Introduction
Hereditary breast cancer (HBC) is an autosomal
dominant familial and early-onset breast cancer syndrome manifested
by a gradual and exponential risk of developing breast and ovarian
cancer. HBC is primarily caused by mutations in the BRCA1
and BRCA2 genes. The BRCA1 protein has a major function in
the DNA repair system (1). The
present study investigated a 33-year-old Chinese female patient
with HBC using targeted next generation sequencing (NGS). Through
genetic testing, one deletion-insertion mutation
(c.311_312delinsAGGTTTGCA) in the BRCA1 gene was detected,
and it was revealed that this was a typical family of inherited
tumors following detection of the highest likelihood of mutation in
the proband's family members. The proband's uncle and uncle's
daughter carry the same mutation as the proband and therefore, it
was considered that the proband's father, who had succumbed, and
the proband's aunt, who was diagnosed with triple-negative breast
cancer (TNBC), may exhibit the same mutation. This
deletion-insertion mutation (c.311_312delinsAGGTTTGCA) causes the
formation of a truncated BRCA1 protein of 109 amino acids instead
of a wild type BRCA1 protein of 1863 amino acids. Hence, this
mutation is a loss-of-function mutation.
Case report
The proband is a 33-year-old Chinese female from
non-consanguineous parents. The proband was diagnosed with breast
cancer and enrolled in the present study. Clinical diagnosis was
based on the patient's clinical history. The diagnosis for the
patient, TNBC, was supported by her previous clinical information.
The present study was performed in the Department of Internal
Medicine, The Fourth Hospital of Hebei Medical University
