expression and OS rate in patients
with BC. As presented in Table III
and Fig. 8, the high expression of
NOD2 (P<0.05), S100A9 (P<0.001), CXCL1 (P<0.05) and CXCR2
(P<0.01) were significantly associated with a poor prognosis in
patients with BC.
| Table III.Association between mRNA expression
of enriched genes and overall survival in patients with bladder
cancer. |
Table III.
Association between mRNA expression
of enriched genes and overall survival in patients with bladder
cancer.
| Gene name | End point | PROBE ID | n | Cox P-value | Hazard ratio |
|---|
| CCL5 | Overall survival
rate | ILMN_1773352 | 165 | 0.112459 | 1.2 |
| SLPI | Overall survival
rate | ILMN_1669650 | 165 | 0.225693 | 1.09 |
| S100A9 | Overall survival
rate | ILMN_1714991 | 165 | 0.000119 | 1.28 |
| CXCL1 | Overall survival
rate | ILMN_1787897 | 165 | 0.013803 | 1.23 |
| TNFRSF1A | Overall survival
rate | ILMN_1685005 | 165 | 0.732444 | 1.08 |
| CCL20 | Overall survival
rate | ILMN_1657234 | 165 | 0.636372 | 0.95 |
| MMP1 | Overall survival
rate | ILMN_1726448 | 165 | 0.279756 | 1.07 |
| NOD2 | Overall survival
rate | ILMN_1762594 | 165 | 0.025039 | 1.45 |
| IL1B | Overall survival
rate | ILMN_1775501 | 165 | 0.374688 | 1.10 |
| CXCR2 | Overall survival
rate | ILMN_1783085 | 165 | 0.006080 | 0.59 |
| CD83 | Overall survival
rate | ILMN_1780582 | 165 | 0.169956 | 1.27 |
| MMP1 | Overall survival
rate | ILMN_1796316 | 165 | 0.114052 | 1.15 |
Discussion
BC is the ninth most common genitourinary
malignancy, globally and resulted in 165,000 mortalities in 2012
(31–33). Understanding the molecular mechanism
of BC is of great importance for diagnosis and treatment. Due to
well-developed microarray and high-throughput sequencing
technology, it is now easier to determine the general genetic
alterations in the progression of diseases, and has been widely
adopted to predict potential diagnosis and therapeutic targets for
BC (34).
In the present study, data were extracted from the
GSE31189 dataset and 457 upregulated and 519 downregulated DEGs
between BC and normal control specimens were identified using
bioinformatics analysis. The upregulated genes were enriched in the
negative regulation of apoptotic processes and the positive
regulation of cell division, while the downregulated genes were
mainly involved in cell proliferation, bicellular tight junction,
mitochondrial matrix and immune responses. Furthermore, by
constructing the PPI, a number of key genes were identified that
may be useful in future therapeutic studies on BC. Notably, key
nodes in the PPI network and genes in the significant modules,
including NOD2, S100A9, CXCL1 and CXCR2, may have
specific contributions to the occurrence and development of BC.
The NOD2 gene, a member of the evolutionarily
conserved Nod-like receptors family, is located on chromosome 16q21
(35,36). Wang et al revealed that the
abnormal expression of NOD2 was highly expressed in primary liver
tumor types, which was associated with a shorter median survival
time (36). In addition, in the
present study, a higher NOD2 mRNA expression was identified in BC,
which was associated with a shorter OS rate.
S100A9, a member of the S100 family of
calcium-binding proteins, is primarily detected in neutrophil
granulocytes and known to serve a function in the innate immune
system (37,38). Previous studies have demonstrated that
the abnormal overexpression of S100A9 is an unfavorable prognostic
factor for carcinogenesis and prognosis in various neoplasms, such
as hypopharyngeal and bladder cancer (39–43).
Additionally, S100A9 is associated with colorectal carcinoma
progression and contributes to colorectal carcinoma cell survival
and migration via the Wnt/β-catenin pathway (37). In the present study, S100A9 was
revealed to be differentially expressed in BC and non-cancerous
urothelial cells and was identified as a key node in the PPI
network constructed using DEGs between these two groups. The
results additionally demonstrated that a high S100A9
expression was associated with a shorter OS rate. Therefore, this
gene may be an essential marker for the diagnosis and prognosis of
BC.
CXCL1, a member of the CXC chemokine family, was
originally characterized by Wang et al (44) and is known to promote the
proliferation of melanoma cells. Previous studies have demonstrated
that CXCL1 may be associated with tumor epithelial-stromal
interactions that facilitate tumor growth and invasion (45–47). In
addition, Wang et al (48)
revealed that CXCL1 derived from tumor-associated lymphatic
endothelial cells drives gastric cancer cells into the lymphatic
system by activating integrin β1/focal adhesion kinase/protein
kinase B (Akt) signaling. Furthermore, Kawanishi et al
(49) revealed that CXCL1 may
modulate the invasive abilities of BC cells, and therefore is a
potential candidate biomarker and therapeutic target for invasive
BC. In the present study, a higher CXCL1 mRNA expression was
identified in patients with BC in the GSE31189 gene expression
profile. Furthermore, PrognoScan analysis results revealed that
high CXCL1 expression was significantly associated with a shorter
OS rate. Due to these findings, the expression level of CXCL1 may
be a useful prognostic marker of BC.
CXCR2, the co-receptor of IL-8 and CXCL1, is an
important therapeutic target in a number of solid tumor types,
including lung, breast, prostate, ovarian, colorectal and liver
cancer (50–55). Xu et al (56) revealed that CXCR2 may promote breast
cancer metastasis and chemoresistance via the suppression of AKT
serine/threonine kinase 1 and activation of cyclooxygenase 2.
Inhibition of CXCR2 may reduce the activity of breast cancer stem
cells and improve the survival of human epidermal growth factor
receptor 2 (HER2)-positive patients in combination with HER2-target
chemotherapies (57). Furthermore,
Gao et al (58) demonstrated
that the CXCL5/CXCR2 axis may promote BC cell migration and
invasion by activating the phosphoinositide 3-kinase/Akt-induced
upregulation of MMP2/MMP9. In the present study, CXCR2 was
increased in BC compared with normal specimens, similar with the
previous studies. However, the BC patients with higher mRNA levels
of CXCR2 were predicted to have a better OS rate. As few studies
have focused on CXCR2, the underlying function of CXCR2 requires
further research.
Altogether, the DEGs identified in the BC urothelial
cells when compared with the normal controls may be involved in
tumorigenesis. The key nodes identified in the PPI network
constructed with these DEGs and genes involved in the significant
module, including NOD2, S100A9 and CXCL1, may be
important in the development of BC, and may provide valuable clues
in order to investigate the pathogenesis of BC. However, further
biological experimental evidence is required in order to confirm
the function of the identified gene in BC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Project of Shanxi Provincial Department of Health (grant
no. 201601070).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XJ and XC participated in the design of the present
study, performed the statistical analysis and drafted the
manuscript. MY and YL performed the study and collected background
information and data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DEG
|
differentially expressed gene
|
|
GO
|
gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
PPI
|
protein-protein interaction
|
References
|
1
|
Hao L, Zhao Y, Li ZG, He HG, Liang Q,
Zhang ZG, Shi ZD, Zhang PY and Han CH: Tumor necrosis
factor-related apoptosis-inducing ligand inhibits proliferation and
induces apoptosis of prostate and bladder cancer cells. Oncol Lett.
13:3638–3640. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li X, Ma X, Tang L, Wang B, Chen L, Zhang
F and Zhang X: Prognostic value of neutrophil-to-lymphocyte ratio
in urothelial carcinoma of the upper urinary tract and bladder: A
systematic review and meta-analys. Oncotarget. 8:62681–62692.