(Shijiazhuang, China).
In February 2015, a mass ~2 cm in diameter was
identified in the patient's left breast. No other abnormalities
were identified during physical examination. The laboratory
examination results were within the normal range. Breast-conserving
surgery and sentinel lymph node biopsy revealed that the left
breast tumor was 1.8 cm in diameter; the breast metaplastic cancer
lacked vascular tumor thrombus metastasis of the sentinel lymph
nodes with negative margins. H&E staining is presented in
Fig. 1A-C, with the following
immunohistochemistry: Estrogen receptor (ER), 0; progesterone
receptor (PR), 0; human epidermal growth factor 2 (HER2), 0; tumor
protein p53 (P53), 60%; Ki-67, 50%; type II topoisomerase (TOMOII),
30%; creatine kinase (CK), +; vimentin (Vim), +; S-100 calcium
binding protein (S-100), +; synuclein (Syn), -; and cluster of
differentiation 56 (CD56), −/+, belonging to TNBC, as shown in the
Fig. 1D-N. The post-operative staging
was IA (T1cN0M0) (2). Pathology
revealed invasive cancer of the left breast with an extensive area
of necrosis. The patient was treated with paclitaxel (90–120 mg/m2)
intravenously for 12 weeks, followed by cyclophosphamide treatment
(200 mg/m2) for 12 weeks.
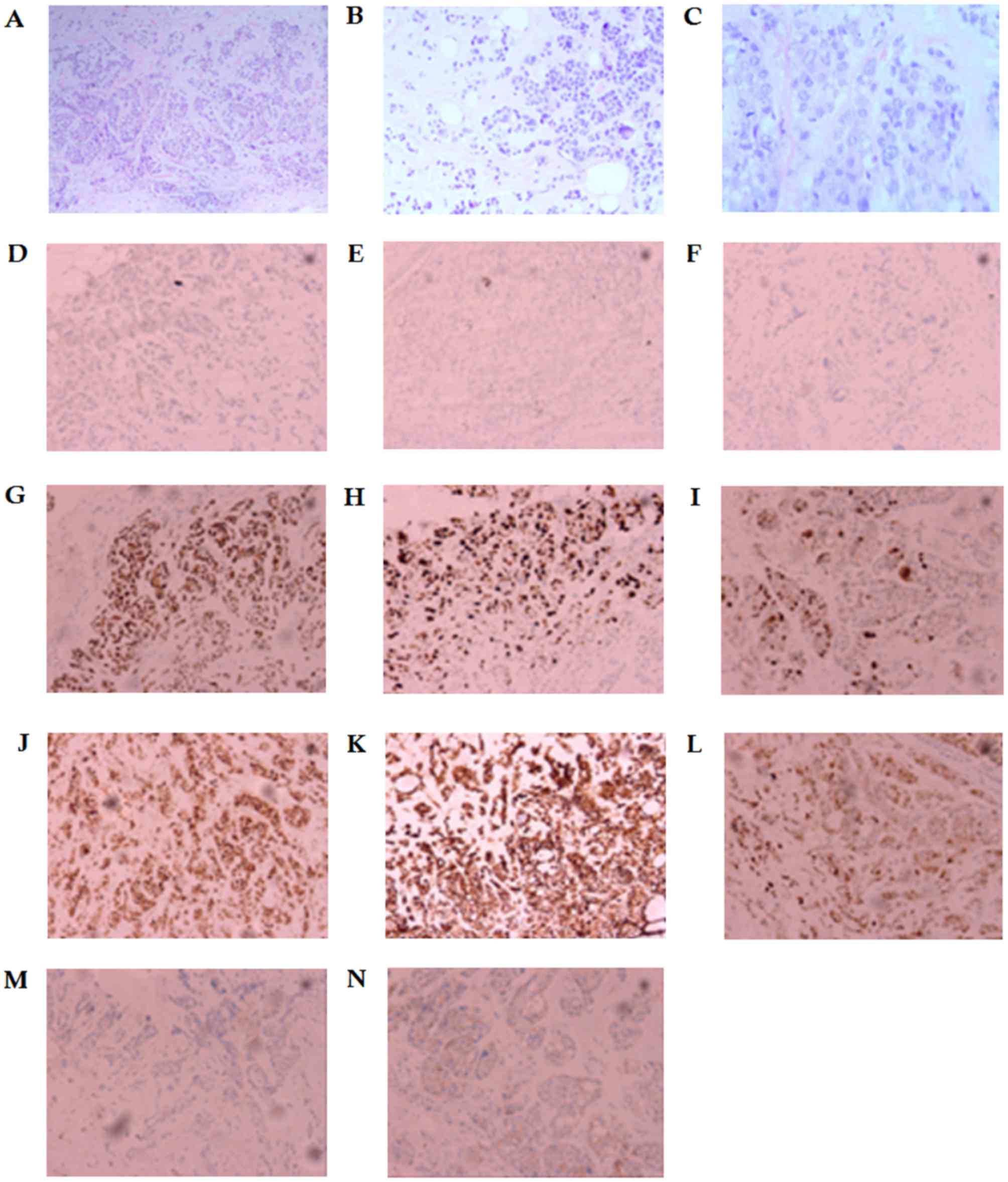 | Figure 1.Immunostaining images. H&E
staining for tumor tissues. (A) Scale bar=100 µm, (B) scale bar=50
µm and (C) scale bar=25 µm. Immunohistochemistry for (D) ER
(negative), (E) PR (negative), (F) HER2 (negative), (G) P53 (60%
positive immunoexpression), (H) Ki-67 (50% positive
immunoexpression), (I) TOPOII (30% positive immunoexpression), (J)
CK(+), (K) Vim(+), (L) S-100(+), (M) Syn(−), and (N) CD56(−/+).
Scale bar=50 µm. ER, estrogen receptor; PR, progesterone receptor;
HER2, human epidermal growth factor receptor 2; P53, tumor protein
p53; TOPOII, type II topoisomerase; CK, creatine kinase; Vim,
vimentin; S-100, S-100 calcium binding protein; Syn, synuclein;
CD56, cluster of differentiation 56. |
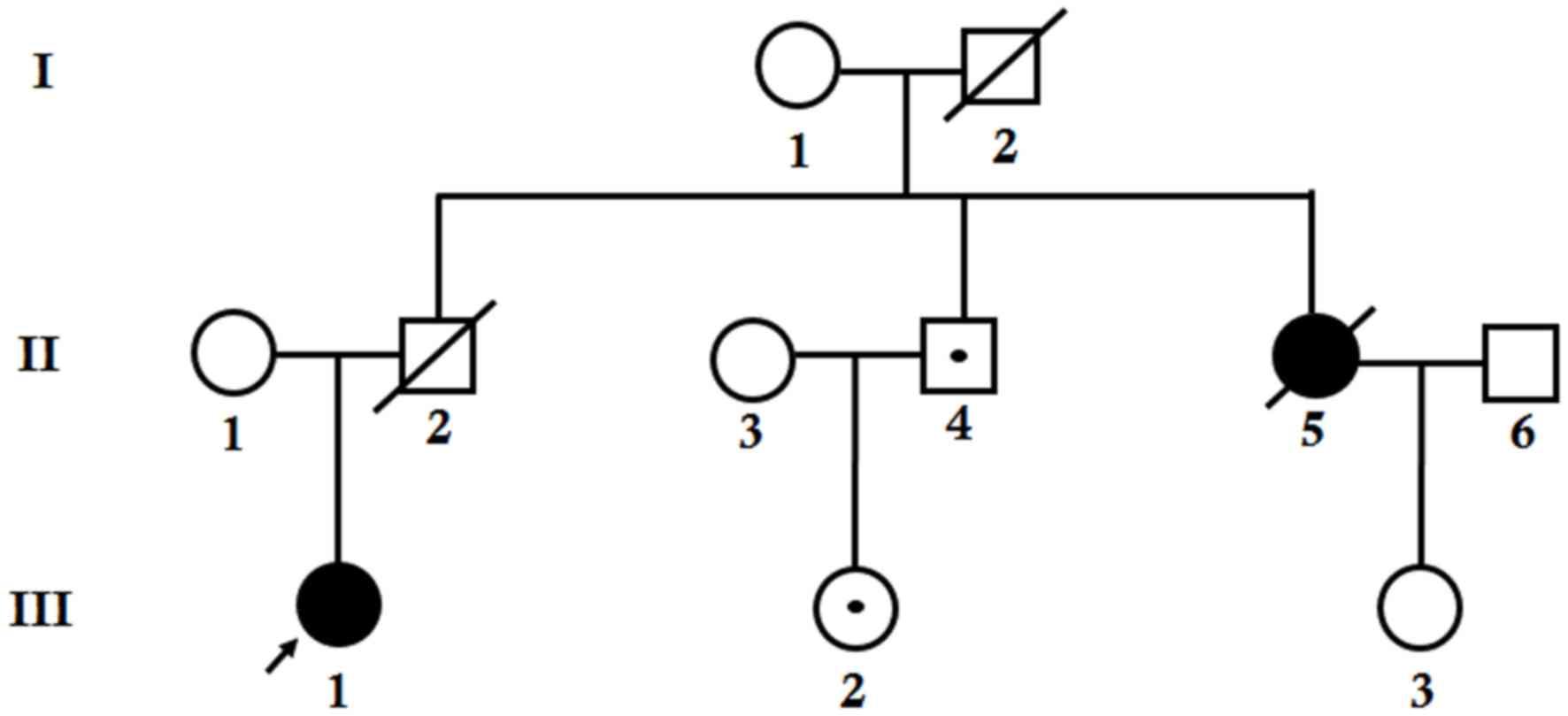
The proband was clinically diagnosed with TNBC at
the age of 31 (Fig. 1O, patient
III-1) and underwent genetic testing at the age of 33. One mutation
in the BRCA1 gene was detected. As detailed knowledge of the
proband's family history was acquired, it was revealed that the
proband's father and grandfather were deceased (Fig. 2, I-2 and II-2, respectively); the
cause and age of mortality were unknown. The proband's mother was
>50 years of age and healthy (Fig.
2, II-1). The proband's aunt was diagnosed with TNBC at the age
of 47 and succumbed at the age of 50 (Fig. 2, II-5). The pathological type of the
proband was the same as that of her aunt. In addition, the proband
has a healthy uncle (Fig. 2,
II-4).
Prior to performing pedigree verification, it was
inferred that there are several hypotheses: i) The proband's aunt
and father each carried an identical mutation in the BRCA1
gene, which was inherited from proband's grandfather or
grandmother; in this situation, the proband's uncle has a 50%
probability of also carrying this mutation; ii) the mutation
exhibited by the proband was inherited from her mother, who was a
carrier that did not suffer from cancer; in which case, the
proband's aunt who had cancer might have been a sporadic case; or
she has other mutations iii) the mutation exhibited by the proband
was not inherited from her parents, but was de novo; the
proband's aunt's breast cancer is not associated with this
mutation.