2016.PubMed/NCBI
|
|
3
|
Peng M, Huang Y, Tao T, Peng CY, Su Q, Xu
W, Darko KO, Tao X and Yang X: Metformin and gefitinib cooperate to
inhibit bladder cancer growth via both AMPK and EGFR pathways
joining at Akt and Erk. Sci Rep. 6:286112016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim HJ, Eoh KJ, Kim LK, Nam EJ, Yoon SO,
Kim KH, Lee JK, Kim SW and Kim YT: The long noncoding RNA HOXA11
antisense induces tumor progression and stemness maintenance in
cervical cancer. Oncotarget. 7:83001–83016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo G, Wang M, Wu X, Tao D, Xiao X, Wang
L, Min F, Zeng F and Jiang G: Long non-coding RNA meg3 inhibits
cell proliferation and induces apoptosis in prostate cancer. Cell
Physiol Biochem. 37:2209–2220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang S, Qing C, Huang Z and Zhu Y: The
long non-coding RNA CCAT2 is up-regulated in ovarian cancer and
associated with poor prognosis. Diagn Pathol. 11:492016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui X, Jing X, Long C, Yi Q, Tian J and
Zhu J: Accuracy of the urine UCA1 for diagnosis of bladder cancer:
A meta-analysis. Oncotarget. 8:35222–35233. 2017.PubMed/NCBI
|
|
8
|
Cui X, Jing X and Wu X: The prognostic
value of long non coding RNAs in cervical cancer: A meta-analysis.
Oncotarget. 8:62470–62477. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jing X, Liang H, Cui X, Han C, Hao C and
Huo K: Long noncoding RNA CCAT2 can predict metastasis and a poor
prognosis: A meta-analysis. Clin Chim Acta. 468:159–165. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cui X, Jing X, Yi Q, Long C, Tan B, Li X,
Chen X, Huang Y, Xiang Z, Tian J and Zhu J: Systematic analysis of
gene expression alterations and clinical outcomes of STAT3 in
cancer. Oncotarget. 9:3198–3213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cui X, Jing X, Yi Q, Long C, Tian J and
Zhu J: Clinicopathological and prognostic significance of SDC1
overexpression in breast cancer. Oncotarget. 8:111444–111455. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo Y, Sheng Q, Li J, Ye F, Samuels DC and
Shyr Y: Large scale comparison of gene expression levels by
microarrays and RNAseq using TCGA data. PLoS One. 8:e714622013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Senchenko VN, Kisseljova NP, Ivanova TA,
Dmitriev AA, Krasnov GS, Kudryavtseva AV, Panasenko GV, Tsitrin EB,
Lerman MI, Kisseljov FL, et al: Novel tumor suppressor candidates
on chromosome 3 revealed by NotI-microarrays in cervical cancer.
Epigenetics. 8:409–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen G, Li Y, Su Y, Zhou L, Zhang H, Shen
Q, Du C, Li H, Wen Z, Xia Y and Tang W: Identification of candidate
genes for necrotizing enterocolitis based on microarray data. Gene.
661:152–159. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shu B, Fang Y, He W, Yang J and Dai C:
Identification of macrophage-related candidate genes in lupus
nephritis using bioinformatics analysis. Cell Signal. 46:43–51.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao K, Shen LS, Li HH, Huang S and Zhang
Y: Systematic-analysis of mRNA expression profiles in skeletal
muscle of patients with type II diabetes: The glucocorticoid was
central in pathogenesis. J Cell Physiol. 233:4068–4076. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen J, Ding J, Wang Z, Zhu J, Wang X and
Du J: Identification of downstream metastasis-associated target
genes regulated by LSD1 in colon cancer cells. Oncotarget.
8:19609–19630. 2017.PubMed/NCBI
|
|
18
|
Urquidi V, Goodison S, Cai Y, Sun Y and
Rosser CJ: A candidate molecular biomarker panel for the detection
of bladder cancer. Cancer Epidemiol Biomarkers Prev. 21:2149–2158.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carvalho BS and Irizarry RA: A framework
for oligonucleotide microarray preprocessing. Bioinformatics.
26:2363–2367. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gene Ontology Consortium, . The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34:(Database
Issue). D322–D326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al: The Gene Ontology (GO) database and informatics resource.