As the grandmother of the proband (Fig. 2, I-1) refused to undergo genetic
testing, and the proband's aunt and father were already deceased,
samples from the proband's mother were tested initially. It was
revealed that the mother of the proband did not carry any mutations
in the BRCA1 gene and therefore, the first hypothesis was
highly suspected. Next, verification was performed with the
proband's uncle, the proband's uncle's daughter (Fig. 2, III-2), and the proband's aunt's
daughter (Fig. 2, III-3). It was
revealed that the proband's uncle and the proband's uncle's
daughter carry the same mutation as the proband. This result
confirms the first hypothesis. The uncle of the proband was 54
years old, the proband's uncle's daughter was 28 years old, and the
two of them were healthy. A risk management scheme was provided for
them to facilitate early cancer detection, prevention and risk of
developing the disease in following generations.
Diagnosis and treatment
In March 26, 2015, the proband received paclitaxel
liposome (180 mg, intravenously, day 1). Due to the presence of
proteinuria, chemotherapy was suspended and radiotherapy was
administered. At the end-point of the study (July 2017), no tumor
recurrence had been observed.
Following written informed consent being obtained, a
sample of the proband's peripheral blood was collected and genomic
DNA was extracted. Targeted NGS was performed with a panel of 21
genes (BRCA1, BRCA2, CHEK2, PALB2, BRIP1, TP53, PTEN, STK11,
CDH1, ATM, BARD1, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PMS1, PMS2,
RAD50, RAD51C) with a target area of 172,959 bp. The coverage
and the depth of the target area was 99.49% and 586.13×
respectively.
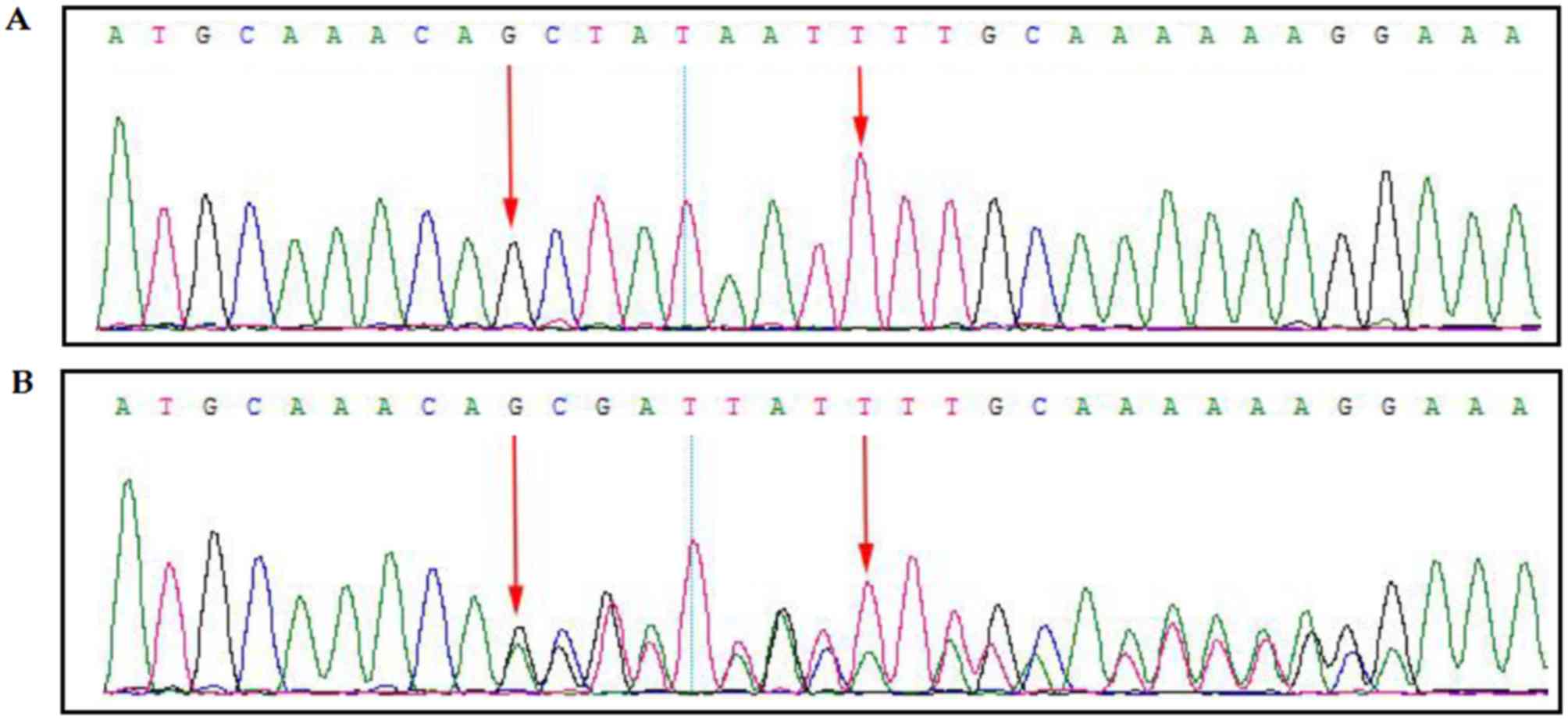
A novel heterozygous mutation
(c.311_312delinsAGGTTTGCA) was identified in the proband.
MutationTaster version 2 (http://www.mutationtaster.org/) predicted that this
mutation causes the formation of a premature stop codon which leads
to a frameshift followed by the formation of a truncated BRCA1
protein (3). This mutation was
confirmed by Sanger sequencing (Fig. 3A
and B); Fig. 3A presents the
wild-type result, while Fig. 3B
presents the proband's result, which revealed the mutation. This
mutation was not present in the Human Gene Mutation database
(www.hgmd.cf.ac.uk) or OMIM (omim.org). The mutation was not identified in the BGI
database, which contains ~30,000 Chinese Han samples (www.genomics.cn/en/).
Discussion
The present study investigated a 33-year-old Chinese
female with HBC. A heterozygous novel heterozygous
deletion-insertion (c.311_312delinsAGGTTTGCA) in the BRCA1
gene was identified in the proband. This mutation causes formation
of a truncated BRCA1 protein with 109 amino acids instead of the
wild-type of the BRCA1 protein with 1,863 amino acids.
Genetic screening for BRCA1 and BRCA2
genes and identification of novel variants serves a key role for
timely diagnosis, proper counseling, successive follow-up and
management of disease (4). By
contrast, a large number of variants of unknown clinical
significance (VUS) in the BRCA1 and BRCA2 genes has
been identified in the patients and their family members by NGS.
Functional characterization of germ-line mutations at
transcriptional or translational levels is required to understand
the dominant negative effect of the mutation in functional
wild-type proteins (5).
BRCA1 gene mutations have a 60–80%
probability of causing breast cancer in females; these mutations
also increase the risk of developing ovarian cancer in females and
prostate cancer in males. Germ-line mutations in the BRCA2
gene are observed in ~35% of families with early-onset breast
cancer in females; these mutations lead to an increased risk of
developing ovarian cancer in females and breast cancer in males
(6). Breast cancer caused by a
mutation in the BRCA1 gene has a higher incidence, higher
mitotic rate and more lymphatic penetrance than sporadic breast
cancer (7). These types of cancer are
more likely to lack expression of ER, PR and HER-2nue receptors,
and to have a somatic mutation in the P53 gene (7–10). In
addition to having an increased risk of developing breast cancer,
BRCA1 or BRCA2 gene mutation carriers have an
increased risk for other types of cancer, including colon,
prostate, pancreatic, melanoma and gastric cancer (11,12). Novel
mutations in the BRCA1 and BRCA2 genes are very rare,
as 2,000 mutations have been discovered in these two genes so far.
The most common mutation forms are small insertions, small
deletions, nonsense mutations, missense mutations, premature
transcription terminations and splicing troubles. Deletion and
insertion mutations lead to a frame shift. According to the Breast
Information Core, the majority of the breast cancer-causing
mutations in the BRCA1 and BRCA2 genes lead to the
production of truncated protein through nonsense, frame shift and
splicing mutations (13).
However, BRCA1 and BRCA2 genes
associated with human breast and ovarian cancer occur with an
autosomal dominant mode of inheritance and late onset of age.