Nucleic Acids Res. 32:(Database Issue). D258–D261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T
and Yamanishi Y: KEGG for linking genomes to life and the
environment. Nucleic Acids Res. 36:(Database Issue). D480–D484.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wishart DS, Knox C, Guo AC, Shrivastava S,
Hassanali M, Stothard P, Chang Z and Woolsey J: DrugBank: A
comprehensive resource for in silico drug discovery and
exploration. Nucleic Acids Res. 34:(Database Issue). D668–D672.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cui X, Jing X, Long C, Tian J and Zhu J:
Long noncoding RNA MEG3, a potential novel biomarker to predict the
clinical outcome of cancer patients: A meta-analysis. Oncotarget.
8:19049–19056. 2017.PubMed/NCBI
|
|
32
|
Gnesin S, Mitsakis P, Cicone F, Deshayes
E, Dunet V, Gallino AF, Kosinski M, Baechler S, Buchegger F, Viertl
D and Prior JO: First in-human radiation dosimetry of
68Ga-NODAGA-RGDyK. EJNMMI Res. 7:432017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cohen T, Ricchiuti D and Memo M: Bladder
cancer that metastasized to the skin: A unique presentation that
signifies poor prognosis. Rev Urol. 19:67–71. 2017.PubMed/NCBI
|
|
34
|
Zhang Y, Fang L, Zang Y and Xu Z:
Identification of core genes and key pathways via integrated
analysis of gene expression and DNA methylation profiles in bladder
cancer. Med Sci Monit. 24:3024–3033. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu J, He C, Xu Q, Xing C and Yuan Y: NOD2
polymorphisms associated with cancer risk: A meta-analysis. PLoS
One. 9:e893402014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X, Yang C, Liao X, Han C, Yu T, Huang
K, Yu L, Qin W, Zhu G, Su H, et al: NLRC and NLRX gene family mRNA
expression and prognostic value in hepatocellular carcinoma. Cancer
Med. 6:2660–2672. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duan L, Wu R, Ye L, Wang H, Yang X, Zhang
Y, Chen X, Zuo G, Zhang Y, Weng Y, et al: S100A8 and S100A9 are
associated with colorectal carcinoma progression and contribute to
colorectal carcinoma cell survival and migration via Wnt/β-catenin
pathway. PLoS One. 8:e620922013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Källberg E, Vogl T, Liberg D, Olsson A,
Björk P, Wikström P, Bergh A, Roth J, Ivars F and Leanderson T:
S100A9 interaction with TLR4 promotes tumor growth. PLoS One.
7:e342072012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
De Veirman K, De Beule N, Maes K, Menu E,
De Bruyne E, De Raeve H, Fostier K, Moreaux J, Kassambara A, Hose
D, et al: Extracellular S100A9 protein in bone marrow supports
multiple myeloma survival by stimulating angiogenesis and cytokine
secretion. Cancer Immunol Res. 5:839–846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yasar O, Akcay T, Obek C and Turegun FA:
Significance of S100A8, S100A9 and calprotectin levels in bladder
cancer. Scand J Clin Lab Invest. 77:437–441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu P, Quan H, Kang J, He J, Luo S, Xie C,
Xu J, Tang Y and Zhao S: Downregulation of calcium-binding protein
S100A9 inhibits hypopharyngeal cancer cell proliferation and
invasion ability through inactivation of NF-κB signaling. Oncol
Res. 25:1479–1488. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang Q, Li X, Chen H, Cao Y, Xiao Q, He Y,
Wei J and Zhou J: IRF7 regulates the development of granulocytic
myeloid-derived suppressor cells through S100A9 transrepression in
cancer. Oncogene. 36:2969–2980. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lim SY, Yuzhalin AE, Gordon-Weeks AN and
Muschel RJ: Tumor-infiltrating monocytes/macrophages promote tumor
invasion and migration by upregulating S100A8 and S100A9 expression
in cancer cells. Oncogene. 35:5735–5745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang D, Yang W, Du J, Devalaraja MN, Liang
P, Matsumoto K, Tsubakimoto K, Endo T and Richmond A:
MGSA/GRO-mediated melanocyte transformation involves induction of
Ras expression. Oncogene. 19:4647–4659. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Verbeke H, Struyf S, Laureys G and Van
Damme J: The expression and role of CXC chemokines in colorectal
cancer. Cytokine Growth Factor Rev. 22:345–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miyake M, Lawton A, Goodison S, Urquidi V,
Gomes-Giacoia E, Zhang G, Ross S, Kim J and Rosser CJ: Chemokine
(C-X-C) ligand 1 (CXCL1) protein expression is increased in
aggressive bladder cancers. BMC Cancer. 13:3222013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Han KQ, Han H, He XQ, Wang L, Guo XD,
Zhang XM, Chen J, Zhu QG, Nian H, Zhai XF and Jiang MW: Chemokine
CXCL1 may serve as a potential molecular target for hepatocellular
carcinoma. Cancer Med. 5:2861–2871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Z, Wang Z, Li G, Wu H, Sun K, Chen J,
Feng Y, Chen C, Cai S, Xu J and He Y: CXCL1 from tumor-associated
lymphatic endothelial cells drives gastric cancer cell into
lymphatic system via activating integrin β1/FAK/AKT signaling.
Cancer Lett. 385:28–38. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kawanishi H, Matsui Y, Ito M, Watanabe J,
Takahashi T, Nishizawa K, Nishiyama H, Kamoto T, Mikami Y, Tanaka
Y, et al: Secreted CXCL1 is a potential mediator and marker of the
tumor invasion of bladder cancer. Clin Cancer Res. 14:2579–2587.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cong L, Qiu ZY, Zhao Y, Wang WB, Wang CX,
Shen HC and Han JQ: Loss of β-arrestin-2 and activation of CXCR2
correlate with lymph node metastasis in non-small cell lung cancer.
J Cancer. 8:2785–2792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Devapatla B, Sharma A and Woo S: CXCR2
inhibition combined with sorafenib improved antitumor and
antiangiogenic response in preclinical models of ovarian cancer.
PLoS One. 10:e01392372015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang T, Tseng C, Zhang Y, Sirin O, Corn
PG, Li-Ning-Tapia EM, Troncoso P, Davis J, Pettaway C, Ward J, et
al: CXCL1 mediates obesity-associated adipose stromal cell
trafficking and function in the tumour microenvironment. Nat
Commun. 7:116742016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ignacio RM, Kabir SM, Lee ES, Adunyah SE
and Son DS: NF-κB-mediated CCL20 reigns dominantly in CXCR2-driven
ovarian cancer progression. PLoS One. 11:e01641892016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao J, Ou B, Feng H, Wang P, Yin S, Zhu
C, Wang S, Chen C, Zheng M, Zong Y, et al: Overexpression of CXCR2
predicts poor prognosis in patients with colorectal cancer.
Oncotarget. 8:28442–28454. 2017.PubMed/NCBI
|
|
55
|
Ding D, Zhang Y, Yang R, Wang X, Ji G, Huo
L, Shao Z and Li X: miR-940 suppresses tumor cell invasion and
migration via regulation of CXCR2 in hepatocellular carcinoma.
Biomed Res Int. 2016:76183422016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xu H, Lin F, Wang Z, Yang L, Meng J, Ou Z,
Shao Z, Di G and Yang G: CXCR2 promotes breast cancer metastasis
and chemoresistance via suppression of AKT1 and activation of COX2.
Cancer Lett. 412:69–80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Singh JK, Farnie G, Bundred NJ, Simões BM,
Shergill A, Landberg G, Howell SJ and Clarke RB: Targeting CXCR1/2
significantly reduces breast cancer stem cell activity and
increases the efficacy of inhibiting HER2 via HER2-dependent and
-independent mechanisms. Clin Cancer Res. 19:643–656. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gao Y, Guan Z, Chen J, Xie H, Yang Z, Fan
J, Wang X and Li L: CXCL5/CXCR2 axis promotes bladder cancer cell
migration and invasion by activating PI3K/AKT-induced upregulation
of MMP2/MMP9. Int J Oncol. 47:690–700. 2015. View Article : Google Scholar : PubMed/NCBI
|