Therefore, genetic screening for patients and their family members
are the key factors for the proper clinical management, accurate
follow-up and understanding of the disease risk for all the family
members. Additionally, prenatal genetic screening, as well as
prenatal diagnosis for the family members with breast and ovarian
cancer, would be a great step in the future in order to reduce the
risk of disease occurrence in the successive generations in a
family with several patients with BRCA1/2-associated breast/ovarian
cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
SB, ZP, DJ designed and coordinated the study. DJ,
QZ, XZ, YC, JL and HJ assessed the clinical findings of the cases.
YW, HH, JW, KL, WC and JX performed the molecular genetic studies
and analyzed the data. YW and WC wrote the draft of the manuscript
with input from the other co-authors. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All study participants provided written informed
consent and the study design was approved by an Institutional
Ethics Review Board of the Department of Internal Medicine, The
Fourth Hospital of Hebei Medical University (Shijiazhuang,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Narod SA and Foulkes WD: BRCA1 and BRCA2:
1994 and beyond. Nat Rev Cancer. 4:665–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carlson RW, Goldstein LJ, Gradishar WJ,
Lichter AS, McCormick B, Moe RE and Theriault RL: NCCN breast
cancer practice guidelines. The National comprehensive cancer
network. Oncology (Williston Park). 10 11 Suppl:S47–S75. 1996.
|
|
3
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Richter S, Haroun I, Graham TC, Eisen A,
Kiss A and Warner E: Variants of unknown significance in BRCA
testing: Impact on risk perception, worry, prevention and
counseling. Ann Oncol. 24 Suppl 8:viii69–viii74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ward AJ and Cooper TA: The pathobiology of
splicing. J Pathol. 220:152–163. 2010.PubMed/NCBI
|
|
6
|
Ayub SG, Rasool S, Ayub T, Khan SN, Wani
KA and Andrabi KI: Mutational analysis of the BRCA2 gene in breast
carcinoma patients of Kashmiri descent. Mol Med Rep. 9:749–753.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Musolino A, Naldi N, Michiara M, Bella MA,
Zanelli P, Bortesi B, Capelletti M, Savi M, Neri TM and Ardizzoni
A: A breast cancer patient from Italy with germline mutations in
both the BRCA1 and BRCA2 genes. Breast Cancer Res Treat.
91:203–205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen W, Pan K, Ouyang T, Li J, Wang T, Fan
Z, Fan T, Lin B, Lu Y, You W and Xie Y: BRCA1 germline mutations
and tumor characteristics in Chinese women with familial or
early-onset breast cancer. Breast Cancer Res Treat. 117:55–60.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lakhani SR, Van De Vijver MJ, Jacquemier
J, Anderson TJ, Osin PP, McGuffog L and Easton DF: The pathology of
familial breast cancer: Predictive value of immunohistochemical
markers estrogen receptor, progesterone receptor, HER-2, and p53 in
patients with mutations in BRCA1 and BRCA2. J Clin Oncol.
20:2310–2318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Phillips KA: Immunophenotypic and
pathologic differences between BRCA1 and BRCA2 hereditary breast
cancers. J Clin Oncol. 18 21 Suppl:107S–112S. 2000.PubMed/NCBI
|
|
11
|
Jancárková N, Zikán M, Pohlreich P,
Freitag P, Matous B and Zivný J: Detection and occurrence of BRCA 1
gene mutation in patients with carcinoma of the breast and ovary.
Ceska Gynekol. 68:11–16. 2003.(In Czech). PubMed/NCBI
|
|
12
|
Dutil J, Colon-Colon JL, Matta JL, Sutphen
R and Echenique M: Identification of the prevalent BRCA1 and BRCA2
mutations in the female population of Puerto Rico. Cancer Genet.
205:242–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pohlreich P, Zikan M, Stribrna J, Kleibl
Z, Janatova M, Kotlas J, Zidovska J, Novotny J, Petruzelka L, Szabo
C and Matous B: High proportion of recurrent germline mutations in
the BRCA1 gene in breast and ovarian cancer patients from the
Prague area. Breast Cancer Res. 7:R728–R736. 2005. View Article : Google Scholar : PubMed/NCBI
|